
Abstract
The zoanthid Parazoanthus axinellae (Schmidt, 1862) is a widespread coral species in the Mediterranean coralligenous assemblages where two morphotypes are found: Slender and Stocky, differing in size, color, and preferred substrate. Due to these marked differences, Slender and Stocky morphotypes were hypothesized to be two species. Here, we used 2bRAD to obtain genome‐wide genotyped single nucleotide polymorphisms (SNPs) to investigate the genetic differentiation between Slender and Stocky morphs, as well as their population structure. A total of 101 specimens of P. axinellae were sampled and genotyped from eight locations along the Italian coastline. In four locations, samples of the two morphotypes were collected in sympatry. 2bRAD genome-wide SNPs were used to assess the genetic divergence between the two morphotypes (1319 SNPs), and population connectivity patterns within Slender (1926 SNPs) and Stocky (1871 SNPs) morphotypes. Marked and consistent differentiation was detected between Slender and Stocky morphotypes. The widely distributed Slender morphotype showed higher population mixing patterns, while populations of the Stocky morphotype exhibited a stronger genetic structure at a regional scale. The strong genetic differentiation observed between P. axinellae Slender and Stocky morphotypes provides additional evidence that these morphs could be attributed to different species, although further morphological and ecological studies are required to validate this hypothesis. Our study highlights the importance of resolving phylogenetic and taxonomic disparities within taxonomically problematic groups, such as the P. axinellae species complex, when performing genetic connectivity studies for management and conservation purposes.
Graphical Abstract
Schematic overview of the main genetic structuring patterns observed in this study. Coral polyps were colored to intuitively associate the reader to Parazoanthus axinellae morphotypes, with orange tones being attributed to the Stocky morphotype, and yellow tones to the Slender morphotype. Bidirectional arrows represent gene flow between coral individuals, with the number and thickness of arrows corresponding to the intensity of gene flow rates. The red dashed line represents the potential reproductive isolation between Slender and Stocky morphs

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetic connectivity data can provide valuable insights into the dynamics of population recovery and replenishment following environmental perturbation events. High connectivity potentially allows local populations to be replenished by immigration or reestablished by colonization (Padrón et al. 2018). Species that exhibit higher rates of gene flow and connectivity may also show greater resilience to environmental stress, as increased local genetic diversity (introduced via gene flow from adjacent populations) can increase the likelihood that some genotypes may adapt to local stressful conditions (Bernhardt and Leslie 2013). Hence, population connectivity data are often used as a proxy for population resilience and are crucial to predict the capacity of a species and/or communities to recover following disturbance events (Gatti et al. 2015; Schlacher et al. 2010).
Prior to the use of connectivity data for conservation, restoration, and management plans, it is essential to fully resolve the taxonomic status of the target species. This is needed because merging population genomics data of sibling species can introduce artifacts/biases in population connectivity models, which can lead to erroneous inferences and misleading conclusions about population resilience (Chenuil et al. 2019; Costantini et al. 2018; Cowen et al. 2007; Pante et al. 2015a, b). The zoanthid Parazoanthus axinellae (Schmidt, 1862), a cnidarian commonly present in the Mediterranean Sea and the North Atlantic Ocean, represents one of such taxonomically problematic groups. This zoanthid is classified within a species complex (Ocaña Vicente et al. 2019), and P. axinellae individuals have distinct (1) morphological characteristics and distribution patterns (Gili et al. 1987); (2) ecological requirements (Ocaña Vicente et al. 2019); (3) biochemical/metabolic profiles (Cachet et al. 2015); and (4) phylogenetic position within the family Parazoanthidae (Villamor et al. 2020). In detail, two distinct morphotypes are found within the P. axinealle species complex (Fig. 1): Slender – with an elongated trunk, longer and thinner tentacles, and a light-yellow color (hereinafter ‘Y’ to denote yellow); and Stocky – with a shorter, thicker trunk and tentacles, and orange color (hereinafter ‘O’ to denote orange) (Cachet et al. 2015; Villamor et al. 2020). The Slender morphotype is found throughout the Mediterranean Sea and in the North Atlantic Ocean, whereas the Stocky morphotype occurs only in the North-West Mediterranean (Gili et al. 1987). In terms of their ecology and life strategies, Slender individuals can be associated with axinellid sponges, whereas Stocky individuals only dwell on rocky substrates (Ocaña Vicente et al. 2019). The morphs also differ metabolically, and Cachet et al. (2015) identified the presence of ‘parazoanthines’ (highly bioactive secondary metabolites) within the Slender morphotype of P. axinellae, which are conversely absent from Stocky individuals. Recent phylogenetic studies examining sequence polymorphism of mitochondrial cytochrome C oxidase subunit I (COI) and nuclear internal transcribed spacer (ITS) gene markers revealed marked and consistent differentiation between Slender and Stocky morphotypes (Villamor et al. 2020), while high gene flow rates were observed within the morphotypes, with the more widespread Slender morph showing more pronounced population connectivity patterns (Villamor et al. 2020).

Morphological differences between a the ‘Slender’ morphotype of Parazoanthus axinellae (Photo: C. Cerrano) and b the ‘Stocky’ morphotype of Parazoanthus axinellae (Photo: C. Cerrano)
This study builds upon the previous work by Villamor et al. (2020) and aims to obtain a deeper understanding of genetic divergence between and within Slender and Stocky morphotypes, by utilizing genome-wide genotyping data. High-throughput shotgun sequencing methods allow for more robust inferences of phylogenetic and population genomic structuring compared to traditional genetic markers—as utilized in Villamor et al. (2020)—due to the greater number of single nucleotide polymorphisms (SNPs) retrieved, typically measured in hundreds or thousands (Everett et al. 2016; Hodel et al. 2017; Reitzel et al. 2013; Schopen et al. 2008). In recent years, next-generation sequencing methods like restriction site associated DNA sequencing (RAD-seq) (Baird et al. 2008; Davey and Blaxter 2010) have been increasingly utilized to investigate species delimitation in corals (Bongaerts et al. 2021; Pante et al. 2015a, b; Quattrini et al. 2019), and this approach fully resolved species boundaries within e.g., (1) the common scleractinian belonging to the Pocillopora genus (Johnston et al. 2017); (2) the deep-sea octocoral Chrysogorgia (Pante et al. 2015a, b); (3) Paragorgia (Herrera and Shank 2016); and (4) Sinularia (Quattrini et al. 2019) genera. While RAD-seq has been successfully applied across numerous coral groups, this method has not been utilized to evaluate cryptic genetic diversity in zoanthid species.
In this study, we obtained 2bRAD-Seq data (methodology for 2bRAD described in Wang et al. 2012) for individuals of the zoanthid P. axinellae to investigate population connectivity patterns among populations of Slender and Stocky morphotypes sampled at eight localities along the Italian coastline. Specifically, the main aims of this study were (1) to determine the genetic differentiation between the two morphotypes; (2) to investigate population connectivity patterns within Slender and Stocky morphotypes; and (3) to propose the use of the obtained population connectivity data for management and conservation purposes of P. axinellae.
Methods
Field sampling
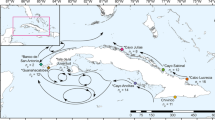
Samples from 12 sampling populations were collected by SCUBA diving at eight localities in the Mediterranean Sea (Fig. 2a), from June 2013 to August 2017. This included three Ligurian locations, from West to East: (1) Alassio (ALA), (2) Portofino (PTF), and (3) Porto Venere (PVEN); three Tyrrhenian locations, from West to East: (4) Olbia, Sardinia (SAR), (5) Giannutri, Tuscany (GIA), and (6) Punta Campanella, Campania (CAM); one location in the northern Adriatic Sea (7) Grado, Friuli (GRA), and one location in the Ionian Sea (8) Gallipoli, Puglia (PUG) (Table 1). Slender individuals were collected from all eight localities, while Stocky individuals (marked with ‘OR’ in the sampling locality code in Table 1) were found in four locations, always in sympatry with the Slender morphotype. Polyps were sampled at least 2 m apart to avoid sampling of clones from the same colony. A total of 68 Slender and 33 Stocky individuals were collected between 10 and 35 m depth (Table 1). All samples were immediately fixed in 80% ethanol and refrigerated at 4 °C until genetic analysis. Field and experimental protocols were approved by the University of Bologna, Italy, and were performed in accordance with its relevant guidelines and regulations. No permit was required for the collection of the specimens.
Genomic DNA extraction, 2bRAD library preparation, and raw data processing
Total genomic DNA (gDNA) was extracted from 101 P. axinellae polyps following a CTAB-based DNA extraction procedure (Winnepenninckx et al. 1993). The quality of gDNA extracts was evaluated on a 0.8% agarose gel stained with GelRed (Invitrogen). DNA was quantified using Qubit® dsDNA BR Assay Kit (ThermoFisher Scientific) to ensure sufficient DNA quantity before 2bRAD library preparation. Genome-wide SNPs were obtained using the 2bRAD genotyping approach (Wang et al. 2012), using modified protocols from Terzin et al. (2021). Briefly, approximately 200 ng of gDNA was digested into uniform 32 bp fragments using the restriction enzyme CspCI (New England BioLabs). DNA digests were then ligated to library-specific adapters and amplified using sample-specific dual barcodes and Illumina adaptors. PCR products were visualized on a 2.0% agarose gel to verify the presence of the expected 160–170 bp target band (i.e., fragment, barcodes, and adaptors included). The target band was excised from the gel and purified using Macherey–Nagel™ NucleoSpin™ Gel and PCR Clean-up Kit (ThermoFischer), following manufacturer protocol. Purified libraries were quantified using Qubit® dsDNA BR Assay Kit and pooled at equimolar concentrations. Pooled libraries were sequenced and demultiplexed by Genomix4Life S.r.l. (Baronissi, Salerno, Italy) on an Illumina HiSeq platform with a single-end 50 bp read module (SR50 High Output mode). To control for potential erroneous variants from library preparation and sequencing, 34 random specimens were sequenced in duplicate to serve as technical replicates for comparison of libraries.
Data analysis – hierarchical design
All bioinformatics analyses were conducted at three levels, using the following hierarchical approach: (1) between ‘Slender and Stocky’ morphotypes (‘Y&O’ level), and within (2) Slender (‘Y’ level) and (3) Stocky (‘O’ level) morphotypes. We applied this hierarchical approach because previous studies found instances where the signal from genetically divergent populations can ‘mask’ fine-scale populations variability (Carreras et al. 2020; Carreras-Carbonell et al. 2006).
Data analysis – quality-filtering and trimming of raw reads
Demultiplexed raw reads were initially checked for various sequence quality metrics using FastQC (v0.11.5) (Andrews 2010). Ligated adapters, CspCI restriction enzyme recognition sites, and low-quality basecalls (Phred score < 20) were trimmed to obtain high-quality 2bRAD tags, following the Eli Meyer pipeline (https://github.com/Eli-Meyer/2bRAD_utilities), and as performed in Boscari et al. (2019) and Terzin et al. (2021).
Data analysis – SNP calling in Stacks v2
SNP genotyping was performed in the Stacks v2 software (version 2.3e) (Rochette et al. 2019). After confirming that technical replicates had identical profiles (data not shown), technical replicates were concatenated for each of the samples sequenced in duplicate to increase sequencing depth, and low coverage samples (data with less than 100,000 quality-filtered 2bRAD tags) were excluded. SNP calling was performed on the remaining samples using the denovo_map.pl pipeline (Rochette et al. 2019) due to a lack of a reference genome for P. axinellae. Stacks loci assembly parameters were set to 3 (for -m—minimum depth of coverage required to create a stack for ustacks; -M – the number of mismatches allowed between stacks within individuals for ustacks; and -n—number of mismatches allowed between stacks between individuals for cstacks). The obtained SNPs were exported in genind (genotypes of individuals) format using the following options from the Stacks v2 Populations program: (1) -R = 0.8 (to retain loci shared across at least 80% of individuals), (2) –write_single_snp (to keep a maximum of one SNP per each locus), (3) -min-maf = 0.01 (to exclude alleles with minimum minor allele frequency below 1%), and (4) -min-mac = 2 (to exclude non-informative monomorphic loci with minimum minor allele count of 1).
Population genomic analysis
SNP data in genind format were imported into R version 3.6.3 (R Core Team 2018) with the R package adegenet (Jombart 2008), and we additionally filtered: (1) loci out of Hardy–Weinberg equilibrium (HWE) in pegas (Paradis 2010) and (2) any remaining missing loci were replaced with mean allele frequency in poppr (Kamvar et al. 2014, 2015). Loci under selection (outliers) were detected and removed in BayeScan (Foll and Gaggiotti 2008) using default parameters, set to: -n (the number of iterations) to 5,000, -thin (thinning interval size) to 10, -nbp (the number of pilot runs) to 20, -pilot (the length of pilot runs) to 5,000, -burn (the burn-in length) to 50,000, and -pr_odds (prior odds for the neutral model) to 10. All downstream analysis was performed using neutral loci only as previous comparisons between neutral and all loci showed similar patterns (Boscari et al. 2019; Terzin et al. 2021; Poliseno et al. 2022).
Overall population statistics parameters, such as the inbreeding coefficient (FIS), and observed (HO) and expected (HE) heterozygosity values, were computed at population level in hierfstat (Goudet 2005). The number of ancestral populations was detected using the LEA R package (Frichot and François 2015). Prior to the LEA analysis, all non-informative monomorphic loci were removed. Admixture coefficients were estimated in LEA at individual and population levels with the sparse non-negative matrix factorization (SNMF) algorithm, as in Jenkins et al. (2019), and integrated into a map as bar plots (‘individual’ level) or pie charts (‘population’ level) following this tutorial: https://github.com/Tom-Jenkins/admixture_pie_chart_map_tutorial.
Additional analyses to explore patterns in the genetic subdivision at the Y&O level were conducted using a hierarchical design with two factors: (1) ‘Morphotype’ (two levels: Slender and Stocky) and (2) ‘Populations’ (12 levels: ALA, ALAOR, CAMP, GUB, GIA, GIAOR, PTF, PUG, PVEN, PVENOR, SAR, and SAROR; see Table 1 for code meaning). Each population was nested within its corresponding morphotype, taking into account that the two morphotypes are distinct populations even if they come from the same locality. These analyses included: (1) discriminant analysis of principal components (DAPC) to explore the genetic structure through the assignment of each individual to its predetermined population a priori, conducted in adegenet; (2) principal coordinates analysis (PCoA) in vegan (Dixon 2003; Oksanen et al. 2007); (3) a hierarchical analysis of molecular variance (AMOVA) in poppr; and (4) construction of a UPGMA (unweighted pair group method with arithmetic mean) phylogenetic tree with bootstrap support inferred from 999 iterations and a cutoff value of p = 50%. The same analyses for Slender and Stocky morphotypes were carried out without deploying a two-level hierarchical approach, to independently test the effect of ‘Populations’ on genetic structuring within each morphotype. PCoA, AMOVA, and UPGMA trees were all computed using Provesti genetic distances.
Results
Selection of suitable loci
Sequencing yielded 1,724,678 ± 1,596,305 (mean ± SD) raw reads (50 bp) per sample, ranging from 4,562 to 9,345,347 sequences per individual (Table S1). After quality filtering, 1,157,808 ± 1,251,355 high-quality 2bRAD tags remained per sample (Table S1), which were then processed for SNP calling. SNP genotyping in Stacks identified 1,788 (Y&O), 2,656 (O), and 2,038 (Y) SNPs, which were then exported from Stacks as three R genind objects. A total of 0 (Y&O), 762 (O), and 108 (Y) loci showed significant departures from HWE following a Benjamini–Yekutieli false discovery rate (FDR) correction for multiple comparisons (Benjamini and Yekutieli 2001). Additionally, BayeScan identified 469 (Y&O), 23 (O), and 4 (Y) outlier loci (Fig. S1; Table S2). By removing loci out of HWE and loci under selection (‘outliers’), we obtained the final ‘neutral loci’ datasets containing 1319 (Y&O), 1871 (O), and 1926 (Y) SNPs, shared across 101 (Y&O), 33 (O), and 68 (Y) individuals. All downstream analyses (LEA, DAPC, PCoA, computation of pairwise FST values, hierarchical AMOVA, and the UPGMA tree) were performed using neutral loci only.
Population structure and connectivity – looking for the ‘true’ number of ancestral populations
The LEA admixture analysis detected 2 (Y&O) (Fig. 2a, b), 3 (Y) (Fig. S2), and 4 (O) (Fig. S3) subpopulation clusters (Ks) at each hierarchical level, respectively. Analysis of both morphotypes combined showed a clear distinction, with Slender (Yellow) and Stocky (Orange) individuals forming clearly differentiated clusters, regardless of sampling locality (Figs. 2–4). Each Stocky population at the ‘O’ level formed a unique cluster (Fig. S3), while the three clusters detected among Slender populations (at the ‘Y’ level) corresponded to (1) the ALA population (Cluster 1, light brown), (2) the PTF population (Cluster 2, violet), (3) and all the remaining populations which grouped within Cluster 3 (brown) (Fig. S2).

Admixture analysis in the LEA R package (Frichot and François 2015) computed for all individuals (at the ‘Y&O’ level, indicating ‘Yellow’ for Slender, and ‘Orange’ for Stocky) to detect the true number of ancestral populations (Ks). a Mean admixture proportions for each site are presented as pie charts and shown on the map. b Individual admixture proportions are shown as bar plots for each individual of the sampled populations. Populations are grouped within their corresponding morphotypes (Slender and Stocky)
A clear and marked genetic differentiation was recorded between Slender and Stocky morphotypes
When using pre-defined clusters based on sampling locations, the DAPC analysis (with 13 PCs as an optimal number of DAPC dimensions identified by the alpha (α) and cross-validation scores, Fig. S10a-b) showed a clear genetic differentiation between Slender and Stocky populations, with the PTF population being particularly isolated from the remaining Slender populations (Fig. 3a). Based on population membership probabilities, the DAPC composite bar plot showed that Slender individuals exhibit panmixia and admixture (except for the PTF population), while the Stocky individuals were assigned to their a priori determined populations with higher membership assignment scores (Fig. 3b). The UPGMA phylogenetic tree also showed two clearly separated clusters (composed by Slender and Stocky individuals, respectively) with 100% bootstrap support (Fig. S4).

Discriminant analysis of principal components (DAPC). a DAPC with populations as prior grouping information. b Stacked bar plots show posterior assignment probabilities of individuals to their predetermined populations and were constructed using previously calculated DAPC population membership assignments, at the ‘Y&O’ level (indicating ‘Yellow’ for Slender, and ‘Orange’ for Stocky)
In addition, PCoA showed separate clustering when all Slender and Stocky populations were analyzed together (Fig. 4a). Pairwise FST values ranged between 0.81 and 0.92 when comparing Slender and Stocky populations, with the corresponding p-values being significant in all Slender/Stocky population comparisons (p < 0.05; Fig. 4b). Furthermore, the hierarchical AMOVA recorded significant genetic differentiation at all three levels of variation: (1) within ‘Populations’ (p = 0.001) (Fig. S5a), (2) between ‘Populations’, nested within ‘Morphotype’ (p = 0.001) (Fig. S5b), and (3) between ‘Morphotypes’, (p = 0.002) (Fig. S5c). As expected, most variance (~ 93.32%) arose from between ‘Morphotypes’, while only ~ 1.44% and ~ 5.24% of variance originated from between ‘Populations’ (nested within ‘Morphotypes’) and within ‘Populations’ levels, respectively (Table S3).

Ordination analysis and FST statistics between Slender and Stocky morphs computed at the ‘Y&O’ level (indicating ‘Yellow’ for Slender and ‘Orange’ for Stocky). a Principal coordinates analysis (PCoA) clustering based on Prevosti genetic distances, with the factor ‘Morphotype’ as a grouping factor. b Heatmap and dendrogram based on Nei FST pairwise distances. Pairwise Nei’s FST values are shown in the lower colored triangle of the heatmap, and the corresponding p values (marked in red when Benjamini–Yekutieli adjusted p value < 0.05) are in the upper triangle. Sampling codes for Slender and Stocky populations are colored in yellow and orange on the right, respectively
Stocky morphotype shows stronger genetic structuring patterns
PCoA (Fig. 5a) and DAPC clustering (Fig. S6a, using 6 PCs as an optimal number of DAPC dimensions identified by the alpha (α) and cross-validation scores, Fig. S10c-d), as well as the UPGMA phylogenetic tree (Fig. S7), revealed discrete clusters for each Stocky population, with the Tyrrhenian populations (SAROR + GIAOR) clustering closely together and separate from the Ligurian populations. Pairwise FST values ranged between 0.27 and 0.31, and each Stocky/Stocky population comparison was identified as statistically significant (Benjamini–Yekutieli (B-Y) adjusted p = 0.02) (Fig. 5b). AMOVA also detected a significant effect of ‘within Populations’ on genetic structuring (Fig. S5d), with the ‘between populations’ level accounting for most of the variance (~ 68.71%) (Table S3).

Ordination analysis and FST statistics within Stocky morphotype, computed at ‘O’ level (indicating ‘Orange’ for Stocky). a Principal coordinates analysis (PCoA) clustering based on Prevosti genetic distances, with populations as a grouping factor. b Heatmap and dendrogram based on Nei’s FST pairwise distances. Pairwise Nei’s FST values are shown in the lower colored triangle of the heatmap, and the corresponding p values (marked in red when Benjamini–Yekutieli adjusted p value < 0.05) are in the upper triangle
A higher degree of admixture and panmixia was observed in the more widely distributed Slender morphotype
In contrast to the Stocky morphotype, PCoA ordination of the Slender populations detected a strong structuring for the PTF and ALA populations, while all the other populations grouped together (Fig. 6a). Pairwise FST values ranged between 0.01 and 0.30, with the PTF population diverging the most from the other Slender populations (FST = 0.25—0.30) (Fig. 6b). The DAPC clustering (with 13 PCs as an optimal number of DAPC dimensions identified by the alpha (α) and cross-validation scores, Fig. S10e-f) detected only two groups: (1) PTF and (2) ALA + CAMP + GUB + GIA + PUG + PVEN + SAR, although the ALA population seemed to diverge from the remaining populations (Fig. S8). UPGMA phylogenetic tree exhibited a high degree of admixture, although PTF and ALA individuals clustered more closely together (Fig. S9). Finally, AMOVA recorded a significant effect of ‘between Populations’ on genetic structuring (Fig. S5e), although most of the variance (~ 78.36%) was at the ‘within Populations’ level, as expected in panmictic populations (Table S3).

Ordination analysis and FST statistics within Slender morphotype, computed at ‘Y’ level (indicating ‘Yellow’ for Slender). a Principal coordinates analysis (PCoA) clustering based on Prevosti genetic distances, with populations as a grouping factor. b Heatmap and dendrogram based on Nei’s FST pairwise distances. Pairwise Nei’s FST values are shown in the lower colored triangle of the heatmap, and the corresponding p values (marked in red when Benjamini–Yekutieli adjusted p value < 0.05) are in the upper triangle
Discussion
Main findings
Our results show that Slender and Stocky morphotypes of Parazoanthus axinellae clearly diverge in their genomic profiles and their populations group into two well-separated clusters. At the ‘within the morph’ level, the two morphotypes also differ in patterns of connectivity and gene flow. Evaluation of the broadly distributed Slender morphotype revealed a high degree of admixture among populations, and seven Slender populations shared similar genomic profiles, except for the PTF population. In contrast, the Stocky morphotype (occurring only in the North-west Mediterranean) displayed stronger patterns of genetic structure compared to the Slender morphotype.
Population genomic structure within morphotypes
When comparing the two morphotypes, overall we observed a lower gene flow and higher genetic diversity (i.e., HO) within the Stocky morphotype, while the Slender morphotype showed higher rates of gene flow and lower observed heterozygosity. Literature often documents that greater genetic diversity increases population resilience and resistance to environmental perturbations (Hughes and Stachowicz 2004; Pauls et al. 2013); however, our results unexpectedly do not accord with this pattern, as the Stocky morph appears to be less resilient to environmental disturbances despite higher genetic diversity. High gene flow can potentially explain the broader distribution of the Slender morphotype throughout the Mediterranean Sea, and experimental studies suggest that the Slender morphotype may also be more resilient to environmental perturbations caused by climate change. Thermotolerance experiments (temperature treatments from 26 °C to 29 °C) were performed on 10 common benthic invertebrates in Mediterranean coralligenous habitats (three anthozoans, six sponges, and one ascidian), documenting that the Stocky morphotype of P. axinellae was the most sensitive of the 10 species, including the widespread Slender morphotype (Gómez-Gras et al. 2019). In detail, the Stocky morphotype showed initial signs of necrosis after 5 days at 26 °C, while the Slender morph did not display any signs of tissue necrosis for 18 days. Thermotolerance decreased precipitously at higher temperatures, with Stocky individuals displaying signs of necrosis only 1–2 days after experimental exposure to increased temperatures (Gómez-Gras et al. 2019). Interestingly, Cachet et al. (2015) have identified the presence of ‘parazoanthines’, highly bioactive secondary metabolites, found within the Slender morphotype of P. axinellae and absent from the Stocky individuals. Differing thermal tolerance between the two morphotypes may be attributed to the presence of highly bioactive secondary metabolites in one morphotype and not the other (Gómez-Gras et al. 2019), although further experimental validation would be needed to confirm that the presence of ‘parazoanthines’ increases the capacity of the Slender morph to cope with environmental changes. In addition, we assert that additional experimental exposure of Slender and Stocky individuals to various stressors (e.g., nutrient enrichment, sedimentation, increased temperature, and pH decrease) would be needed to identify the major environmental threats to each morphotype, which is critical to underpin future conservation programs for this zoanthid.
Furthermore, it is important to understand the complex dynamics of connectivity and genetic diversity at local spatial scales (DuBois et al. 2022), as different populations may exhibit different resilience and resistance thresholds to environmental disturbance. Within the Tyrrhenian region, the Slender population from Portofino (PTF; Ligurian Sea) was identified to be isolated the most. The Portofino area has been previously documented to present a barrier to gene flow for several species of sessile invertebrates, including: (1) Corallium rubrum (Linnaeus, 1758) (Costantini et al. 2007); (2) Paramuricea clavata (Risso, 1827) (Mokhtar-Jamaï et al. 2011); and (3) Patella caerulea (Linnaeus, 1758) (Villamor et al. 2018), which seems to be caused by the presence of a large-scale separation of currents that occur in the region (Rossi et al. 2014). Therefore, the Portofino Slender population could be more susceptible to environmental stress compared to the other populations, although experimental validation would be needed to confirm this hypothesis.
Are Stocky and Slender morphotypes two separate species?
All analyses performed in this study at the ‘Y&O’ level provided strong evidence that Slender and Stocky morphotypes represent two well-separated clusters with clearly distinct genomic profiles. Such strong genomic differentiation provides additional evidence that Slender and Stocky morphotypes might represent two separate species, as previously hypothesized by Cachet et al. (2015) based on differing metabolomic profiles, and by Villamor et al. (2020) based on molecular observations. In our study, this strong Slender/Stocky contrast is visible from the (1) DAPC analysis (Fig. 3a), (2) PCoA plot (Fig. 4a), the (3) UPGMA phylogenetic tree (Fig. S4), and most importantly (4) the extremely high FST values (Fig. 4b) that we detected in this study (ranging between 0.81 and 0.92 for each of the Slender/Stocky population comparisons); all of which have clearly identified that all individuals agglomerate into two well-supported clades, congruent with distinct morphologies.
A recent study by Villamor et al. (2020) investigated the phylogeny within the Parazoanthidae family by including publicly available COI and ITS gene sequences of allopatric and congeneric zoanthids, alongside COI and ITS data from Slender and Stocky morphs. Interestingly, the genetic differentiation between Slender and Stocky morphotypes was higher compared to some instances where Slender and Stocky individuals were compared to other allopatric species within the Parazoanthidae family (Villamor et al. 2020). In detail, Slender P. axinellae individuals were more similar to (1) P. anguicomus (Norman, 1869) (COI, ITS); (2) P. capensis (Duerden, 1907) (ITS); (3) P. swiftii (Duchassaing de Fonbressin and Michelotti, 1860) (COI); and (4) Umimayanthus parasiticus (Duchassaing de Fonbressin and Michelotti, 1860) (COI) compared to the Stocky morphotype (Villamor et al. 2020). The Stocky morphotype was genetically more similar to the (1) Pacific shallow-water P. elongatus (McMurrich, 1904) (COI, ITS); (2) P. juan-fernandezii (Carlgren, 1922) (ITS); and to the (3) deep Atlantic P. aliceae (Carreiro-Silva, Ocaña, Stanković, Sampaio, Porteiro, Fabri, and Stefanni, 2017) (COI), compared with the Slender morphotype (Villamor et al. 2020). As COI and ITS sequences diverged more between Slender and Stocky morphotypes compared to the aforementioned (1) Slender/congeners and (2) Stocky/congeners contrasts, it is clear why Villamor et al. (2020) also suggest that these two morphs may represent two separate species. Publicly available RAD-Seq datasets on zoanthids can be integrated with our data to investigate if Slender and Stocky morphs will cluster more closely together, or with the ‘outgroup’ zoanthid species from an independent study. To our knowledge, the only available RAD-Seq dataset on zoanthids is from a golden coral Savalia savaglia (Poliseno et al. 2022), and such meta-analysis could confirm the taxonomic status of the P. axinellae species complex.
Further, it would be particularly interesting to use molecular data to investigate the origin of Slender and Stocky morphotypes. Villamor et al. (2020) hypothesized that the Slender P. axinellae could have an Atlantic-Mediterranean origin due to its similarity with other Atlantic Parazoanthus species (P. anguicomus and P. capensis) which are interestingly also able to colonize sponges—just like the Slender morphotype (Sinniger and Häussermann 2009). The mysterious origin of the Stocky morphotype is more difficult to infer, as this morphotype shared similar genomic profiles with both shallow-water Pacific (P. elongatus and P. aliceae) and deep-sea Atlantic (P. aliceae) zoanthids within the Parazoanthidae family (Villamor et al. 2020). These molecular similarities coincide with some ecological features shared across these species. For instance, the Pacific P. elongatus and P. aliceae are mainly found on rocky substrate rather than on sponges, similarly to the Stocky morphotype (Sinniger and Häussermann 2009). However, P. elongatus and P. aliceae are morphologically distinct from the P. axinellae species complex, which is in contrast with the high COI and ITS gene similarities observed across these species in Villamor et al. (2020), all of which further validates that P. axinellae is indeed a taxonomically problematic species complex.
Concluding remarks and implications for future research
Future multidisciplinary and integrative studies are a crucial prerequisite to successfully resolving the taxonomic and phylogenetic status of the Slender and Stocky P. axinellae morphotypes, thus confirming the molecular findings observed in our study and Villamor et al. (2020). So far, a few studies have investigated reproductive strategies in P. axinellae. The Stocky morphotype of P. axinellae was documented to rely predominantly on fission for propagation (which implies a low dispersal capacity), whereas sexual reproduction via spawning plays a minor role in species reproduction (Garrabou 1999). Previati et al. (2010) studied the reproduction in Stocky individuals, recording that oocyte and spermatocyte maturation periods varied between summer and autumn and depending on the locality, which also aligns with findings from Garrabou (1999). Future experimental studies to delineate species relationships between Slender and Stocky morphs could: (1) aim to determine if reproduction between Slender and Stocky individuals will result in hybrid offspring, if hybrids are viable, and if further reproduction between hybrids will produce fertile offspring in the F2 generation; (2) conduct metabolomic and biochemical studies to investigate the differences in chemical profiles between the two morphotypes, with particular focus on the parazoanthines identified in the Slender morphothype by Cachet et al. (2015); (3) reconstruct the genomes of the two morphotypes to assess how Slender and Stocky morphotypes differ in their gene content; (4) analyze the (meta)transcriptomes of the two morphotypes to investigate if the morphotypes differ in their acclimation mechanisms to environmental shifts (Pratlong et al. 2015); and (5) perform a natural comparative experiment to investigate multiple aspects of the biology of the host and the host-associated microbiome. These future perspectives could deepen the understanding of the two morphotypes to identify specific characteristics that support a clear distinction at the species level.
From a monitoring/management perspective, we propose that the Stocky morphotype should be prioritized in terms of conservation due to lower gene flow rates between Stocky populations observed in our study, as also suggested by Gómez-Gras et al. (2019). However, identifying if Slender and Stocky morphotype are two separate species is critical before genetic connectivity data can be effectively implemented in conservation and management plans for this widespread and iconic zoanthid.
Data Availability
Raw fastq files, metadata spreadsheets, and all the code utilized in the analysis can be found at the following link: https://datadryad.org/stash/share/4qDFinqqu0zamNx3Q3zJE0l1EBVd_RFerEsaaQQ6wlA.
References
Andrews S (2010) Babraham bioinformatics-FastQC a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29(4):1165–1188
Bernhardt JR, Leslie HM (2013) Resilience to climate change in coastal marine ecosystems. Ann Rev Mar Sci 5:371–392. https://doi.org/10.1146/annurev-marine-121211-172411
Bongaerts P, Cooke IR, Ying H, Wels D, den Haan S, Hernandez-Agreda A, Brunner CA, Dove S, Englebert N, Eyal G (2021) Morphological stasis masks ecologically divergent coral species on tropical reefs. Cur Biol 31:2286–2298. https://doi.org/10.1016/j.cub.2021.03.028
Boscari E, Abbiati M, Badalamenti F, Bavestrello G, Benedetti-Cecchi L, Cannas R, Cau A, Cerrano C, Chimienti G, Costantini F (2019) A population genomics insight by 2b-RAD reveals populations’ uniqueness along the Italian coastline in Leptopsammia pruvoti (Scleractinia, Dendrophylliidae). Div Distrib 25:1101–1117. https://doi.org/10.1111/ddi.12918
Cachet N, Genta-Jouve G, Ivanisevic J, Chevaldonné P, Sinniger F, Culioli G, Pérez T, Thomas OP (2015) Metabolomic profiling reveals deep chemical divergence between two morphotypes of the zoanthid Parazoanthus axinellae. Sci Rep 5:1–9. https://doi.org/10.1038/srep08282
Carreras C, García-Cisneros A, Wangensteen OS, Ordóñez V, Palacín C, Pascual M, Turon X (2020) East is East and West is West: Population genomics and hierarchical analyses reveal genetic structure and adaptation footprints in the keystone species Paracentrotus lividus (Echinoidea). Div Dist 26:382–398. https://doi.org/10.1111/ddi.13016
Carreras-Carbonell J, Macpherson E, Pascual M (2006) Population structure within and between subspecies of the Mediterranean triplefin fish Tripterygion delaisi revealed by highly polymorphic microsatellite loci. Mol Ecol 15:3527–3539. https://doi.org/10.1111/j.1365-294X.2006.03003.x
Chenuil A, Cahill AE, Délémontey N, Salliant Du, du Luc E, Fanton H (2019) Problems and Questions Posed by Cryptic Species. A Framework to Guide Future Studies. In: Casetta E, Marques da Silva J, Vecchi D (eds) From Assessing to Conserving Biodiversity: Conceptual and Practical Challenges. Springer, Berlin, pp 77–106. https://doi.org/10.1007/978-3-030-10991-2_4
Costantini F, Fauvelot C, Abbiati M (2007) Fine-scale genetic structuring in Corallium rubrum: Evidence of inbreeding and limited effective larval dispersal. Mar Ecol Progr Ser 340:109–119. https://doi.org/10.3354/meps340109
Costantini F, Ferrario F, Abbiati M (2018) Chasing genetic structure in coralligenous reef invertebrates: Patterns, criticalities and conservation issues. Sci Rep 8:5844. https://doi.org/10.1038/s41598-018-24247-9
Cowen R, Gawarkiewicz G, Pineda J, Thorrold S, Werner F (2007) Population connectivity in marine systems: an overview. Oceanogr. https://doi.org/10.5670/oceanog.2007.26
Davey JW, Blaxter ML (2010) RADSeq: next-generation population genetics. Brief Funct Genomics 9(5–6):416–423
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930
DuBois K, Pollard KN, Kauffman BJ, Williams SL, Stachowicz JJ (2022) Local adaptation in a marine foundation species: Implications for resilience to future global change. Glob Change Biol 28:2596–2610. https://doi.org/10.1111/gcb.16080
Everett MV, Park LK, Berntson EA, Elz AE, Whitmire CE, Keller AA, Clarke ME (2016) Large-scale genotyping-by-sequencing indicates high levels of gene flow in the deep-sea octocoral Swiftia simplex (Nutting 1909) on the west coast of the United States. PLoS ONE 11:e0165279. https://doi.org/10.1371/journal.pone.0165279
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Frichot E, François O (2015) LEA: An R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929
Garrabou J (1999) Life-history traits of Alcyonium acaule and Parazoanthus axinellae (Cnidaria, Anthozoa), with emphasis on growth. Mar Ecol Progr Ser 178:193–204
Gatti G, Bianchi CN, Parravicini V, Rovere A, Peirano A, Montefalcone M, Massa F, Morri C (2015) Ecological change, sliding baselines and the importance of historical data: lessons from combing observational and quantitative data on a temperate reef over 70 years. PLoS ONE 10:e0118581. https://doi.org/10.1371/journal.pone.0118581
Gili J-M, Pages F, Barange M (1987) Zoantarios (Cnidaria, Anthozoa) de la costa y de la plataforma continental catalanas (Mediterráneo occidental). Misc Zool 11:13–24
Gómez-Gras D, Linares C, de Caralt S, Cebrian E, Frleta-Valić M, Montero-Serra I, Pagès-Escolà M, López-Sendino P, Garrabou J (2019) Response diversity in Mediterranean coralligenous assemblages facing climate change: insights from a multispecific thermotolerance experiment. Ecol Evol 9:4168–4180. https://doi.org/10.1002/ece3.5045
Goudet J (2005) Hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Herrera S, Shank TM (2016) RAD sequencing enables unprecedented phylogenetic resolution and objective species delimitation in recalcitrant divergent taxa. Mol Phyl Evol 100:70–79. https://doi.org/10.1016/j.ympev.2016.03.010
Hodel RG, Chen S, Payton AC, McDaniel SF, Soltis P, Soltis DE (2017) Adding loci improves phylogeographic resolution in red mangroves despite increased missing data: comparing microsatellites and RAD-Seq and investigating loci filtering. Sci Rep 7:1–14. https://doi.org/10.1038/s41598-017-16810-7
Hughes AR, Stachowicz JJ (2004) Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. Proc Natl Acad Sci USA 101:8998–9002. https://doi.org/10.1073/pnas.0402642101
Jenkins TL, Ellis CD, Triantafyllidis A, Stevens JR (2019) Single nucleotide polymorphisms reveal a genetic cline across the north-east Atlantic and enable powerful population assignment in the European lobster. Evol Appl 12:1881–1899. https://doi.org/10.1111/eva.12849
Johnston EC, Forsman ZH, Flot J-F, Schmidt-Roach S, Pinzón JH, Knapp IS, Toonen RJ (2017) A genomic glance through the fog of plasticity and diversification in Pocillopora. Sci Rep 7:5991. https://doi.org/10.1038/s41598-017-06085-3
Jombart T (2008) adegenet: A R package for the multivariate analysis of genetic markers. Bioinf 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Gen. https://doi.org/10.3389/fgene.2015.00208
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Mokhtar-Jamaï K, Pascual M, Ledoux J-B, Coma R, Féral J-P, Garrabou J, Aurelle D (2011) From global to local genetic structuring in the red gorgonian Paramuricea clavata: the interplay between oceanographic conditions and limited larval dispersal. Mol Ecol 20:3291–3305. https://doi.org/10.1111/j.1365-294X.2011.05176.x
Ocaña Vicente O, Moro L, Herrera R, Çinar M, Hernández A, Cuervo J, Herrero R (2019) Parazoanthus axinellae: a species complex showing different ecological requierements. Rev Acad Canar Cienc 31:1–24
Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, Suggests M (2007) The Vegan Package. Comm Ecol Pack 10:719
Padrón M, Costantini F, Bramanti L, Guizien K, Abbiati M (2018) Genetic connectivity supports recovery of gorgonian populations affected by climate change. Aq Cons: Mar Fresh Ecosyst 28:776–787. https://doi.org/10.1002/aqc.2912
Pante E, Abdelkrim J, Viricel A, Gey D, France SC, Boisselier M-C, Samadi S (2015a) Use of RAD sequencing for delimiting species. Heredity 114:450–459
Pante E, Puillandre N, Viricel A, Arnaud-Haond S, Aurelle D, Castelin M, Chenuil A, Destombe C, Forcioli D, Valero M, Viard F, Samadi S (2015b) Species are hypotheses: avoid connectivity assessments based on pillars of sand. Mol Ecol 24:525–544. https://doi.org/10.1111/mec.13048
Paradis E (2010) pegas: An R package for population genetics with an integrated–modular approach. Bioinf 26:419–420. https://doi.org/10.1093/bioinformatics/btp696
Pauls SU, Nowak C, Bálint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol 22:925–946. https://doi.org/10.1111/mec.12152
Poliseno A, Terzin M, Costantini F, Trainito E, Mačić V, Boavida J, Perez T, Abbiati M, Cerrano C, Reimer JD (2022) Genome-wide SNPs data provides new insights into the population structure of the Atlantic-Mediterranean gold coral Savalia savaglia (Zoantharia: Parazoanthidae). Ecol Gen Genom 25:100135. https://doi.org/10.1016/j.egg.2022.100135
Pratlong M, Haguenauer A, Chabrol O, Klopp C, Pontarotti P, Aurelle D (2015) The red coral (Corallium rubrum) transcriptome: a new resource for population genetics and local adaptation studies. Mol Ecol Res 15:1205–1215. https://doi.org/10.1111/1755-0998.12383
Previati M, Palma M, Bavestrello G, Falugi C, Cerrano C (2010) Reproductive biology of Parazoanthus axinellae (Schmidt, 1862) and Savalia savaglia (Bertoloni, 1819) (Cnidaria, Zoantharia) from the NW Mediterranean coast. Mar Ecol 31:555–565. https://doi.org/10.1111/j.1439-0485.2010.00390.x
Quattrini AM, Wu T, Soong K, Jeng M-S, Benayahu Y, McFadden CS (2019) A next generation approach to species delimitation reveals the role of hybridization in a cryptic species complex of corals. BMC Evol Biol 19:1–19. https://doi.org/10.1186/s12862-019-1427-y
R Core Team, and R. Version. (2018) 3.6. 3. R Foundation for Statistical Computing: Vienna, Austria
Reitzel AM, Herrera S, Layden MJ, Martindale MQ, Shank TM (2013) Going where traditional markers have not gone before: Utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol Ecol 22:2953–2970. https://doi.org/10.1111/mec.12228
Rochette NC, Rivera-Colón AG, Catchen JM (2019) Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol Ecol 28:4737–4754. https://doi.org/10.1111/mec.15253
Rossi V, Ser-Giacomi E, López C, Hernández-García E (2014) Hydrodynamic provinces and oceanic connectivity from a transport network help designing marine reserves. Geophys Res Lett 41:2883–2891. https://doi.org/10.1002/2014GL059540
Schlacher TA, Rowden AA, Dower JF, Consalvey M (2010) Seamount science scales undersea mountains: New research and outlook. Mar Ecol 31:1–13. https://doi.org/10.1111/j.1439-0485.2010.00396.x
Schopen GCB, Bovenhuis H, Visker M, Van Arendonk JAM (2008) Comparison of information content for microsatellites and SNPs in poultry and cattle. Anim Genet 39:451–453. https://doi.org/10.1111/j.1365-2052.2008.01736.x
Sinniger F, Häussermann V (2009) Zoanthids (Cnidaria: Hexacorallia: Zoantharia) from shallow waters of the southern Chilean fjord region, with descriptions of a new genus and two new species. Org Div Evol 9:23–36. https://doi.org/10.1016/j.ode.2008.10.003
Terzin M, Paletta MG, Matterson K, Coppari M, Bavestrello G, Abbiati M, Bo M, Costantini F (2021) Population genomic structure of the black coral Antipathella subpinnata in Mediterranean Vulnerable Marine Ecosystems. Coral Reefs 40:751–766. https://doi.org/10.1007/s00338-021-02078-x
Villamor A, Costantini F, Abbiati M (2018) Multilocus phylogeography of Patella caerulea (Linnaeus, 1758) reveals contrasting connectivity patterns across the Eastern-Western Mediterranean transition. J Biogeogr 45:1301–1312. https://doi.org/10.1111/jbi.13232
Villamor A, Signorini LF, Costantini F, Terzin M, Abbiati M (2020) Evidence of genetic isolation between two Mediterranean morphotypes of Parazoanthus axinellae. Sci Rep 10:1–11
Wang S, Meyer E, McKay JK, Matz MV (2012) 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat Met 9:808–810
Winnepenninckx B, Backeljau T, De Wachter R (1993) Extraction of high molecular weight DNA from molluscs. Trends Gen 9:407. https://doi.org/10.1016/0168-9525(93)90102-n
Acknowledgements
This study was part of the Italian Ministry of University and Research (MIUR) PRIN project “Coastal bio-constructions: structure, function, and management”. Laboratory activities were also supported by the project ”Reef ReseArcH – Resistance and resilience of Adriatic mesophotic biogenic habitats to human and climate change threats” (Call 2015; Prot. 2015J922E4; 2017–2020) funded by MIUR, and by the European project SEAMoBB: ”Solutions for Semi-Automated Monitoring of Benthic Biodiversity” funded by ERA-Net Mar-TER-A. GC was supported by the Italian Ministry of Education, University and Research (PON 2014–2020, Grant AIM 1807508-1, Linea 1). A special thank you goes to Dr. Alexandro Palmieri for his immeasurable help in the Server setup and informatics aspects.
Funding
Open access funding provided by Alma Mater Studiorum - Università di Bologna within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
MT, AV, MA, and FC research interests are focused on the connectivity patterns of benthic organisms and habitats. Author contributions: AV, FC, and MA conceived the ideas and collected the data; GB, LBC, CC, GC, SF, GI, FM, LP, MP, ET, and RS contributed to sampling; AV, LM, VB and MGP performed the laboratory work; EB, LC, and LZ provided technical advice; and MT analyzed the data and together with FC and KM led the writing. All authors revised the article critically and approved the final version to be published.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Terzin, M., Villamor, A., Marincich, L. et al. 2bRAD reveals fine-scale genetic structuring among populations within the Mediterranean zoanthid Parazoanthus axinellae (Schmidt, 1862). Coral Reefs 43, 357–370 (2024). https://doi.org/10.1007/s00338-023-02456-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-023-02456-7