
Abstract
Using the solution casting method, a blend of polyvinyl alcohol/chitosan (PVA/Cs) blend was prepared with different amounts of polypyrrole (PPy) to enhance the structural and electrical conductivity of the films. The miscibility and interaction between PVA/Cs were confirmed by FT-IR measurements by shifting and disappearing some bands suggesting that the PVA functional group interacted with every other functional group on the Cs side chain. For PVA/Cs/PPy, some band intensities varied, indicating the interaction between PPy and PVA/Cs. XRD showed that the addition of PPy to the blend leads to a noticeable decrease in the intensity of the diffraction peak at 2θ = 20.0° confirming the interactions have occurred between the PVA/Cs mixture and PPy. The UV–visible spectra indicate that increasing the amounts of PPy leads to a dramatic decrease in the energy band gap and an increase in the Urbach energy due to the creation of new energy levels that emerged between conduction and valance bands. After PPy was introduced, the polaron and bipolaron transition peaks at 416, 465, and 560 nm become apparent in the PL spectrum. The dielectric and the electrical properties were reported. The values of ε′ and ε′′ were stronger at low frequencies confirming the possibilities of interface polarization processes. The tan δ behavior diagrams have exhibited one peak trend in all samples shift toward higher frequencies as the temperature and PPy rise. The presence of the peaks was explained based on the electrical conductivity mechanism and dielectric behavior. The Cole–Cole plot displays a half one semicircle shape that explains the absence of contact effects. This semicircle expands as both temperature and PPy increase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polymer blending can be considered a superior method used to overcome the disadvantages combined with the use of individual polymers or to obtain new materials with unique characteristics employed for various applications [1, 2]. In addition, using low-cost blend polymers helped to improve both of physical and chemical properties of polymers. Poly(vinyl alcohol) (PVA) is one of the most essential synthetic and biodegradable polymers, and it is widely used in many fields [3, 4]. It is a semi-crystalline structure and the presence of active hydroxyl groups in its structure is the reason for its bonding and its ability to dissolve in water at temperatures according to the molecular weight. PVA is also characterized by its ability to form films alone or with other polymers [5,6,7,8].
Chitosan (CS) is a natural polysaccharide and has gained much attention due to its low cost and easy processing. CS is considered a biopolymer and a biomaterial with a distinct ability to form films alone or with other polymers. The CS is widely used in biomedical applications for its outstanding non-toxicity, biodegradability, antibacterial, and wound healing [9]. CS is used in drug delivery systems [10, 11]. CS is considered a biocompatible polymer with many polymers like PVA. The characteristic of polymer blend can be improved further by adding a filler which may be inorganic material or organic material like polymers. Despite these critical properties, PVA/CS hybrid matrix shows poor electrical and optical properties and still needs improvement. By using the superior physical properties of nanoparticles for bioelectronic and optoelectronic applications [12,13,14], it is possible to create metal-free bio-composite materials with advanced properties. Although the PVA/CS polymer blend has many essential properties, the structural, optical, and electrical properties still need to be improved [10, 12, 15,16,17,18]. Sweah et al. [19] improved the electrical properties of PVA/Cs by the addition of hydroxyethylcellulose and resorcinol monosodium salt hydrate. Ceja et al. [20] modulated the electrical properties of PVA/Cs by the addition of gold nanoparticles and single-wall carbon nanotubes carboxylic acid functionalized. Aziz et al. [21] developed the electrical properties of PVA/Cs by the addition of ammonium iodide. Yousof et al. [22] improved the electrical properties of PVA/Cs by the addition of ammonium bromide. Tommalieh et al. [23] edited the electrical properties of PVA/Cs by the addition of gold nanoparticles using laser ablation method.
Self-conductive polymers have attracted much interest as advanced materials. Polypyrrole (PPy) is one of the most important polymers due to its ease of preparation with potential witnesses and chemical stability besides it has an excellent electrical conductivity. PPy is prepared by chemical oxidation of pyrrole monomer in an organic solvent under certain conditions. The presence of positively charged nitrogen atoms in the matrix for the PPy matrix leads to the ability of PPy to absorb some organic and inorganic anions during ion exchange [24,25,26,27,28]. Therefore, pure PPy can be added to some polymers or polymeric blends. PPy is a promising polymer due to its many commercial applications and properties. It can be used as a biosensor, supercapacitor, polymer battery, electronic device, membrane function, and electrocatalyst.
The electrical properties of PVA/Cs can be enhanced by adding polypropylene (PPy) with different concentrations to take advantage of its properties such as electrical and optoelectronic applications and obtain a metal-free composite with advanced properties. This work reports the effect of additive different weight ratios of polypyrrole (PPy) with good electrophilic properties. On the change in the structural, optical, and electrical properties of the PVA/CS blend using X-ray spectroscopy and UV–Vis spectroscopy, and Fourier transforms infrared (FT-IR) spectroscopy. The electrical conductivity before and after adding polypyrrole at room temperature and over a range of temperatures was studied in detail.
Experimental work
Materials
Poly(vinyl alcohol) (PVA) has a molecular weight of 1.15 × 105 g/mol and has degree of hydrolysis of 98.6% and was purchased from Alpha Chemika, India. High molecular weight chitosan (Cs) obtained from Alamia firm for chemicals, Egypt, has a molecular weight of 140.000 with degree of acetylation of 81.2%, and polypyrrole (PPy) purchased from Sigma–Aldrich doped with proprietary organic acids and 5 wt % solutions in water).
Sample preparation
To make the PVA/Cs polymer solution, a specific weight quantity (2 gm) of PVA was dissolved in distilled water (50 ml) with a stirrer at 65 °C until completely dissolved. The Cs powder (2 gm)was dissolved in distilled water and acetic acid at 65 °C until a homogeneous solution. To make a homogeneous PVA/Cs (50/50 wt%) polymer solution, the two solutions were added to each other under vigorous stirring for about 2 h. After that, PPy solution with different ratios (1, 3, 5, 7, and 10 wt%) was added drop by drop to the PVA/Cs solution, which was then cast in a petri dish and dried at 45 °C for 24 hours.
Measurement techniques
The FT-IR analysis was taken with a Bruker VERTEX 80 (Germany) in the range 4000–500 cm−1 with resolution 4 cm−1. PANalyticalX'Pert Pro XRD measurements were worked at 45 kV with target Cu-K and secondary monochromator Holland radiation at 0.1540 nm in 2 theta ranges 5 ≤ 2θ ≤ 80°. The spectrophotometer Jasco V-630 UV–VIS (Japan) was used to examine ultraviolet–visible absorption spectra. Photoluminescence spectra in the range from 290 to 700 nm excited at 325 nm of the produced samples were obtained at room temperature using a spectrofluorometer (Jasco, FP-6500, Japan). The AC and the dielectric were measured using broadband dielectric spectroscopy (BDS)-type (concept 40) novocontrol high-resolution alpha analyzer with Quatro temperature controllers in frequency range of 10–1–107 Hz.
Results and discussion
FT-IR analysis
Figure 1 represents the FT-IR of pure PVA, pure Cs, and their blend sample (PVA/Cs). As seen in the PVA spectrum, there is broadband at 3268 cm−1 which is related to the stretching vibration of the OH group, while the bending vibration of the OH group is seen at 1327 cm−1. Bands at 2939 cm−1 and 2907 cm−1 correspond to the asymmetric and symmetric stretching vibration of the CH group, respectively. The band at 1713 cm−1 is noticed for the C=O group (residual of acetate group). The band that appeared at 1656 cm−1 is associated with so-called “bound” water, which can overlap with the C=O bonding group. The bending vibration of the CH2 group is seen at 1416 cm−1. The CH wagging vibration is observed at 1377 cm−1 and 1238 cm−1. The C–O stretching vibration is seen as a shoulder at 1141 cm−1. The high intense band at 1087 cm−1 is related to the C=O stretching vibration. The rocking vibration of the CH2 group and the stretching vibration of the C–C group is seen at 916 cm−1 and 842 cm−1, respectively [29,30,31,32,33].

FT-IR spectrum of pure PVA, pure Cs, and PVA/Cs polymer blend
For pure chitosan (Cs), there is broadband at 3245 cm−1 which corresponded to the overlapping of the stretching vibration of N–H and O–H groups. The stretching vibration of the C–H group is observed at 2878 cm−1. There are two characteristics bands in Cs polymer at 1633 cm−1 and 1537 cm−1 which are associated with the amide Ι and amide ΙΙ vibrations. The CH bending vibration is seen at 1405 cm−1, and the wagging vibration of CH2 is seen at 1255 cm−1. The C–O stretching vibrations are observed at 1152 cm−1, 1067 cm−1, and 1020 cm−1. The CH bending out of the plane of the ring monosaccharides is seen at 897 cm−1. The out-of-plane vibration of the N–H group is shown at 650 cm−1 [9, 11, 19,20,21,22]. Figure 2 represents the FT-IR of the PVA/Cs blend and PVA/Cs doped with different concentrations of PPy. For PVA/Cs, it can be observed that the spectrum of PVA/Cs comprises the bands of both PVA and CS. The interaction between PVA and Cs is confirmed by disappearing bands at 1255 cm−1, 1152 cm−1, and 1020 cm−1. It is noticed that there is shifting in the band at 1537 cm−1, 1416 cm−1, 1377 cm−1, 916 cm−1 and 897 cm−1. Also, there is a noticeable change in the intensity of the bands. All these results approved the miscibility between PVA and Cs polymers. For PVA/Cs incorporated with different ratios of PPy, it is observed that there is a change in the intensities of some bands regarded that the PVA functional group interacted with all functional groups on the side chain of Cs. This reaction weakened the complexation between PVA/Cs and PPy.

FT-IR of the PVA/Cs blend and PVA/Cs doped with different concentrations of PPy
XRD
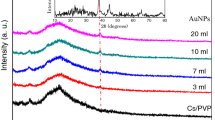
Figure 3a and b represents the X-ray diffraction (XRD) patterns of pure PVA, Cs, and PVA/Cs blend incorporated with different ratios of PPy. The amorphous structure form of chitosan was shown by a very broad diffraction peak at 2θ = 21.5° with another peak at 2θ = 11.8° [34, 35]. The semi-crystalline nature of PVA is evidenced by the presence of diffraction peaks at 2θ = 19.6°, 23.0°, and 40.4° [36,37,38]. The XRD of the chitosan/PVA film exhibits only one diffraction peak at 2θ = 20.0°, whereas the other diffraction peaks of both PVA and chitosan have vanished, indicating the compatibility and miscibility between PVA and Cs. According to the previous literature, PPy has only a broad diffraction peak at 15° < 2θ < 30°. For PVA/Cs incorporated with different ratios of PPy, the diffraction peak at 2θ = 20.0° shifted toward lower theta degree, and its intensity decreased also the diffraction peak of PPy coupled with the diffraction peak of PVA/Cs which revealed the interaction between PVA/Cs and PPy, and the addition of PPy increases the amorphous nature of the blend system causing the increase in the conductivity of the samples.

a X-ray diffraction of the PVA/Cs blend and b PVA/Cs doped with different concentrations of PPy
UV–Vis measurement
Figure 4 represents the UV–Vis spectra of all prepared samples. As seen, PVA spectrum does not show any absorption peak in the UV–Vis region, while the Cs spectrum has a sharp absorption peak at 202 nm due to the chromophoric group of Cs. For PVA/Cs, there is an absorption peak at 201 nm, and this peak is broader than the Cs absorption peak, and this confirmed the complexation between PVA and Cs. By increasing the weight ratio of PPy into the PVA/Cs blend, there is a shift in the peak at 201 nm toward a longer wavelength. Also, there is a new peak that appeared at 228 nm, the π–π* interband transition, i.e., the transition from the valence band to the conduction band of the PPy neutral state, which is suggestive of PPy creation. It is noticed that at a higher weight ratio of PPy, there are broad peaks at 230 nm and 385 nm which are characteristic peaks of PPy. This suggested the interaction between PVA/Cs and PPy.

UV–Vis spectra of PVA, Cs, PVA/Cs blend, and PVA/Cs doped with different concentrations of PPy
The fluctuation of the absorption coefficient (α) with photon energy \({({\text{h}}\upsilon )}\) is a unique factor that provides the most relevant optical information for substance identification, and it can be calculated using equation [9]:
where d is the film thickness and A is the absorbance. Figure 5 shows the relation between absorption coefficient and bandgap energy. It gives an extrapolation of the basic absorption edge values, and their values are listed in Table 1 from the point of intersection of the linear part of the curve with the zero point with the values of the x-axis. As seen, values of absorption edges decrease by increasing ratios of PPy which revealed the formation of the conjugate bond system induced by breaking and reconfiguration of the bonds, which suggests a change in structural modification of PVA/Cs.

Relation between absorption coefficient and photon energy of PVA, Cs, PVA/Cs blend, and PVA/Cs doped with different concentrations of PPy
The absorption of photons will be associated with the occurrence of localized tail states in the forbidden gap for optical transitions generated by photons of energy \({{\text{h}}\upsilon }\) < band gap energy (Eg). The Urbach tail, which is the width of this tail, is a measure of the defect levels in the forbidden bandgap. The width of the Urbach tail was calculated using the following formula where Eu is the band tail energy and α0 is constant [39, 40].
Figure 6 represents the relation between ln α and \({{\text{h}}\upsilon }\), and the reciprocal slope of the straight line of this relation gives the value of band tail energy. Table 1 shows that band tail energy increases with increasing PPy weight ratio which revealed the development of disruption, ionic complexes, and deformities in PVA/Cs due to enhanced interaction between functional groups of PVA/Cs and N–H. The increase in band tail energy shows that PPy introduces localized states within the forbidden energy bandgap. The presence of such localized states may operate as a charge carrier trap.

Relation between ln (α) and photon energy of PVA, Cs, PVA/Cs blend, and PVA/Cs doped with different concentrations of PPy
Tauc’s expression can be used to calculate the optical energy gap of the materials based on the absorbance measurements [41].
where Eg is the optical bandgap energy, and the exponent n is an empirical index. B is a constant that depends on the material structure. The value of n is determined by the type of absorption electronic transition. For direct allowed, direct forbidden, indirect allowed, and indirect forbidden, they can use the values 1/2, 3/2, 2, and 3 correspondingly. Figure 7 shows the relation between (αhʋ)1/2 against photon energy, and as shown in Table 1, the values of bandgap energy decrease with increasing PPy. This is because of the creation of new energy levels that emerged between the conduction and valance bands as a result of the inclusion of PPy, reducing the bandgap of the composites.

Relation between (αhʋ)1/2 against photon energy of PVA, Cs, PVA/Cs blend, and PVA/Cs doped with different concentrations of PPy
PL analysis
The photoluminescence (PL) spectra of pure PVA/Cs doped with different concentrations of PPy in the range from 290 to 700 nm excited at 325 nm are shown in Fig. 8. The emission spectrum of pure PVA/Cs showed a broad peak at 369–487 nm centered at 412 nm which are attributed to π → π* electronic transitions. After the addition of PPy, the peaks at 416, 465, and 560 nm are observed and attributed due to polaron and bipolaron transition [42].

Photoluminescence (PL) emission spectra of PVA/Cs blend and PVA/Cs doped with different concentrations of PPy
AC electrical conductivity
Figure 9a–e depicts the variation of the real part of the dielectric constant (ε′) with the frequency (Log f) at various temperatures (30, 60, 90, and 120 °C). The values of the ε′ at high temperatures are abnormally high. Also, the ε′ gradually decreases as frequency increases. The Maxwell–Wagner model is used to explain the higher values of ε′. The Maxwell–Wagner effect can be used to explain the accumulation of interfacial charge in a blend of two polymers with distinct dielectrics. Maxwell theorized that a dielectric material’s heterogeneous structure (interfacial polarization) could result in dielectric dispersion. This stratified model is conceivable but irrelevant in the case of heterogeneous mixtures of spherical particles (grain boundaries), as the distinction between a dispersion medium and a dispersion phase is omitted from this specimen. Wagner proposed the “interpolarization theory of slightly dispersed grain boundaries scattering.” According to this model, the structure of the material consists of the good grains separated by narrow, highly resistant sheets (granular boundaries). The voltage applied to the sample decreases across the grain boundary, leading to the formation of space charge polarization at the grain boundary. The conductivity of the sample and the accessible free charges on the grain boundary affect the charge carrier polarization. The fact that the electron transfer between the ions never follows the changing of the applied external field beyond that frequency which causes the observable decrease in ε′ values at high frequency, but at low frequencies, the temperature has a significant effect on the dielectric constant [43].

a–e: Variation of the real part (ε′) with the frequency (Log f) at various temperatures (30, 60, 90, and 120 °C)
Increasing the temperature on the samples causes a major rise at the lower frequencies (in the range of frequency from 0.1 to 4.3 Hz), but a rapid drop in the relatively higher-frequency region starting from the frequency range of 100 Hz with a constant value close to zero until the end of the frequency. This phenomenon can be explained by the fact that at applied temperatures, charge carriers are unable to position themselves in the direction of the applied field, and hence contribute to polarization and dielectric constant (ε′). As the temperature increases, bound charge carriers gain enough thermal energy for excitation and can more easily obey changes in the external field, resulting in a greater contribution to polarization and an increase in the ε′ of the samples.
Figure 10 depicts the variation of dielectric loss (ε′′) for the produced samples over a range of frequencies and temperatures. The dielectric loss (ε′′) reduces with increasing frequency, as shown in the graph. When the field is applied to alternating current and during half a cycle, the charge carriers have enough time to travel over a microscopic distance and accumulate at the interfaces between the sample and the electrodes in the low-frequency range, causing very large values of the dielectric loss (ε′′). Charges are transported on microscopic dimensions, and charges are accumulated at the limits of conduction parameters in the samples. In the higher-frequency range, there is practically no time for charges to start at the sample electrode interfaces. Therefore, the electric charges cannot follow the electric field change in the high-frequency region, so the polarization mechanisms of the molecular structure only contribute to the polarization effect. The dielectric loss (ε′′) values in the samples are in the range of 110–1740 at 30 °C and 8.4 × 104–1.21 × 106 at 120 °C, measured at low frequency. Generally, the values of ε′ and ε′′ are stronger at low frequencies, which is due to the possibilities of interface polarization processes, because interface states cannot follow the AC signal at higher frequencies. The Maxwell–Wagner model [44] and space charge polarization are responsible for these frequency dispersions in ε′ and ε′′.

a–e: Variation of the imaginary part (ε″) with the frequency (Log f) at various temperatures (30, 60, 90, and 120 °C)
The dielectric loss tangent (tan δ) against frequency (Log f) of the prepared samples at a range of temperatures is shown in Fig. 11. The dielectric loss tangent (tan δ) is calculated as tan δ = ε′′/ε′ [45]. The diagrams have exhibited a peaking trend in all samples. Temperature and the ratio of PPy added in the PVA/Cs blend affect the observed peaks. The peaks shift toward higher frequencies as the temperature and as the PPy rise. It is also worth noting that the peak’s height drops as the PPy additive increases and temperature rises. The presence of these observed peaks was explained based on the electrical conductivity mechanism and dielectric behavior [42, 45]. And we notice that as the temperature increases, the peak shifts toward a higher frequency, and this shows that the chance of jumping per unit time increases. The orientation of the peak toward higher frequencies is also affected with increasing PPy content and with increasing temperature until the jump probability decreases with increasing PPy content.

a–e: Loss tangent (tan δ) against frequency (Log f) at various temperatures (30, 60, 90, and 120 °C)
The dielectric modulus (M*) is defined as a function of the dielectric permittivity (ε*) as [45, 46]:
where M′ and M″ are the real and imaginary parts of M*. At different temperatures, Fig. 12a–e depicts the real M′ part of the electric modulus as a function of frequency. The M′ values at lower frequencies are very small, tending to zero, implying that electrode polarization has been removed, whereas the M′ values increase with increasing frequency, and it reaches a maximum value at high frequency, due to the distribution of relaxation processes over a wide range of frequencies. The apparent dispersion is primarily attributable to conductivity relaxation over a broad frequency range, indicating the possibility of a relaxation time, which should be matched by a losses peak within the plot of the imaginary part of electric modulus versus frequency. There is no peak in the M′ diagram, suggesting that M′ indicates the material’s ability to store energy. The decrease in M′ values as temperature increases is observed and attributed to the mobility in the polymer component and the increase in charge carriers. It is well-known that charge carriers and molecular dipoles have easier at high temperatures. Figure 13a–e depicts the imaginary M′′ part of the electric modulus as a function of frequency. The development of loss peaks is seen. At lower frequencies, M′′ approaches zero, showing that electrode polarization phenomena play a minor role, even though electrode polarization can induce an apparent increase in the dielectric constant at low frequencies. A high-impedance layer on the electrode surface is the source of the abnormality. This can be produced by poor contact between the electrode surface and the specimen, which is exacerbated by the accumulation of electrolysis products, for example. At low frequencies, any small conduction through the specimen has enough time to transfer all of the applied fields across the very thin electrode layers, resulting in a large increase in measured capacitance. However, well-defined peaks are detected at high frequencies, and the broad and asymmetric nature of peaks on both sides of the maxima indicate non-Debye behavior. In polymer nanocomposite models, jump conduction is a relaxation process, since particle movement is accompanied by a jump process that returns the particle to the vacancy it left behind. As indicated by a global dynamic reaction, the related relaxation process is not strongly non-Debye. The range of charge carriers that can go great distances is shown to the left of the peak. The range in which carriers are confined to potential wells that are mobile over short distances is determined by the region to the right of the peak. The relaxation frequency associated with each peak is what determines the most likely conductivity relaxation time for ions. With increasing temperature, the peak frequency moves to higher frequencies. The energy loss in the irreversible conduction process is related to the M′′.

a–e: Real M′ part against frequency (Log f) at various temperatures (30, 60, 90, and 120 °C)

a–e: Imaginary M′′ part against frequency (Log f) at various temperatures (30, 60, 90, and 120 °C)
The Cole–Cole diagram (relationship between the real part (M′) and the imaginary part (M″)) is shown in Fig. 14a–c. The diagram depicts a half one semicircle shape that explains the absence of contact effects and the Debye model relaxation method. (Most polymeric composites are characterized by Debye relaxation, which can be described by the presence of a single peak in the Cole–Cole diagram.) Here, the grain size or the mobility of free charges caused by interactions between the components in the prepared samples is credited with this model. The recorded semicircle expands as the temperature and as the addition of PPy increase.

a–e: Cole–Cole diagram between the real part (M') and the imaginary part (M") at various temperatures (30, 60, 90, and 120 °C)
Conclusions
In the present work, polyvinyl alcohol/chitosan composites doped with different concentrations of polypropylene (PPy) were synthesized and characterized using different techniques. The structure, optical, and electrical properties of the PVA/Cs-PPy composite films were studied. The additive of PPy to the PVA/Cs blend causes a decrease in the intensity of the X-ray diffraction peaks indicating that interactions can occur between PVA/Cs-PPy. The UV–visible spectra show the absorption was increased dramatically with the decrease in the energy bandgap. The UV–visible band at 228 nm (the transition between the π–π* band) was attributed to the transition from the valence band to the conduction band. The values of bandgap energy decrease with the increase in PPy due to the creation of new energy levels resulting from the inclusion of PPy. The dielectric parameters and the electrical properties were reported. The values of ε′ and ε′′ were obviously at lower frequencies confirming the possibilities of interface polarization processes, and the values are low at higher frequencies because interface states cannot follow the AC signal. The loss tangent (tan δ) behavior diagrams have exhibited one peak trend in all samples shift toward higher frequencies as the temperature and PPy rise. The presence of these observed peaks was explained based on the electrical conductivity mechanism and dielectric behavior. The Cole–Cole plot (M′ with M″ relation) contains a half one semicircle shape that explains the absence of contact effects and the Debye model relaxation method. The recorded semicircle expands as the temperature and as the addition of PPy increase. So, PVA/Cs/ 10 wt% PPy is the best composition that helps to enhance the electrical properties.
References
Kamoun EA, Chen X, Mohy Eldin MS, Kenawy ERS (2015) Crosslinked poly(vinyl alcohol) hydrogels for wound dressing applications: a review of remarkably blended polymers. Arab J Chem. 8:1–14. https://doi.org/10.1016/j.arabjc.2014.07.005
Dorigato A (2021) Recycling of polymer blends. Adv Ind Eng Polym Res 4:53–69. https://doi.org/10.1016/j.aiepr.2021.02.005
Elashmawi IS, Menazea AA (2019) Different time’s Nd:YAG laser-irradiated PVA/Ag nanocomposites: structural, optical, and electrical characterization. J Mater Res Technol 8:1944–1951. https://doi.org/10.1016/j.jmrt.2019.01.011
Ali FM (2019) Structural and optical characterization of [(PVA:PVP)-Cu2+] composite films for promising semiconducting polymer devices. J Mol Struct 1189:352–359. https://doi.org/10.1016/j.molstruc.2019.04.014
Farea MO, Abdelghany AM, Meikhail MS, Oraby AH (2020) Effect of cesium bromide on the structural, optical, thermal and electrical properties of polyvinyl alcohol and polyethylene oxide. J Mater Res Technol 9:1530–1538. https://doi.org/10.1016/j.jmrt.2019.11.078
Zidan HM, El-Ghamaz NA, Abdelghany AM, Waly AL (2018) Photodegradation of methylene blue with PVA/PVP blend under UV light irradiation. Spectrochim Acta - Part A Mol Biomol Spectros. 199:220–227. https://doi.org/10.1016/j.saa.2018.03.057
Elashmawi IS, Abdelrazek EM, Hezma AM, Rajeh A (2014) Modification and development of electrical and magnetic properties of PVA/PEO incorporated with MnCl2. Phys B Condens Matter 434:57–63. https://doi.org/10.1016/j.physb.2013.10.038
Elashmawi IS, Hakeem NA, Abdelrazek EM (2008) Spectroscopic and thermal studies of PS/PVAc blends. Phys B Condens Matter 403:3547–3552. https://doi.org/10.1016/j.physb.2008.05.024
Abdelghany AM, Menazea AA, Ismail AM (2019) Synthesis, characterization and antimicrobial activity of chitosan/polyvinyl alcohol blend doped with Hibiscus Sabdariffa L. extract. J Mol Struct 1197:603–609. https://doi.org/10.1016/j.molstruc.2019.07.089
Kaur R, Goyal D, Agnihotri S (2021) Chitosan/PVA silver nanocomposite for butachlor removal: fabrication, characterization, adsorption mechanism and isotherms. Carbohydr Polym 262:117906. https://doi.org/10.1016/j.carbpol.2021.117906
Abdelrazek EM, Elashmawi IS, Labeeb S (2010) Chitosan filler effects on the experimental characterization, spectroscopic investigation and thermal studies of PVA/PVP blend films. Phys B Condens Matter 405:2021–2027. https://doi.org/10.1016/j.physb.2010.01.095
Menazea AA, Ismail AM, Awwad NS, Ibrahium HA (2020) Physical characterization and antibacterial activity of PVA/chitosan matrix doped by selenium nanoparticles prepared via one-pot laser ablation route. J Mater Res Technol 9:9598–9606. https://doi.org/10.1016/j.jmrt.2020.06.077
Meikhail MS, Abdelghany AM, Awad WM (2018) Role of CdSe quantum dots in the structure and antibacterial activity of chitosan/poly ɛ-caprolactone thin films. Egypt J Basic Appl Sci 5:138–144. https://doi.org/10.1016/j.ejbas.2018.05.003
Abou El-Reash YG, Abdelghany AM, Elrazak AA (2016) Removal and separation of Cu(II) from aqueous solutions using nano-silver chitosan/polyacrylamide membranes. Int J Biol Macromol 86:789–798. https://doi.org/10.1016/j.ijbiomac.2016.01.101
Shabeeba A, Ismail YA (2022) Chitosan/polypyrrole hybrid film as multistep electrochemical sensor: sensing electrical, thermal and chemical working ambient. Mater Res Bull 152:111817. https://doi.org/10.1016/j.materresbull.2022.111817
Janani B, Okla MK, Abdel-Maksoud MA, AbdElgawad H, Thomas AM, Raju LL, Al-Qahtani WH, Khan SS (2022) CuO loaded ZnS nanoflower entrapped on PVA-chitosan matrix for boosted visible light photocatalysis for tetracycline degradation and anti-bacterial application. J Environ Manag 306:114396. https://doi.org/10.1016/j.jenvman.2021.114396
Nezhadali A, Koushali SE, Divsar F (2021) Synthesis of polypyrrole - chitosan magnetic nanocomposite for the removal of carbamazepine from wastewater: absorption isotherm and kinetic study. J Environ Chem Eng 9:105648. https://doi.org/10.1016/j.jece.2021.105648
Elkomy GM, Mousa SM, Abo Mostafa H (2016) Structural and optical properties of pure PVA/PPY and cobalt chloride doped PVA/PPY films. Arab J Chem 9:S1786–S1792. https://doi.org/10.1016/j.arabjc.2012.04.037
Sweah ZJ, Malik FH (2020) Preparation and study the electrical properties of chitosan, hydroxyethylcellulose, and polyvinyl alcohol polymer blend doped with different ratios of 4-(2-Pyridylazo)Resorcinol Monosodium Salt Hydrate. Muthanna J Pure Sci 8:49–55. https://doi.org/10.52113/2/08.01.2021/49-55
Ceja I, González-Íñiguez KJ, Carreón-Álvarez A, Landazuri G, Barrera A, Casillas JE, Fernández-Escamilla VVA, Aguilar J (2020) Characterization and electrical properties of PVA films with self-assembled chitosan-AuNPs/SWCNT-COOH nanostructures. Materials (Basel). https://doi.org/10.3390/ma13184138
Aziz SB, Marf AS, Dannoun EMA, Brza MA, Abdullah RM (2020) The study of the degree of crystallinity, electrical equivalent circuit, and dielectric properties of polyvinyl alcohol (PVA)-based biopolymer electrolytes electrical equivalent circuit, and dielectric. Polymers 12(10):1–17. https://doi.org/10.3390/polym12102184
Yusof YM, Illias HA, Kadir MFZ (2014) Incorporation of NH4Br in PVA-chitosan blend-based polymer electrolyte and its effect on the conductivity and other electrical properties. Ionics (Kiel) 20:1235–1245. https://doi.org/10.1007/s11581-014-1096-1
Tommalieh MJ, Ibrahium HA, Awwad NS, Menazea AA (2020) Gold nanoparticles doped polyvinyl alcohol/chitosan blend via laser ablation for electrical conductivity enhancement. J Mol Struct 1221:128814. https://doi.org/10.1016/j.molstruc.2020.128814
Wang XY, Heng LP, Yang NL, Xie Q, Zhai J (2010) Preparation of polypyrrole/polyvinylalcohol (PPy/PVA) composite foam electrode material. Chin Chem Lett 21:884–887. https://doi.org/10.1016/j.cclet.2010.01.005
Diani J, Gall K (2006) Finite strain 3D thermoviscoelastic constitutive model for shape memory polymers. Polym Eng Sci 46(4) 486–492. https://doi.org/10.1002/pen.20497
Simamora P, Manullang M, Munthe J, Rajagukguk J (2018) The structural and morphology properties of Fe3O4 /Ppy nanocomposite. J Phys Conf Ser 1120:1–7. https://doi.org/10.1088/1742-6596/1120/1/012063
Jha S, Bhavsar V, Tripathi D (2021) Dielectric properties of MWCNT dispersed conducting polymer nanocomposites films of PVA-CMC-PPy. Mater Today Proc 45:4824–4829. https://doi.org/10.1016/j.matpr.2021.01.294
Hryniewicz BM, Gil IC, Vidotti M (2022) Enhancement of polypyrrole nanotubes stability by gold nanoparticles for the construction of flexible solid-state supercapacitors. J Electroanal Chem 911:116212. https://doi.org/10.1016/j.jelechem.2022.116212
Kaur R, Singh KP, Tripathi SK (2022) Electrical, linear and non-linear optical properties of MoSe2/PVA nanocomposites as charge trapping elements for memory device applications. J Alloys Compd 905:164103. https://doi.org/10.1016/j.jallcom.2022.164103
Haleem N, Khattak A, Jamal Y, Sajid M, Shahzad Z, Raza H (2022) Development of poly vinyl alcohol (PVA) based biochar nanofibers for carbon dioxide (CO2) adsorption. Renew Sustain Energy Rev 157:112019. https://doi.org/10.1016/j.rser.2021.112019
Al-Bayati JH, Abood HMA, Al-Shakarchi EK (2022) Synthesis, characterization and electrical studies of AlN:U/IL on some properties of PVA. Mater Today Proc. https://doi.org/10.1016/j.matpr.2022.01.146
Zhao YT, Zhang K, Zeng J, Yin H, Zheng W, Li R, Ding A, Chen S, Liu Y, Wu W, Jing Z (2022) Immobilization on magnetic PVA/SA@Fe3O4 hydrogel beads enhances the activity and stability of neutral protease. Enzym Microb Technol 157:110017. https://doi.org/10.1016/j.enzmictec.2022.110017
Mir FA, Ahmad PA, Ullah F, Naik MM, Ghayas B (2020) Structural, optical & diode studies of PVA-Coumarin composite. Optik (Stuttg) 221:165344. https://doi.org/10.1016/j.ijleo.2020.165344
Amruth K, Abhirami KM, Sankar S, Ramesan MT (2022) Synthesis, characterization, dielectric properties and gas sensing application of polythiophene/chitosan nanocomposites. Inorg Chem Commun 136:109184. https://doi.org/10.1016/j.inoche.2021.109184
Chen H, Yu Z, Ye J, Yang G, Chen L, Pang Y, Zhu L, Tang J, Liu Y (2022) Chitosan functionalized hexagonal boron nitride nanomaterial to enhance the anticorrosive performance of epoxy resin. Colloids Surf A Physicochem Eng Asp 645:128941. https://doi.org/10.1016/j.colsurfa.2022.128941
Elashmawi IS, Al-Muntaser AA, Ismail AM (2022) Structural, optical, and dielectric modulus properties of PEO/PVA blend filled with metakaolin. Opt Mater (Amst) 126:112220. https://doi.org/10.1016/j.optmat.2022.112220
Elashmawi IS, Abdel Baieth HE (2012) Spectroscopic studies of hydroxyapatite in PVP/PVA polymeric matrix as biomaterial. Curr Appl Phys 12:141–146. https://doi.org/10.1016/j.cap.2011.05.011
Zidan HM, Abdelrazek EM, Abdelghany AM, Tarabiah AE (2019) Characterization and some physical studies of PVA/PVP filled with MWCNTs. J Mater Res Technol 8:904–913. https://doi.org/10.1016/j.jmrt.2018.04.023
Soliman TS, Zaki MF, Hessien MM, Elkalashy SI (2021) The structure and optical properties of PVA-BaTiO3 nanocomposite films. Opt Mater (Amst) 111:110648. https://doi.org/10.1016/j.optmat.2020.110648
Abdelhamied MM, Atta A, Abdelreheem AM, Farag ATM, El Sherbiny MA (2021) Oxygen ion induced variations in the structural and linear/nonlinear optical properties of the PVA/PANI/Ag nanocomposite film. Inorg Chem Commun 133:108926. https://doi.org/10.1016/j.inoche.2021.108926
Heiba ZK, Mohamed MB, Imam NG (2017) Fine-tune optical absorption and light emitting behavior of the CdS/PVA hybridized film nanocomposite. J Mol Struct 1136:321–329. https://doi.org/10.1016/j.molstruc.2017.02.020
Abo El Ata AM, Attia SM, Meaz TM (2004) AC conductivity and dielectric behavior of CoAlxFe2-xO4. Solid State Sci 6:61–69. https://doi.org/10.1016/j.solidstatesciences.2003.10.006
Elashmawi IS, Ismail AM (2022) Study of the spectroscopic, magnetic, and electrical behavior of PVDF/PEO blend incorporated with nickel ferrite (NiFe2O4) nanoparticles. Polym Bull. https://doi.org/10.1007/s00289-022-04139-9
Singh AP, Pandey OP, Sharma P (2022) Impedance spectroscopy and magnetic studies on Co2Z ferrite sintered with SiO2 and Bi2O3 additives. Mater Chem Phys 277:1–9. https://doi.org/10.1016/j.matchemphys.2021.125574
Wang J, Lu Z, Chen Z (2019) The novel effects of Cu-deficient on the dielectric properties and voltage–current nonlinearity in CaCu3Ti4O12 ceramics. Mater Sci Eng B Solid-State Mater Adv Technol 243:1–7. https://doi.org/10.1016/j.mseb.2019.03.010
Bakış Y, Auwal IA, Ünal B, Baykal A (2016) Maxwell-Wagner relaxation in grain boundary of BaBixLaxYxFe12−3xO19 (0.0 ≤ x ≤ 0.33) hexaferrites. Compos Part B Eng. 99:248–256. https://doi.org/10.1016/j.compositesb.2016.06.047
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
ISE discussed the results, analyzed the samples, and revised the final of the manuscript before submission. AMI analyzed the data and discussed the results and collected the data. AMA prepared the samples, discussed the results, and revised the manuscripted before submission.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elashmawi, I.S., Ismail, A.M. & Abdelghany, A.M. The incorporation of polypyrrole (PPy) in CS/PVA composite films to enhance the structural, optical, and the electrical conductivity. Polym. Bull. 80, 11379–11399 (2023). https://doi.org/10.1007/s00289-022-04611-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-022-04611-6