
Abstract
Animal models of diabetes, such as db/db mice, are a useful tool for deciphering the genetic background of molecular changes at the initial stages of disease development. Our goal was to find early transcriptomic changes in three tissues involved in metabolism regulation in db/db mice: adipose tissue, muscle tissue and liver tissue. Nine animals (three per time point) were studied. Tissues were collected at 8, 12 and 16 weeks of age. Transcriptome-wide analysis was performed using mRNA-seq. Libraries were sequenced on NextSeq (Illumina). Differential expression (DE) analysis was performed with edgeR. The analysis of the gene expression profile shared by all three tissues revealed eight upregulated genes (Irf7, Sp100, Neb, Stat2, Oas2, Rtp4, H2-T24 and Oasl2) as early as between 8 and 12 weeks of age. The most pronounced differences were found in liver tissue: nine DE genes between 8 and 12 weeks of age (Irf7, Ly6a, Ly6g6d, H2-Dma, Pld4, Ly86, Fcer1g, Ly6e and Idi1) and five between 12 and 16 weeks of age (Irf7, Plac8, Ifi44, Xaf1 and Ly6a) (adj. p-value < 0.05). The mitochondrial transcriptomic profile also changed with time: we found two downregulated genes in mice between 8 and 12 weeks old (Ckmt2 and Cox6a2) and five DE genes between 12 and 16 weeks of age (Mavs, Tomm40L, Mtfp1, Ckmt2 and Cox6a2). The KEGG pathway analysis showed significant enrichment in pathways related to the autoimmune response and cytosolic DNA sensing. Our results suggest an important involvement of the immunological response, mainly cytosolic nucleic acid sensing, and mitochondrial signalling in the early stages of diabetes and obesity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Animal models play an important role in understanding the aetiology of diseases as well as their multisystemic effects (Okechukwu 2018). In this study, we used a genetically induced mouse model of type 2 diabetes with db/db mice, which have a mutation in the leptin receptor gene (LepR) and are characterised by obesity, decreased insulin receptor sensitivity and elevated levels of blood glucose (Burke et al. 2017).
It is already known that metaflammation, a process in which macrophages become polarised towards pro-inflammatory phenotypes, thus resulting in the production of pro-inflammatory cytokines, plays a crucial role in the development of obesity. Moreover, according to recent studies, the activation of the immune response stems from the accumulation of nucleic acids and their recognition by damage-associated molecular patterns (DAMPs). It has also been shown that cGas and STING activation is increased in mice fed a high-fat diet and that it is due to the release of mitochondrial DNA in adipose tissue.
Our goal was to find very early transcriptional changes during the development of obesity in the db/db mouse model. We analysed mice at 8, 12 and 16 weeks of age to track changes in the transcriptomic profile in three metabolically active tissues: adipose tissue, liver tissue and muscle tissue. We confirmed that sterile inflammation is the underlying cause of diabetes and obesity in db/db mice and showed that the activation of the cGas-STING pathway and the resulting upregulation of Irf7 takes place as early as at 8 weeks of age. Moreover, we found that as obesity develops after 12 weeks of age, the activation of Irf7 is muted, possibly to regulate the metabolic response, as loss of Irf7 was shown to decrease glucose tolerance. This observation also confirms that the activation of the immune response is a very early event in the development of the disease development. Tissue-wise, the most pronounced changes were observed in liver tissue, which shows that it is not only immune cells and adipocytes that contribute to obesity-mediated pathogenesis. Our data show the whole transcriptomic profile of young obese mice and underline the importance of sterile inflammation as the earliest cause of the disease.
Materials and methods
Animals and tissue collection
Nine Dock7 <m> + / + Lepr <db> /J males (db/db mice) were purchased from Charles River. The animals were acclimated for 2 weeks at the age of 6 weeks. The mice were kept in temperature-controlled rooms 21 (± 0.5 to ± 5%), humidity 55% on a 12-h light/12-h dark cycle and had ad libitum access to pelleted food and filtered water. The animals were randomly allocated into groups and examined at three stages of life: 8 weeks, 12 weeks and 16 weeks. Three animals were analysed at each time point. On the day of sacrifice, liver, muscle and adipose tissue were immediately snap-frozen or collected in RNAlater solution and stored at − 80 °C. All procedures were approved by the Local Ethics Commission for Animal Experiments in Łódź, Poland.
RNA isolation and sequencing
Total RNA was isolated using a Maxwell 16 Instrument (Promega) from the following groups: 8-week-old db/db mice (n = 3; animals A, B, C), 12-week-old mice (n = 3; animals D, E, F) and 16-week-old mice (n = 3; animals G, H, I). The quality and quantity were checked with Quantus (Promega) and Tape Station (Agilent). 500 ng of total RNA was used to prepare libraries according to SENSE mRNA-Seq Library Preparation Kit (Lexogen). The libraries were sequenced on NextSeq (Illumina).
Bioinformatic and statistical analysis
The bioinformatic analysis consisted of trimming using the Cutadapt tool (Martin 2011), mapping sequences to the GRCm38 mouse reference genome with the STAR aligner (Dobin et al. 2013) and counting the mapped mRNA reads using HT-Seq (Putri et al. 2022). Differential expression analysis was performed with the aid of edgeR package in R (Robinson et al. 2010). Analysis of the KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (Gene Ontology) pathways was also performed. Analysis of mitochondrial gene expression was performed on genes reported to be associated with mitochondrial functioning listed in MitoCarta (Rath et al. 2021).
Immunofluorescence and imaging
Frozen sections were air-dried for several minutes and fixed in 4% PFA (for 15 min), permeabilised in 1% (v/v) Triton-X in PBS (for 10 min) and blocked with 1% BSA blocker (for 1.5 h). An overnight incubation with primary antibody (cGAS—#703149, Invitrogen, 1:200, + 4 °C or IRF7—#MA541165, Thermo Fischer, 1:100, + 4 °C) was followed by secondary antibody incubation (#96886, Abcam, conjugated with DyLight 650, 1:500, 2 h, RT). The sections were co-stained with MitoView (#70054, Biotium, 1:1000) or dsDNA Dye (#E2670, Promega, 1:400) for 30 or 20 min, respectively. Immunofluorescence-labelled sections were mounted with VectaShield® HardSet™ Mounting Medium with DAPI (#H-1500-10, Vector Laboratories) and analysed on Olympus FluoView1200 scanning confocal laser microscope using the 40 × or 60x (oil immersion) objective lens. A 473-nm diode laser, a 635-nm diode laser and a 405-nm diode laser were used to excite green (MitoView, dsDNA Dye), far-red (DyLight 650) and blue (DAPI) fluorescence, respectively. At least three images per sample were collected. Each image was divided into 36 ROIs. These were used to calculate the fluorescence intensity per image.
For lipid visualisation, the sections were fixed in 4% PFA, washed with dH2O, rinsed with 60% isopropanol and stained with 4.5 g/l Oil Red O (#O0625-25G, Sigma Aldrich) for 15 min. Excess dye was removed by 60% isopropanol rinsing, followed by a running tap water wash. The nuclei were counterstained with Harris haematoxylin and slides were mounted. High-resolution bright-field images were taken (under 20 × magnification) using an Olympus IX83 microscope and were analysed using CellSens Dimension software (Olympus). Three images per sample were collected. Each image was divided into 36 ROIs. At least 27 ROIs were used to calculate the amount of lipid accumulation by analysing the stained surface area in μm2.
Results
The aim of the study was to observe the early events of diabetes and obesity development at the transcriptomic level. Three metabolically important tissues were analysed: adipose tissue, liver tissue and muscle tissue.
Nine db/db mice were analysed over an 8-week period. Time point 1 represents 8-week-old animals (marked as A, B and C). Time point 2 represents the transcriptional profile of 12-week-old animals (mice labelled D, E, and F), while time point 3 represents 16 week-old mice (animals G, H, I). A diagram of the experiment is shown in Fig. 1.

The design of the experiment. The study focused on 3 time points each representing different animal age. Time point 1: 8-week-old animals, time point 2: 12-week-old animals, time point 3: 16-week-old animals. Three metabolically active tissues: adipose tissue, liver tissue and muscle tissue were analyzed
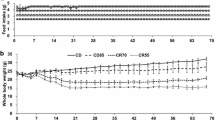
The animals were weighed weekly and their blood glucose levels (fasting state) and other biochemical parameters were checked, as shown in Table S1.
RNA profiles of adipose tissue, muscle tissue and liver tissue were analysed at three time points. The RNA expression profiles were specific to each tissue and maintained throughout the analysed time points, as shown by the clusters in Fig. S1.
Analysis of the gene expression profile shared by all three tissues over the course of the experiment
Regardless of the transcriptional uniqueness of the tissues, our goal was to check whether there is a trend in gene expression which would be shared by all three tissues; thus, we searched for genes with different expression between different time points in all the selected tissues. We identified eight statistically significant genes which were upregulated between time points 2 and 1 (Table 1). No differentially expressed genes were found between time points 3 and 2.
Both analyses, however, yielded a substantial number of nominally significant genes with differential expression. These were subjected to KEGG pathway analysis, which led to the discovery of 21 pathways between time points 2 and 1 (Fig. 2) and two between time points 3 and 2: oxidative phosphorylation (mmu00190) and diabetic cardiomyopathy (mmu05415) (both were also enriched in the first analysis). The GO enrichment for these comparisons is shown in Table S2.

KEGG pathways significantly enriched between time points 2 and 1 (12- vs 8-week-old mice) in all tissues analysed
Analysis of unidirectional, tissue-specific changes in gene expression over the course of the experiment
Next, we wanted to check which tissue undergoes the greatest unidirectional change in gene expression over the course of the experiment. The PCA analysis showed a trend for directionality for liver tissue only (Fig. S2), where we found 507 genes with unidirectional change (Table S3). Interestingly, the KEGG pathway analysis revealed no common pathway for them. However, 30 genes were associated with mitochondria (Table 2).
The analysis of adipose tissue and muscle did not show any statistically significant genes with unidirectional change in expression, while the pathway analysis found two enriched pathways for adipose tissue (oxidative phosphorylation [mmu00190] and diabetic cardiomyopathy [mmu05415]) and seven pathways for muscle (protein digestion and absorption [mmu04974], platinum drug resistance [mmu01524], ECM-receptor interaction [mmu04512], focal adhesion [mmu04510], amoebiasis [mmu05146], African trypanosomiasis [mmu05143], and human papillomavirus infection [mmu05165]) (Table S4).
Analysis of tissue-specific changes between the time points
Finally, we decided to compare the expression profiles between the time points within each tissue. No statistically significant differences in gene expression were found in the adipose tissue. We found five genes in the muscle tissue that differed between time points 3 and 2, while no changes were found between time points 2 and 1 (Table 3). In the liver tissue, we identified nine genes that differed between time points 2 and 1, six that differed between time points 3 and 1, and five that differed between time points 3 and 2 (Table 3).
When we looked specifically into mitochondrial gene expression, we found two genes whose expression was significantly downregulated between time points 2 and 1, and five differentially regulated genes between time points 3 and 2 (Table 4).
The KEGG pathway analysis for adipose tissue showed a massive immunologic response to viral infection. Pathways that were common to both analyses (time points 2 vs 1, and time points 3 vs 2) included Epstein-Barr virus, coronavirus, hepatitis C, measles, herpes simplex virus 1, type 1 diabetes, RIG-like receptor signalling and human papillomavirus infection (Table S5). There were also several pathways specific to particular time points. For time points 2 vs 1, we found the cytosolic DNA sensing pathway, oxidative phosphorylation, cardiac muscle contraction, diabetic cardiomyopathy, DNA adducts and metabolism of xenobiotics by cytochrome P450. Only three pathways were uniquely characteristic of time points 3 vs 2: the pathways for toxoplasmosis, cell adhesion molecules and C-type lectin receptor signalling (Table S5).
In muscle tissue, pathways involved in the autoimmune response were the most enriched both in the analysis of time points 2 vs 1 and time points 3 vs 2. Pathways specific to time points 2 vs 1 included the anti-viral response (Kapsi sarcoma-associated herpesvirus infection, herpes simplex virus 1 infection, human immunodeficiency virus 1 infection, and human papillomavirus infection), the NOD-like receptor signalling pathway, cell adhesion and cell senescence. Interestingly, more specific changes were observed when we compared time points 3 and 1, namely enrichment of protein digestion and adsorption, focal adhesion, the AGE-RAGE signalling pathway in diabetic complications and the TNF signalling pathway, phagosome, apoptosis and cytokine-cytokine receptor interaction (Table S5).
In liver tissue, similarly to muscle, the pathways specific to time points 3 vs 2 fully overlapped with those from time points 2 vs 1. These were natural killer cell mediated cytotoxicity, antigen processing and presentation, graft-versus-host disease and autoimmune responses (autoimmune thyroid disease and type 1 diabetes mellitus). The pathways characteristic of time points 2 vs 1 were those associated with immunologic responses (Leishmaniosis, toxoplasmosis and Staphylococcus aureus, human cytomegalovirus, human T-cell leukaemia virus 1 and Kaposi sarcoma-associated herpes virus infection), the NOD-like receptor pathway, cytosolic DNA sensing, cytokine-cytokine receptor interaction and the chemokine pathway and pathways involved in steroid biosynthesis and metabolism (Fig. 3 and Table S5).

KEGG pathways significantly enriched between time points 2 and 1 (12- vs 8-week-old mice) in liver tissue
Imaging
Since most of the transcriptomic analyses showed that the immunologic response (response to viral infection and cytosolic DNA sensing) and mitochondrially associated genes play a key role in the development of diabetes, we decided to perform mitochondrial imaging and immunostaining for Irf7 and cGas in the liver and muscle tissues of all mice studied.
The pattern of Irf7 protein expression reflected that observed at the RNA level, as it increased between 8 and 12 weeks of age and decreased between 12 and 16 weeks of age. This trend, even though not statistically significant (time point 1 vs 2 p = 0.302, time point 2 vs 3 p = 0.155 in liver tissue; time point 1 vs 2 p = 0.385, time point 2 vs 3 p = 0.698 in muscle tissue) was found in both liver and muscle tissue (Fig. 4A).

Expression of Irf7 in liver tissue (A), cGas in liver tissue (B) and cGas in muscle (C) (in red) at different time points. Sections were stained with MitoView (for Irf7) or dsDNA Dye (for cGAS) (in green)
The expression pattern of cGas showed a steady statistically insignificant increase in liver tissue (time point 1 vs 2 p = 0.801, time point 2 vs 3 p = 0.881) (Fig. 4B), while it tended to decrease in muscle between 12 and 16 weeks of age (time point 1 vs 2 p = 0.469, time point 2 vs 3 p = 0.037) (Fig. 4C).
We also performed Oil Red O staining to check liver lipid accumulation. Our analysis revealed a non-statistically significant decrease in lipid staining at 12 weeks of age compared to 8 weeks of age (p = 0.078). Lipid accumulation increased between 12 and 16 weeks of age (p = 0.132).
Discussion
Our study aimed to find early transcriptomic changes in three tissues involved in metabolism regulation in db/db mice: adipose tissue, muscle tissue and liver tissue. Our results clearly show that a dysregulated immune response along with mitochondrial dysfunction are fundamental to the development of obesity and diabetes as early as at 12 weeks of age.
In our analysis of all sequenced tissues, we found that dysregulation of Irf7, a master regulator of the interferon response, is the major driver of the development of diabetes. Other upregulated genes between ages 8 and 12 weeks were Oas2, Stat2, Oasl2 and Sp100, all of which are transcriptional targets of Irf7 that are also activated by foreign DNA or RNA particles. Both Oas2 and Stat2 can be activated by dsRNA, leading to the degradation of viral RNA and the activation of RIG-I pattern recognition receptors (Choi et al. 2015; Li et al. 2022). Oasl, a member of the Oas family, also interacts with RIG-I and enhances antiviral activity. It has also been shown to act as a negative regulator of the cGAS-STING signalling pathway, which is activated by foreign DNA (Ghosh et al. 2019). Another upregulated gene, Sp100 is a known inhibitor of double-stranded DNA viruses, e.g. HSV (Stepp et al. 2013). This corroborates the observation that between 8 and 12 weeks of age, the response to the development of diabetes and obesity depends on pattern recognition receptors (PRRs) and is interferon-centred.
We noted an upregulation of Irf7 in liver tissue between 8 and 12 weeks of age, along with increased expression of cluster of genes known to be induced by it – Ly6a, Ly6e, Ly6g6d – all of which are located next to MHC genes (Khodadoust et al. 1999; Long et al. 2011; Wahadat et al. 2018). The upregulation of Irf7 between the two time points was also seen at the protein level. Moreover, we noted an upregulation of Pld4, a phospholipase with 5'- > 3' DNA exonuclease activity, which can digest single-stranded DNA (Gavin et al. 2018). Pld4 expression is also associated with M1 macrophage expression, which increases the inflammation response (Gao et al. 2017). Pld4-deficient mice have inflammatory disease and an exaggerated TLR9 response, while Pld4- and Pld3-knockdown mice die before the age of 21 days due to lethal liver inflammation (Gavin et al. 2021). We believe that Pld4 upregulation results from an increase of cytosolic nucleic acids, which activate TLR9 PAMP. This cytosolic DNA is sensed by the cGAS-STING pathway, which is also upregulated by IFN. In fact, our immunofluorescence experiment showed a steady increase of cGAS expression in the cytoplasm of liver samples. The only gene which was downregulated between 8 and 12 weeks of age was Idi1. Idi1 is an enzyme necessary for cholesterol synthesis. Its downregulation might result from an increase in ROS production and lead to lower cholesterol synthesis (Sun et al. 2014). Our Oil Red O staining showed a non-statistically significant decrease in lipid staining in liver at the age of 12 weeks compared to 8-week-old mice.
It is interesting that the interferon-associated immune response is muted after 12 weeks of life. We saw a downregulation of Irf7 expression along with its target genes, Ly6a and Ifi44, which are known to inhibit viral replication (Hallen et al. 2007; Busse et al. 2020). This is likely a way to manage increased inflammation. It has been shown that Irf7 KO ob/ob mice gain less weight than wild-type controls (Wang et al. 2013). The downregulation of Irf7 was also seen in immunofluorescence analysis even though the change was not statistically significant. Moreover, we observed a decrease in Xaf1, which is known to contribute to the stabilisation of Irf7 (Jeong et al. 2018). Xaf1 is also an inducer of apoptosis inhibited by another Ifi family member, Ifi6 (Qi et al. 2015). We also found a decrease in the expression of Plac8, which is believed to regulate thermogenesis in brown fat tissue and to influence the size of white adipose tissue cells (Jimenez-Preitner et al. 2011; Lee et al. 2018).
A similar effect was found in muscle tissue, where Irf7 and Xaf1 are downregulated in 16-week-old mice (compared to 12-week-old mice). The downregulation of Rtp4, the interferon stimulated gene (ISG), might stem from lower Irf7 expression. However, Rtp4 is also known to act as a negative regulator of TBK1 and IFN (He et al. 2020). The results also show downregulation of Oasl2, which may reduce RIG activation, but can also serve as an activator of the cGAS-STING signalling pathway. In fact, our immunofluorescence analysis show an upregulation of cGAS in muscle. Finally, our results indicate a downregulation of Pfkfb3, which is a rate-limiting enzyme responsible for commitment towards glycolysis. It is an activator of 6-phosphofructo-1-kinase. Xiang et al. showed that elevated glycolytic metabolism in muscle tissue leads to reduced obesity and IR, thus its downregulation would exacerbate disease development. Pfkfb3 also leads to increased lipid metabolism (Xiang et al. 2021). Moreover, Zhu et al. showed that haematopoietic cell specific disruption of Pfkfb3 aggravates the effects of HFD and leads to increased inflammation (Zhu et al. 2021).
The observed results are closely associated with mitochondrial functioning. The analysis of MitoCarta genes revealed an upregulation of Mavs, a mitochondrial antiviral factor, between 12 and 16 weeks of age. Mavs is an adaptor protein activated by extracellular RNA and responsible for the downstream activation of the TBK1 and IRF3 pathways. Moreover, we found increased expression of the mitochondrial transporter Tomm40L, which interacts with Mavs and recruits IRF3 to the mitochondria (Jacobs and Coyne 2013), inducing IRF3 phosphorylation, dimerisation and nuclear translocation and allowing a full interferon response (Liu et al. 2015). Moreover, under normal conditions, Mavs interacts with mitofusin Mfn-1 and inhibits Mavs-mediated induction of type I IFN (Sandhir et al. 2017). This interaction is apparently distorted, as we found upregulation of Mtfp1, which is responsible for mitochondrial fission and increased cell death (Morita et al. 2017; Aung et al. 2017).
The changes in mitochondrial functioning are also evident from the downregulation of Cox6a2 and Ckmt2. Cox6a2 is part of complex IV of the electron transport chain. Mice lacking Cox6a2 have elevated energy expenditure (likely due to increased uncoupling via UCPs), reduced complex IV activity and increased ROS production (Quintens et al. 2013; Nagai et al. 2021). Ckmt2 synthesises creatine phosphate (PCr) from creatine and ATP produced by mitochondria. PCr is a better transporter of high-energy phosphate. It also liberates mitochondrial ADP to increase the rate of mitochondrial respiration. However, it has also been found that in the futile creatine cycle, a process in which creatine is phosphorylated and then dephosphorylated, energy is dissipated without being used for any mechanical work (Greenhill 2021). This affects thermogenesis, which is an important factor in countering obesity and diabetes. Downregulation of Ckmt2 might thus disturb thermogenesis as well as decrease mitochondrial membrane potential and increase ROS production (Park et al. 2021). On the other hand, PCr depletion has recently been shown to decrease ATP levels and NLRP3 inflammasome activation (Billingham et al. 2022).
In conclusion, the sterile inflammation associated with severe mitochondrial metabolic changes is the underlying reason for the development of diabetes and obesity in db/db mice as early as in 12-week-old animals. This is consistent with the hypothesis that the mitochondrial genome contributes to the aetiology and progression of inflammation [lately reviewed (Marchi et al. 2022)].
Data availability
The data sets generated and analysed during the current study are available from the corresponding authors upon request.
References
Aung LHH, Li R, Prabhakar BS, Li P (2017) Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J Cell Mol Med 21:3394–3404. https://doi.org/10.1111/jcmm.13250
Billingham LK, Stoolman JS, Vasan K et al (2022) Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol 23:692–704. https://doi.org/10.1038/s41590-022-01185-3
Burke SJ, Batdorf HM, Burk DH et al (2017) db / db mice exhibit features of human type 2 diabetes that are not present in weight-matched C57BL/6J mice fed a western diet. J Diabetes Res 2017:1–17. https://doi.org/10.1155/2017/8503754
Busse DC, Habgood-Coote D, Clare S et al (2020) Interferon-induced protein 44 and interferon-induced protein 44-like restrict replication of respiratory syncytial virus. J Virol. https://doi.org/10.1128/JVI.00297-20
Choi UY, Kang J-S, Hwang YS, Kim Y-J (2015) Oligoadenylate synthase-like (OASL) proteins: dual functions and associations with diseases. Exp Mol Med 47:e144–e144. https://doi.org/10.1038/emm.2014.110
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Gao L, Zhou Y, Zhou S-X et al (2017) PLD4 promotes M1 macrophages to perform antitumor effects in colon cancer cells. Oncol Rep 37:408–416. https://doi.org/10.3892/or.2016.5216
Gavin AL, Huang D, Huber C et al (2018) PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat Immunol 19:942–953. https://doi.org/10.1038/s41590-018-0179-y
Gavin AL, Huang D, Blane TR et al (2021) Cleavage of DNA and RNA by PLD3 and PLD4 limits autoinflammatory triggering by multiple sensors. Nat Commun 12:5874. https://doi.org/10.1038/s41467-021-26150-w
Ghosh A, Shao L et al (2019) Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. Immunity 50:51-63.e5. https://doi.org/10.1016/j.immuni.2018.12.013
Greenhill C (2021) Unravelling the molecular basis of futile creatine cycling. Nat Rev Endocrinol 17:381–381. https://doi.org/10.1038/s41574-021-00513-1
Hallen LC, Burki Y, Ebeling M et al (2007) Antiproliferative activity of the human IFN-α-inducible protein IFI44. J Interf Cytokine Res 27:675–680. https://doi.org/10.1089/jir.2007.0021
He X, Ashbrook AW, Du Y et al (2020) RTP4 inhibits IFN-I response and enhances experimental cerebral malaria and neuropathology. Proc Natl Acad Sci 117:19465–19474. https://doi.org/10.1073/pnas.2006492117
Jacobs JL, Coyne CB (2013) Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol 425:5009–5019. https://doi.org/10.1016/j.jmb.2013.10.007
Jeong S-I, Kim J-W, Ko K-P et al (2018) XAF1 forms a positive feedback loop with IRF-1 to drive apoptotic stress response and suppress tumorigenesis. Cell Death Dis 9:806. https://doi.org/10.1038/s41419-018-0867-4
Jimenez-Preitner M, Berney X, Uldry M et al (2011) Plac8 is an inducer of C/EBPβ required for brown fat differentiation, thermoregulation, and control of body weight. Cell Metab 14:658–670. https://doi.org/10.1016/j.cmet.2011.08.008
Khodadoust MM, Khan KD, Bothwell AL (1999) Complex regulation of Ly-6E gene transcription in T cells by IFNs. J Immunol 163:811–819
Lee H, Kim J-I, Park J-S et al (2018) CRISPR/Cas9-mediated generation of a Plac8 knockout mouse model. Lab Anim Res 34:279. https://doi.org/10.5625/lar.2018.34.4.279
Li P, Liu Y, Song R et al (2022) RNA 2’-O-methyltransferase fibrillarin facilitates virus entry into macrophages through inhibiting type i interferon response. Front Immunol. https://doi.org/10.3389/fimmu.2022.793582
Liu S, Cai X, Wu J et al (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. https://doi.org/10.1126/science.aaa2630
Long KK, Montano M, Pavlath GK (2011) Sca-1 is negatively regulated by TGF-βlin myogenic cells. FASEB J 25:1156–1165. https://doi.org/10.1096/fj.10-170308
Marchi S, Guilbaud E, Tait SWG et al (2022) Mitochondrial control of inflammation. Nat Rev Immunol. https://doi.org/10.1038/s41577-022-00760-x
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10. https://doi.org/10.14806/ej.17.1.200
Morita M, Prudent J, Basu K et al (2017) mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell 67:922-935.e5. https://doi.org/10.1016/j.molcel.2017.08.013
Nagai Y, Matsuoka T, Shimo N et al (2021) Glucotoxicity-induced suppression of Cox6a2 expression provokes β-cell dysfunction via augmented ROS production. Biochem Biophys Res Commun 556:134–141. https://doi.org/10.1016/j.bbrc.2021.03.148
Okechukwu IB (2018) Introductory chapter: animal models for human diseases, a major contributor to modern medicine. Experimental animal models of human diseases - an effective therapeutic strategy. InTech, London
Park N, Marquez J, Garcia MVF et al (2021) Phosphorylation in novel mitochondrial creatine kinase tyrosine residues render cardioprotection against hypoxia/reoxygenation injury. J Lipid Atheroscler 10:223. https://doi.org/10.12997/jla.2021.10.2.223
Putri GH, Anders S, Pyl PT et al (2022) Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38:2943–2945. https://doi.org/10.1093/bioinformatics/btac166
Qi Y, Li Y, Zhang Y et al (2015) IFI6 inhibits apoptosis via mitochondrial-dependent pathway in dengue virus 2 infected vascular endothelial cells. PLoS ONE 10:e0132743. https://doi.org/10.1371/journal.pone.0132743
Quintens R, Singh S, Lemaire K et al (2013) Mice deficient in the respiratory chain gene Cox6a2 are protected against high-fat diet-induced obesity and insulin resistance. PLoS ONE 8:e56719. https://doi.org/10.1371/journal.pone.0056719
Rath S, Sharma R, Gupta R et al (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 49:D1541–D1547. https://doi.org/10.1093/nar/gkaa1011
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Sandhir R, Halder A, Sunkaria A (2017) Mitochondria as a centrally positioned hub in the innate immune response. Biochim Biophys Acta 1863:1090–1097. https://doi.org/10.1016/j.bbadis.2016.10.020
Stepp WH, Meyers JM, McBride AA (2013) Sp100 provides intrinsic immunity against human papillomavirus infection. Mbio. https://doi.org/10.1128/mBio.00845-13
Sun Y, Zhang Y, Li N et al (2014) Exposure to high levels of glucose increases the expression levels of genes involved in cholesterol biosynthesis in rat islets. Exp Ther Med 8:991–997. https://doi.org/10.3892/etm.2014.1812
Wahadat MJ, Bodewes ILA, Maria NI et al (2018) Type I IFN signature in childhood-onset systemic lupus erythematosus: a conspiracy of DNA- and RNA-sensing receptors? Arthritis Res Ther 20:4. https://doi.org/10.1186/s13075-017-1501-z
Wang X-A, Zhang R, Zhang S et al (2013) Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am J Physiol Metab 305:E485–E495. https://doi.org/10.1152/ajpendo.00505.2012
Xiang C, Zhang Y, Chen Q et al (2021) Increased glycolysis in skeletal muscle coordinates with adipose tissue in systemic metabolic homeostasis. J Cell Mol Med 25:7840–7854. https://doi.org/10.1111/jcmm.16698
Zhu B, Guo X, Xu H et al (2021) Adipose tissue inflammation and systemic insulin resistance in mice with diet-induced obesity is possibly associated with disruption of PFKFB3 in hematopoietic cells. Lab Investig 101:328–340. https://doi.org/10.1038/s41374-020-00523-z
Acknowledgements
The authors would like to thank Prof. Dr hab. Zbigniew Arent and Dr Laura Pardyak for making the cryostat available for this research. This research was supported in part by PLGrid Infrastructure. The bioinformatic analysis was performed using Prometheus (AGH, Krakow, Poland).
Funding
This study was funded by the Polish Diabetes Association under the grant “Mitochondria in metabolic diseases – the role in obesity” (to ALS).
Author information
Authors and Affiliations
Contributions
AHLS, JW, AB, MD and MŁW were involved in the acquisition and interpretation of the data; AHLS and PPW contributed to the design of the study; AHLS, MTS, AL and JW analysed and interpreted the data; AHLS wrote the paper; GL revised the text, AHLS and PPW are the guarantors of this study. All authors contributed to the critical revision of the manuscript and approved its publication.
Corresponding authors
Ethics declarations
Competing interest
Agnieszka Ludwig-Słomczyńska has received honoraria from RYVU Therapeutics. Anna Ledwoń has received honoraria from Consid.
Ethical approval
This study was approved by the Local Ethics Commission for Animal Experiments in Lodz, Poland (ŁB113).
Consent to publication
All authors contributed to the critical revision of the manuscript and approved its publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Ludwig-Słomczyńska, A.H., Seweryn, M.T., Wiater, J. et al. Cytosolic nucleic acid sensing and mitochondrial transcriptomic changes as early triggers of metabolic disease in db/db mice. Mamm Genome 35, 68–76 (2024). https://doi.org/10.1007/s00335-023-10026-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-023-10026-z