
Abstract
Insufficient mechanical properties of hydroxyapatite-based composites prompted the search for new and effective solutions for dental applications. To improve the mechanical properties without losing the remineralization potential, the use of hybrid fillers was proposed. The first of them was based on the formation of hydroxyapatite (HA) layer on the surface of SYLOID®244 silica. The second of the investigated fillers was created by simultaneous synthesis of nanoparticles from precursors of HA and silica. The obtained fillers were extensively characterized by spectral methods including X-ray Diffractometry (XRD), Fourier-Transform Infrared Spectroscopy (FT-IR), and X-ray fluorescence (XRF), as well as by Scanning Electron Microscopy (SEM)/Energy Dispersive Spectroscopy (EDS). Tests using probiotic microorganisms were an important part of the analysis, indicating that there was no potential interaction of the materials with microflora. The tests of degree of conversion, depth of cure, opacity, sorption, solubility, flexural and compressive strength, and the remineralizing potential also showed that the composites with nano-sized silica/HA showed better mechanical properties than the composites with HA alone or commercial silica and at the same time the remineralization remained at the desired level. Thus, the proposed composite has a high application potential in the creation of implants and dental materials.
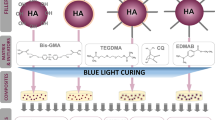
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Calcium phosphates (CPs) are highly biocompatible [1,2,3,4], osteoconductive [2,3,4], and non-toxic [2] ceramic materials. Due to their excellent biological properties, calcium phosphates arouse wide interest from the point of view of medical applications, mainly as components of systems for bone regeneration [1], or a part of an artificial bone [5]. In dentistry, they found application as components of products inducing remineralization, i.e. toothpaste, mouth rinses, chewing gums, etc. [3]. According to their remineralizing potential, understood as the ability to release calcium and phosphate ions, it is not surprising that attempts are made to apply calcium phosphates as bioactive fillers in resin-based composites to obtain restorative materials able to remineralize the surrounding tissues. Additionally, they are also cheaper than other currently used fillers [3]. The most widely examined CP filler is amorphous calcium phosphate (ACP) [6]. It is somehow surprising as ACP is highly soluble in water, which is both an advantage and disadvantage of this calcium phosphate. On the one hand, this high water solubility provides a high amount of ion released. However, on the other hand, it results in a weak mechanical strength of an ACP-filled composite. Moreover, the high ion release capacity is only apparently advantageous, as it lasts shortly (2–3 months) [6]. The most obvious choice, but not so well-examined as ACP, is hydroxyapatite (HA). This choice is obvious due to the chemical similarity of synthetic HA to natural HA that builds teeth and bones [7,8,9,10]. Our previous research proved that HA can release calcium ions when incorporated into the organic matrix of methacrylic resin-based composites (RBC) [11]. Ca2+ release from HA-filled composites is long-term, as it lasts for at least 16 weeks [11]. Despite doubts on the potential mismatch of refractive indices between HA and methacrylic resins, the degree of conversion (DC), as well as depth of cure (DOC) of HA-filled composites are high [12]. In general, the main concern of CP-filled composites is related to the weak mechanical properties [6, 13]. Our previous research on mechanical properties of HA- and tricalcium phosphate (TCP)-filled RBC showed that both flexural and compressive strength should be improved [14]. The flexural strength of these composites was far from ISO 4049:2019 requirements, i.e. lower than 80 or even 50 MPa [15]. Also, compressive strength was below the values established as typical for RBC i.e. below 200–300 MPa, and far from the compressive strength of enamel (384 MPa) and dentin (295 MPa) [7].
In the current research, to overcome this issue, we propose to obtain hybrid fillers that combine HA and silica. Different types of inert silica are the most widely used fillers in dental composites [16, 17]. Among others, silica is responsible for the excellent mechanical properties of such inactive composites. Our most recent study showed that the application of SYLOID®244 that consists of silica particles with high internal porosity and surface area, and narrow particle size distribution, results in the obtaining of RBC with extremely high compressive strength [18].
Therefore, combining bioactive HA showing the remineralizing potential with mechanically strong silica is highly justified. However, in the current research, the preparation of hybrid HA-silica fillers does not rely on the physical mixing them together, which could result in the lack of homogeneity. These hybrid fillers will be obtained in two different ways. The first consists of the preparation of the HA layer on the SYLOID®244 surface, obtaining, therefore, mechanically strong silica particles with an active HA surface layer. This method was used with success by our team to prepare zeolite and montmorillonite fillers with HA layer in the previous research [19,20,21]. All the obtained materials proved their remineralizing potential understood as the ability to release calcium ions with some weakness laying in their mechanical properties [19,20,21]. However, the prediction is that the silica particles will ensure the mechanical properties improvement of such type of fillers. This hypothesis is to be tested within the current research.
The second way of the preparation of a hybrid HA-silica filler that will be applied within this research is co-synthesis of silica and HA in a one-step method that was previously described by Hao et. al. [22]. It is predicted that this way of HA-silica filler preparation will ensure the acquisition of a homogeneous mixture of mechanically strong silica and bioactive HA particles.
The aim of this study is to obtain hybrid HA-silica fillers by using two different methods, characterize and compare their basic properties, apply them as fillers in RBC, and examine the physicochemical properties of the obtained composites. The hypothesis of the research is that the hybrid fillers will ensure the improvement of the mechanical properties of the composites in comparison with HA-filled RBC with maintaining the remineralization potential of HA. To test this hypothesis, also the reference composite with only HA filler with the constant volume fraction will be prepared and examined as a reference. The properties of the composites according to the preparation methods of hybrid HA-silica fillers also will be compared.
Materials and methods
Sodium hydroxide (NaOH), tetraethyl orthosilicate (98%; TEOS), sodium phosphate dibasic (99%, Na2HPO4), tris(hydroxymethyl)aminomethane (99.8%; Tris), hydroxyapatite (HA), bisphenol A glycerolate dimethacrylate (Bis-GMA), triethylene glycol dimethacrylate (95%; TEGDMA), camphorquinone (97%; CQ), ethyl 4-(dimethylamino)benzoate (≥ 99%; EDMAB), potassium hydroxide (≥ 85%, KOH), acetic acid (≥ 99.7%, CH3COOH) were supplied by Sigma-Aldrich (Germany). Hydrochloric acid (36 − 8%, HCl) was obtained from Avantor (Poland). Calcium chloride of high purity (CaCl2) and potassium phosphate monobasic (min. 99%, KH2PO4) were obtained from Chempur (Poland). SYLOID®244 was supplied by GRACE (USA).
Fillers preparation
Synthesis of silica-hydroxyapatite nanoparticles (n-SiO2-HA)
The synthesis was carried out according to Hao et. al. [22]. The scheme of the synthesis is presented in the Fig. 1. In the first stage, 31.5 mL of a 0.2 M NaOH aqueous solution was added into 432 mL of water under stirring. Next, 0.574 g of Na2HPO4 was added to the solution. After 30 min of stirring at 70 °C, 4.05 mL of TEOS was added dropwise under vigorous stirring. Afterwards, 9 mL of solution containing 0.672 g of CaCl2 was added. The reaction was kept at 70 °C for 4 h. The product was filtered and washed with H2O. The last stage was calcination at 400 °C.

The scheme of n-SiO2-HA synthesis
Preparation of silica (SYLOID®244) with hydroxyapatite layer (SYL-HA)
The methodology of hydroxyapatite mineralization was similar to that presented in our previous works [19,20,21]. The scheme of the synthesis is presented in Fig. 2. SYLOID®244 (5 g) was immersed into 100 mL of 200 mM CaCl2/Tris − HCl solution (pH 7.4) for 0.5 h. Then, the powder was filtered and washed with distilled water. In the second step, the material was placed into 100 mL of 120 mM Na2HPO4 solution for the same period of time (0.5 h). This process, consisting of two steps, was repeated five times. The last stage was calcination at 400 °C.

The scheme of SYL-HA preparation
Fillers characterization
X-ray diffractometry (XRD)
Prepared fillers were examined by the X-ray diffraction method in the range of 2 θ from 10 to 70 using a Bruker D8 Advance diffractometer with Johanson monochromator and LynxEye detector. Samples were measured in polymethyl methacrylate cuvette λ Cu Kα1 = 15406 Å. XRD analysis was used to determine the presence of crystalline hydroxyapatite in the synthesized materials.
Fourier-transform infrared spectroscopy (FT-IR)
FT-IR analysis of all materials was carried out using Vertex70 spectrometer, Bruker Optics. Fillers were studied by using a single reflection, diamond ATR crystal. The tests were performed in the spectral range of 4000 − 600 cm−1 with a resolution of 4 cm−1 and 32 scans for signal accumulation. FTIR analysis was used to determine the presence of characteristic bands for silica and hydroxyapatite in the synthesized materials.
Scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS)
SEM images were recorded using TESCAN VEGA 3 SEM. SEM toll was equipped with an EDS XFlash detector 610 M, Bruker Nano GmbH. EDS was used to conduct elemental analysis of the samples. The final concentration of each element was obtained by taking the average of measurements at different spots (at least 8). SEM analysis was used to determine the textural properties of the fillers. EDS analysis was used to determine the amount of given elements and their distribution.
X-ray fluorescence (XRF)
The chemical compositions of samples were measured by X-ray fluorescence (XRF, FISCHERSCOPE® X-RAY XDV®-SDD, Fischer Technology). In order to improve the precision of XRF analysis due to data scattering, 10 measurements for each sample were carried out. The Ca–Si-P wt% was estimated and then their's oxides calculated. XRF analysis was used to determine the content of given elements in the structure of fillers.
X-ray photoelectron spectroscopy (XPS)
The XPS analyses were carried out in a PHI VersaProbeII Scanning XPS system using monochromatic Al Kα (1486.6 eV) X-rays focused to a 100 µm spot and scanned over the area of 400 µm × 400 µm. The photoelectron take-off angle was 45° and the pass energy in the analyzer was set to 117.50 eV (0.5 eV step) for survey scans and 46.95 eV (0.1 eV step) to obtain high energy resolution spectra for the C 1 s, O 1 s, P 2p, Si 2p, Ca 2p and Na 1 s regions. A dual beam charge compensation with 7 eV Ar + ions and 1 eV electrons were used to maintain a constant sample surface potential regardless of the sample conductivity. All XPS spectra were charge referenced to the unfunctionalized, saturated carbon (C–C) C 1 s peak at 285.0 eV. The operating pressure in the analytical chamber was less than 2 × 10–9 mbar. Deconvolution of spectra was carried out using PHI MultiPak software (v.9.9.2). Spectrum background was subtracted using the Shirley method. The XPS analysis was used to determine the content of individual bonds in the obtained materials.
Nitrogen adsorption/desorption measurements
The BET surface area and pore parameters of obtained zeolites were determined by the nitrogen adsorption isotherm technique by means of an ASAP 2420 analyzer (Micromeritrics). Before experiments, the samples were outgassed at 200 °C in a vacuum chamber.
Microbiological study
Firstly, the 100 mg of every filler was mixed with 10 mL of MRS broth and incubated for 7 days at 30 ºC. Then the samples were collected to conduct microbiological analysis. For that Lactobacillus paracasei 3039 (from Polish Microorganisms Collection, Wroclaw, Poland) was used. The strain was cultivated in MRS broth (BTL Sp. z o.o., Łódź, Poland) for 24 h at 30 ºC, centrifuged and suspended in fresh MRS broth to obtain OD600 nm ca. 1.0. and added to MRS broth after filler incubation in ratio 1:3 (v/v).
Evaluation of microbial cells’ viability was performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT). Total membrane permeability was tested by colorimetric measurements of the uptake of crystal violet solution by microbial cells. Cell surface hydrophobicity was analyzed by measuring the adsorption of Congo red dye on the surface of microbial cells. In all microbiological tests, the reference sample was the culture without any filler added [23].
Composites preparation
Three composites with different fillers were prepared. Each composite contained only one filler: HA, n-SiO2-HA, or SYL-HA. The volume fraction of a filler was constant, 55%. This volume fraction was established according to the handling properties of composite: it is the maximum filler content that does not worsen the mixing comfort of the uncured paste. To prepare the examined composites, the components of the organic matrix were mixed together, i.e. Bis-GMA and TEGDMA (3:2). 0.5% of each component of the initiating system (CQ and EDMAB) was added to the mixture. After thorough mixing, the appropriate amount of filler was added to maintain its 55% volume load. All components were further mixed to obtain a homogeneous paste. Then, the paste was placed into PTFE molds with the specific dimensions for each subsequent test (descriptions below), protected with the PET foil from both sides, and cured for 20 s from both sides (except samples for depth of cure) with the use of dental LED lamp (DB-685, Coxo, the wavelength of the emitted light: 420–480 nm, emitted power > 1100 mW·cm−2, diode power: max 5 W).
Composites characterization
Degree of conversion (DC)
DC values were examined following the previously described method [24] using FT-IR Spectroscopy (Vertex70, Bruker Optics) in the ATR mode (diamond crystal). The tests were performed in the spectral range of 4000−600 cm−1 with a resolution of 4 cm−1 and 32 scans for signal accumulation. For each material, at least three samples of uncured paste and at least six samples (ϕ 4 mm, 4 mm thick) of cured composites were examined. DC was calculated from the following equation:
where Rpolymer and Rmonomer are the ratios of the integral intensities of the band assigned to double methacrylic bonds (1638 cm−1) reacting during the polymerization, to the integral intensities of the band assigned to C = C bonds in the aromatic ring that do not react during polymerization (1608 cm−1), recorded in the polymer (after 20 s of curing) and monomer (before curing) spectra, respectively. Mean values and standard deviations were calculated.
Depth of cure (DOC)
DOC values were examined according to the ISO 4049:2019 [15] method. For this purpose, at least three samples of each material were prepared (ϕ 4 mm, multiple 4 mm thickness). The samples were cured of only one side. Immediately after curing, the part of the uncured material was scratched with the use of a spatula. Then, the thickness of the remaining material was measured with the digital caliper (± 0.02 mm) in four places of the sample and divided by two. Mean values and standard deviations were calculated.
Opacity
The opacity was examined with the use of DT-145 colorimeter (Anticorr) for the samples prepared for SP and SL. Opacity was measured by taking the measure of the samples on a white and black background and background subtraction. Mean values and standard deviations were calculated.
Sorption (SP) and solubility (SL)
SP and SL values were examined after the 14-days incubation at 37 °C in deionized water (20 mL) and demineralizing solution (mL) according to the procedure described in ISO 4049:2019 [15]. The demineralizing solution was composed of 2.2 mM CaCl2, 2.2 mM KH2PO4, and 0.05 M CH3COOH. Its pH was regulated to 4.4 with the use of 1 M KOH. It is a typical solution that is commonly used to imitate cariogenic conditions [25]. This solution was applied to examine the impact of such aggressive conditions on the SP and SL values, as well as other properties of the examined composites. Six samples of each material (ϕ 15 mm, 1 mm thick) were prepared and measured with the electronic caliper (± 0.02 mm) in four places. Samples were stored at 37 °C in the presence of silica gel and weighed each day until the constant mass (± 0.1 mg in 24 h) was obtained (m1). Then, three samples of each material were placed in deionized water, and three in the demineralizing solution for 14 days at 37 °C. After the incubation time, samples were surface dried with the blotting paper and weighed after 60 s (m2). Then, samples were stored at 37 °C in the presence of silica gel and weighed each day until the constant mass (± 0.1 mg in 24 h) was obtained (m3). SP and SL were calculated according to the following equations:
where V is the volume of a sample.
Mean values and standard deviations were calculated.
Flexural and compressive strength
Flexural and compressive strength were examined using the universal testing machine Zwick Z010 TN ProLine (Zwick Roell). Test parameters were selected on the basis of ISO 4049:2019 [15] for 3-point bend test (flexural strength) and [14] for compressive strength. At least ten samples of each material were prepared. Crosshead speed was set as 0.75 mm·min−1 for flexural strength and 1 mm·min−1 for compressive strength. In both cases, the initial force was 1 kgF. Samples for the flexural strength were rectangular (25 × 2 × 2 mm), while for the compressive strength they were cylindrical (ϕ 4 mm, 4 mm thick). The distance between the supports in the flexural strength tests was 20 mm. Mean values and standard deviations were calculated.
Calcium release
The concentrations of calcium released from the examined composites were examined in the solutions (deionized water and demineralized solution) after the incubation of samples for SP and SL. For this purpose, a combined ion-selective calcium electrode perfectION (Mettler Toledo) was applied. Three blank samples of deionized water and three blank samples of demineralizing solution stored in the same conditions as the examined solutions (the same vessels, 14 days at 37 °C) were also prepared and analyzed accordingly. For each 20 mL of the examined solution, 0.4 mL of ionic strength adjustment (TISAB) was added. According to the differences in the composites samples masses, the obtained concentration values were recalculated on the average sample mass, i.e. 350 mg.
Statistical analysis
The statistically significant differences in biological properties (cell viability, cell adhesion to Congo red, and cell membrane permeability) of the fillers were examined with the use of one-way variance analysis at a significance level of 0.05.
All results obtained for composites were subjected to statistical analysis. To examine the statistically significant differences in the values of DC, DOC, opacity, SP, SL, flexural strength, compressive strength, and Ca2+ concentrations one-way variance analysis was carried out at the significance level of 0.05. No statistically significant differences in the values of each parameter were denoted by the same capital letters in the figures or tables. Additionally, Student’s t-tests (α = 0.05) were conducted to examine the effect of the storing solution (water or demineralizing solution) in case of SP, SL, and Ca2+ release. No statistically significant differences in the values of each parameter were denoted by the same lower cases in the figures or tables. All calculations were performed with the use of Statistica 13.1 software (TIBCO Software).
Results and discussion
Fillers
The first technique that confirms the effectiveness of creating hydroxyapatite-silica hybrid fillers is XRD (Fig. 3). The peaks seen for n-SiO2-HA and SYL-HA are in line with those of commercial hydroxyapatite (HA). Of course, they are not as sharp as in HA but this is due to the fact that the hydroxyapatite content is much lower in these fillers. In addition, broader peaks derived from hydroxyapatite may indirectly indicate the production of nanohydroxyapatite [26, 27]. Another difference is the presence of a broad peak in the range 15–30 θ, which is characteristic of amorphous silica [28].

X-ray diffraction patterns of HA, n-SiO2-HA, and SYL-HA
The efficiency of the filler synthesis is also visible in the FTIR spectra (Fig. 4). The most characteristic band proving the presence of hydroxyapatite is the band for vibrations from phosphate groups (PO43−) [29, 30]. This band is visible at 1021 cm−1 for HA, while at ~ 1030 cm−1 for n-SiO2-HA and SYL-HA. For silica-hydroxyapatite fillers, the band is much wider and this is due to the overlap of the phosphate band with the stretching vibration band of Si–O-Si. Despite the overlap of the two bands, the sharp band for PO43− is clearly visible.

FT-IR spectra of HA, n-SiO2-HA, and SYL-HA fillers
The morphology of all fillers was assessed on the basis of SEM images (Fig. 5.). The hydroxyapatite (HA) filler particles are non-uniform in shape and size. There are visible cuboidal and plate-shaped particles. In the case of the n-SiO2-HA filler, single particles cannot be distinguished, only large clusters of nanoparticles are visible. Particles of uniform size can be observed for the SYL-HA filler. The most important information obtained from the SEM images is that both in n-SiO2-HA and SYL-HA there are no distinct areas that would indicate the formation of separate silica and hydroxyapatite.

SEM images of HA, n-SiO2-HA, and SYL-HA fillers
Even more detailed information on the distribution of hydroxyapatite and silica was obtained by EDS analysis (Fig. 6). As can be seen in the case of both synthesized fillers, both components (hydroxyapatite and silica) are evenly distributed as evidenced by the distribution of Ca, P, and Si. It is not surprising that commercial hydroxyapatite (HA) has the highest calcium (36%) and phosphorus (16%) content. Interestingly, both synthesized fillers, despite different methods of their preparation, have similar contents of calcium, phosphorus and silicon. It is very important that there are no places where a very large amount of silicon or calcium and phosphorus is visible. The lack of such places indirectly proves the high homogeneity of the obtained fillers. Obtaining fillers with an even distribution of both elements is important because the composite prepared from them should have the same mechanical properties and remineralization potential in each place.

SEM images of fillers (first row). Elemental mapping of the same regions indicating spatial distribution of calcium (second row), phosphorus (third row), and silicon (fourth row). The values represent the content of elements in the filler (by weight)
XRF results also indicate a similar calcium content in the n-SiO2-HA and SYL-HA samples (Table 1). Due to this, the same amounts of remineralizing agent (Ca2+) can be released from both new fillers. In commercial hydroxyapatite, the content of calcium and phosphorus is higher than in the obtained fillers.
Surface concentrations of chemical bonds obtained from fitting XPS data for all analyzed samples are listed in Table 2. Analogously to the previous analyzes, it can be seen that the main component of the samples is silica. There are also traces of sodium and carbon in the sample.
High resolution XPS spectra of samples are presented on the Figs. 7 and 8.

High resolution XPS spectra for n-SiO2-HA

High resolution XPS spectra for SYL-HA
The Si 2p spectra in both samples show one doublet structure (doublet separation p3/2 – p1/2 equals 0.6 eV) with main 2p3/2 line centered at 103.2 eV which indicate Si4+ type compounds found in SiO2 [31, 32]. The P 2p spectra show one doublet structure (doublet separation p3/2 – p1/2 equals 0.8 eV) with main 2p3/2 line centered at 133.2 eV which indicate presence of phosphate groups PO43−, which occur in the structure of hydroxyapatite [31]. The Ca 2p spectra show one doublet structure (doublet separation p3/2 – p1/2 equals 3.6 eV) with main 2p3/2 line centered at 347.4 eV which indicate presence of Ca2+ oxidation state in Ca3(PO4)2 which also indicates the presence of hydroxyapatite [31]. The Na 1 s spectra were fitted with single line centered at 1072.1 eV which indicate Na+ oxidation state of sodium [31]. As can be seen, the amount of sodium is traceable and comes from the substrates used in the synthesis. The O 1 s spectrum was fitted with three components: first line centered at 531.1 eV which points out the existence of O-P type oxygen in phosphate group and/or O = C organic type bonds from contamination, second line centered at 532.7 indicate presence of either O-Si type bonds in silicates and/or O-C organic species from contamination and last line found at 534.3 eV which originates from either -OH and/or adsorbed H2O [31, 33]. The C 1 s spectra can be fitted with three components: first line at 285.0 eV indicate presence of aliphatic carbon, second line centered at 286.4 eV indicating presence of C-O groups and last line found at 289.2 eV which originates from either C = O or O-C = O groups [34]. All detected lines for carbon are typical for organic contamination present on surface of air-handled samples.
SYL-HA filler has a larger surface area, as well as a pore volume, than n-SiO2-HA. (Table 3). However, larger pores are present in the n-SiO2-HA. Comparing the results with the commercial hydroxyapatite, the surface area of which is about 30 m2·g−1 and the pore volume 0.22 cm3·g−1, it can be noted that the increase in these values for the obtained fillers is closely related to the presence of silica which has a more developed surface area [35].
Cells of the probiotic strain Lactobacillus paracasei 3039 were analyzed for changes in metabolic activity (cell viability), adhesion properties, and cell membrane permeability (Table 4). The collected results clearly indicate that the tested fillers, i.e. hydroxyapatite and silica-hydroxyapatite materials, did not show toxic effects on microorganisms and no negative effects on the other tested parameters were observed. Thus, a negative effect on probiotic bacteria can be excluded.
Interactions between Lactobacilli and hydroxyapatite and derivatives based on it have been studied, although the question of toxicity has not been analyzed and authors have usually focused on cell adhesion to the material [36, 37]. However, the information on ion release from hydroxyapatite required studying this issue to be sure that all tested fillers are safe against probiotic bacteria that are present in the gastrointestinal tract and may be exposed to ions released e.g. in the oral cavity [38].
Composites
As the filler type is the only variable in the composition of the tested composites, in order to simplify the reading of the results, the names of the composites come from the type of the applied filler.
Typically, DC values of Bis-GMA-based composites are in the range of 52–75%, while the 55% value is considered the lower limit for the occlusal restorations [39]. All examined composites show mean DC values far above 55% (Fig. 9).

Mean values of DC of examined composites (error bars present the values of standard deviation; dotted line presents the critical value for occlusal restorations (55%) [40]; the same capital letters (A) mean no statistically significant differences (p > 0.05) in values of DC)
Mean values (± SD) of this parameter are 76.0 (± 8.2), 82.7 (± 14.4), and 74.9 (± 6.2)% for HA, n-SiO2-HA, and SYL-HA, respectively. There are no statistically significant differences in the DC values between the examined composites, so it can be stated that in this case, the effect of the filler type is negligible.
The second parameter related to the curing of the composites that were examined is DOC. Its values are presented in Fig. 10. DOC examination consists of scratching of the uncured part of the composite that was cured of only one side. This one-sided light exposure causes that DOC values differentiate better the effect of the applied filler on the curing.

Mean values of DC of examined composites (error bars present the values of standard deviation; dotted lines present the ISO 4049:2019 requirements [15]; the same capital letters (A, B, C) mean no statistically significant differences (p > 0.05) in values of DOC)
Mean DOC values (± SD) are 3.41 (± 0.09), 5.07 (± 0.18), and 2.77 (± 0.32) mm for HA, n-SiO2-HA, and SYL-HA, respectively. First of all, all these composites meet ISO 4049:2019 criteria for opaque (1 mm) and other restorative materials (1.5 mm) in terms of DOC [15]. The differences observed between the materials are statistically significant (p < 0.05). The highest DOC is observed in n-SiO2-HA, while the smallest depth shows SYL-HA. These differences probably result from different optical properties of the applied fillers, especially the refractive index, as well as particle size. Higher resin and filler refractive index differences result in lower light transmittance through the hardened paste, which causes lower DOC values, as well as higher opacity of the composite [40]. A clear correlation between DOC and opacity is observed (Table 5). The lowest opacity shows n-SiO2-HA composite, while the highest opacity is observed for SYL-HA. The differences in the opacity values are statistically significant (p < 0.05). Lower opacity corresponds with the higher DOC.
The amount of the light that is transmitted through the irradiated material and DOC values depend also on the fillers particle size. DOC became shallower when the filler particle size increases [41]. The obtained results are consistent with this finding, as the n-SiO2-HA filler is nano-sized, while HA and SYL-HA fillers are micro-sized.
The maximum values of SP and SL defined by ISO 4049:2019 are ≤ 40 and ≤ 7.5 μg·mm−2, respectively [15]. All examined composites show higher values of SP (Table 5), thus not meeting the conditions of the ISO. There are no statistically significant differences in the SP values depending on the filler type, as well as solution i.e. deionized water or demineralizing agent. No effect of the solvent type is optimistic, as it was predicted that the cariogenic demineralizing solution act more aggressively than the water.
SL values of all examined composites are lower than 7.5 μg·mm−2, but they are mostly negative (Table 5). It means that their m3 was higher than m1, so the materials strongly absorbed the fluids from their surroundings during the incubation. This absorption prevails over the solubility. No effect of the solvent type was observed, which, one more time, is a positive observation. The effect of the filler type can be observed. HA-filled composite shows lower SL absolute values in both solvents. Probably the presence of the silica in n-SiO2-HA and SYL-HA results in the stronger irreversible bonding of the water by the composites. It is not surprising, as silica is strongly hydrophilic. To overcome this weak mass stability of the examined composites in the future, fillers silanization or hydrophobization is recommended.
Minimum flexural strength values for class 2, group 1 polymer-based restorative materials that are intended to restore the occlusal and other surfaces of teeth are defined by ISO 4049:2019 as 80 and 50 MPa, respectively [15]. Class 2, group 1 restorations are defined as materials cured intra-orally that require the external source of energy, e.g. blue light-emitting lamp [15], therefore, the examined materials fit into this criterion. Flexural strength values of the examined composites are presented in Fig. 11.

Mean values of flexural strength of all examined composites (error bars present the values of standard deviation; dotted lines present the ISO 4049:2019 requirements [15]; the same capital letters (A, B) mean no statistically significant differences (p > 0.05) in values of flexural strength)
Mean values (± SD) of flexural strength of examined composites are 71.56 (± 6.27), 53.08 (± 1.54), and 60.37 (± 1.54) for HA, n-SiO2-HA, and SYL-HA, respectively. HA-filled composite show statistically significantly higher value of this parameter than silica-HA-filled composites (n-SiO2-HA and SYL-HA). There are no difference in flexural strength between n-SiO2-HA and SYL-HA (p > 0.05).
It may be related to the smaller particle size of silica-HA fillers (nano for n-SiO2-HA and 2–3 μm for Syloid 244) compared to HA (c.a. 10 μm). Smaller filler particles create a larger contact surface with the components of the organic matrix. It causes the "saturation" of the resin and forming the cracks on the filler/matrix interface during tests where the bending load is applied [42]. Unfortunately, none of the examined materials is appropriate for the restoration of occlusal surfaces of teeth, as their flexural strength values are lower than 80 MPa. However, they all meet the criterion for the restoration of non-occlusal surfaces exceeding the value of 50 MPa.
The compressive strength of dentin and enamel is 297 and 384 MPa, respectively [7]. Mean values (± SD) of this parameter of examined composites are 292.58 (± 18.82), 402.27 (± 29.93), and 269.27 (± 27.96) for HA, n-SiO2-HA, and SYL-HA, respectively (Fig. 12). There are no statistically significant differences in compressive strength between HA and SYL-HA, while the value of this parameter obtained for n-SiO2-HA is significantly higher.

Mean values of compressive strength of all examined composites (error bars present the values of standard deviation; dotted lines present the compressive strength of dentin and enamel; the same capital letters (A, B) mean no statistically significant differences (p > 0.05) in values of compressive strength)
Considering the mean values of compressive strength, together with the standard deviations it can be cautiously assumed that HA and SYL-HA compressive strengths are very close to the compressive strength of dentin. However, the values of this parameter for both mentioned materials are far lower than the compressive strength of enamel. Therefore, in terms of this parameter, both materials could be considered as appropriate to restore dentin, but inappropriate to restore enamel. The compressive strength of n-SiO2-HA, which is higher than the compressive strength of both discussed tissues, can ensure success in restoring enamel and dentin. The higher value of compressive strength of n-SiO2-HA in comparison with HA and SYL-HA results probably from the smaller particle size of n-SiO2-HA. In general, it is a rule that nanocomposites show higher compressive strength than composites with greater particles [7]. The values of compressive strength show positive correlation with the values of DOC and opacity, i.e. materials showing lower opacity show higher compressive strength and DOC.
Mean values (± SD) of calcium concentration detected in the deionized water after incubation the samples of the examined materials are 1.94 (± 0.27), 0.63 (± 0.33), and 1.13 (± 0.13) ppm for HA, n-SiO2-HA, and SYL-HA, respectively (Fig. 13). HA shows a significantly (p < 0.05) higher amount of calcium released during incubation in deionized water, while the differences between both silica-HA-filled composites (n-SiO2-HA and SYL-HA) are statistically insignificant (p > 0.05). It is not surprising that HA-filled composite is the most effective in the Ca2+ releasing as the only filler in this composite is HA. In contrast, both remaining composites contain also silica, so the amount of calcium-bearing filler is obviously smaller. In all cases, incubation of the samples in the demineralizing solution that imitates the cariogenic environment results in higher calcium concentrations. These differences are statistically significant for all examined materials (different lower cases). Mean values (± SD) of calcium concentration detected in the demineralizing solution after incubation the samples of the examined materials are 7.13 (± 1.64), 7.06 (± 0.35), and 6.62 (± 0.62) ppm for HA, n-SiO2-HA, and SYL-HA, respectively (Fig. 11). There are no statistically significant differences (p > 0.05) in the values of Ca2+ concentrations between all examined materials. Higher concentrations of calcium in demineralizing solution in comparison with deionized water results probably from the presence of phosphate ions in the demineralizing solution. The research proves that PO43− ions in the solution increase the solubility of Ca2+ ions included in the hydroxyapatite. This phenomenon is explained by the formation of complexes of calcium phosphates such as Ca(HPO4)(H2PO4)− and Ca(H2PO4)OH [43]. This increase of the Ca2+ solubility is relatively small compared to the total weight of the composite, therefore, it is not reflected in the SL values. Since HA is bounded to silica only by physical interactions in silica-hydroxyapatite materials, the formation of complexes with phosphates contained in the demineralization solution probably exceeds the strength of these interactions, resulting in the equalization of Ca2+ ion release capacity by all tested composites.

The concentration of calcium ions released by the examined composites recalculated on the average sample mass, i.e. 350 mg (mean values, error bars present the values of standard deviation; the same capital letters (A, B, C) mean no statistically significant differences (p > 0.05) in values of Ca2+ concentrations – calculated separately for deionized water and demineralizing solution; the same lower cases (a, b, c, d, e, f) mean no statistically significant differences (p > 0.05) in values of Ca.2+ concentrations – calculated in pairs in which the type of the solution was set as variable, while the material type was constant)
The increased ability of tested materials to release Ca2+ in demineralizing solution in comparison with deionized water should be considered beneficial. It means that the cariogenic environment activates the anticaries mechanisms, understood as the ability to Ca2+ release with no significant mass loss of the composite.
Conclusions
Both silica-hydroxyapatite hybrid fillers were prepared successfully. Their homogeneity and non-toxicity were proved allowing, therefore, the application in the potential dental restorations. Among these two prepared hybrid fillers, nano-sized silica that was co-synthesized with HA (n-SiO2-HA) proved to be better than SYLOID®244 with HA layer (SYL-HA). RBC filled with n-SiO2-HA showed better DOC and compressive strength in comparison with HA-filled RBC, simultaneously maintaining the remineralization potential understood as the ability to release calcium ions. The mechanism of action of these fillers with remineralization potential can be simple explained with phenomenon of the dissolution of hydroxyapatite, which takes place to a greater extent in the caries environment, what is beneficial for potential patients’ treatment. Therefore, the research hypothesis is partially confirmed. However, it has to be also partially rejected as the flexural strength of n-SiO2-HA composite was lower than that of the HA composite. This property has to be further improved as it is still too low to meet the ISO 4049:2019 criterion for the occlusal restorations. In the context of using the second hybrid filler (SYL-HA), the research hypothesis must be rejected in its entirety, as SYL-HA-filled RBC showed inferior mechanical properties in comparison with HA-filled RBC. Despite that the mass stability (SP and SL) during incubation of all examined composites was the same, it has to be also improved as it is lower than acceptable. However, mixing the fillers with different particle sizes or silanization may improve the flexural strength of the composites. As all materials were rather hydrophilic, the filler’s surface hydrophobization may in turn result in the mass stability improvement. Therefore, since the obtained results are very perspective, further experiments are required to finally verify effectiveness of studied materials.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Dorozhkin SV, Epple M (2002) Biological and medical significance of calcium phosphates. Angew Chem Int Edit 41:3130–3146
Dorozhkin SV (2010) Bioceramics of calcium orthophosphates. Biomaterials 31:1465–1485
Dorozhkin SV (2013) Calcium orthophosphates in dentistry. J Mater Sci – Mater M 24:1335–1363
Xie C, Lu H, Li W, Chen FM, Zhao YM (2012) The use of calcium phosphate-based biomaterials in implant dentistry. J Mater Sci – Mater M 23:853–862
Malidarre RB, Akkurt I, Malidarreh PB, Arslankaya S (2022) Investigation and ANN-based prediction of the radiation shielding, structural and mechanical properties of the Hydroxyapatite (HAP) bio-composite as artificial bone. Radiat Phys Chem 197:110208
Braga RR (2019) Calcium phosphates as ion-releasing fillers in restorative resin-based materials. Dent Mater 35:3–14
Sakaguchi RL, Powers JM (eds) (2012) Craig’s Restorative Dental Materials Thirteenth. Elsevier, Philadelphia
Laurance-Young P, Bozec L, Gracia L, Rees G, Lippert F, Lynch RJM, Knowles JC (2011) A review of the structure of human and bovine dental hard tissues and their physicochemical behaviour in relation to erosive challenge and remineralisation. J Dent 39:266–272
Meyers MA, Chen PY, Lin AYM, Seki Y (2008) Biological materials: Structure and mechanical properties. Prog Mater Sci 53:1–206
Low IM, Duraman N, Mahmood U (2008) Mapping the structure, composition and mechanical properties of human teeth. Mat Sci Eng C 28:243–247
Okulus Z, Buchwald T, Voelkel A (2016) Calcium release from experimental dental materials. Mat Sci Eng C 68:213–220
Z. Okulus, T. Buchwald, A. Voelkel, (2015) Characterization of light-cured, dental-resin-based biocomposites, J. Appl. Polym. Sci. 132 42812 (1–10).
E. Ali Abou Neel, A. Aljabo, A. Strange, S. Ibrahim, M. Coathup, A.M. Young, L. Bozec, V. Mudera, (2016) Demineralization–remineralization dynamics in teeth and bone. Int J Nanomed 11:4743–4763
Okulus Z, Voelkel A (2017) Mechanical properties of experimental composites with different calcium phosphates fillers. Mat Sci Eng C 78:1101–1108
ISO 4049:2019: Dentistry-Polymer-based restorative materials.
Pratap B, Gupta RK, Bhardwaj B, Nag M (2019) Resin based restorative dental materials: characteristics and future perspectives. Jpn Dent Sci Rev 55:126–138
Habib EG, Wang R, Wang Y, Zhu M, Zhu XX (2016) Inorganic fillers for dental resin composites - present and future, ACS Biomater. Sci Eng 2:1–11
Sandomierski M, Buchwald Z, Buchwald T, Voelkel A (2021) Silica-filled methacrylic composites with extremely high compressive strength. J Mech Behav Biomed 116:104319
Okulus Z, Sandomierski M, Zielińska M, Buchwald T, Voelkel A (2019) Zeolite fillers for resin-based composites with remineralizing potential, spectrochim. Acta A 210:126–135
Buchwald Z, Sandomierski M, Voelkel A (2020) Calcium-rich 13X zeolite as a filler with remineralizing potential for dental composites, ACS biomater. Sci Eng 6:3843–3854
Sandomierski M, Buchwald Z, Voelkel A (2020) Calcium montmorillonite and montmorillonite with hydroxyapatite layer as fillers in dental composites with remineralizing potential. Appl Clay Sci 198:105822
Hao X, Hu X, Zhang C, Chen S, Li Z, Yang X, Liu H, Jia G, Liu D, Ge K, Liang X-J, Zhang J (2015) Hybrid mesoporous silica-based drug carrier nanostructures with improved degradability by hydroxyapatite. ACS Nano 9:9614–9625
Pacholak A, Smułek W, Zgoła-Grześkowiak A, Kaczorek E (2019) Nitrofurantoin—microbial degradation and interactions with environmental bacterial strains. Int J Env Res Pub He 16:1526
Okulus Z, Buchwald T, Szybowicz M, Voelkel A (2014) Study of a new resin-based composites containing hydroxyapatite filler using Raman and infrared spectroscopy. Mater Chem Phys 145:304–312
Buchwald T, Buchwald Z (2019) Assessment of the Raman spectroscopy effectiveness in determining the early changes in human enamel caused by artificial caries. Analyst 144:1409–1419
Zhang J, Zhao Y, Palosz B (2007) Comparative studies of compressibility between nanocrystalline and bulk nickel. Appl Phys Lett 90:1–3
Londoño-Restrepo SM, Jeronimo-Cruz R, Millán-Malo BM, Rivera-Muñoz EM, Rodriguez-García ME (2019) Effect of the nano crystal size on the X-ray diffraction patterns of biogenic hydroxyapatite from human, bovine, and porcine bones. Sci Rep 9:5915
Maddalena R, Hall C, Hamilton A (2019) Effect of silica particle size on the formation of calcium silicate hydrate[C-S-H] using thermal analysis, Thermochim. Acta 672:142–149
Zima A (2018) Hydroxyapatite-chitosan based bioactive hybrid biomaterials with improved mechanical strength. Spectrochim Acta A 193:175–184
Coelho CC, Grenho L, Gomes PS, Quadros PA, Fernandes MH (2019) Nano-hydroxyapatite in oral care cosmetics: characterization and cytotoxicity assessment. Sci Rep 9:11050
Wagner AD, Naumkin, AV Kraut-Vass A, Allison JW, Powell CJ, Rumble JRJ, NIST Standard Reference Database 20, in, http:/srdata.nist.gov/xps/, 2003
Wagner CD, Passoja DE, Hillery HF, Kinisky TG, Six HA, Jansen WT, Taylor JA (1982) Auger and photoelectron line energy relationships in aluminum–oxygen and silicon–oxygen compounds. J Vac Sci Technol 21:933–944
Biesinger MC, Lau LWM, Gerson AR, Smart RSC (2010) Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl Surf Sci 257:887–898
High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database (Beamson, G.; Briggs, D.), Journal of Chemical Education, 70 A25 (1993)
Adamska K, Szubert M, Voelkel A, Okulus Z (2013) Characterisation of hydroxyapatite surface modified by poly(ethylene glycol) and poly(hydroxyethyl methacrylate) grafting. Chem Pap 67(4):429–436
Jalasvuori H, Haukioja A, Tenovuo J (2012) Probiotic Lactobacillus reuteri strains ATCC PTA 5289 and ATCC 55730 differ in their cariogenic properties in vitro. Arch Oral Biol 57(12):1633–1638
Venegas SC, Palacios JM, Apella MC, Morando PJ, Blesa MA (2006) Calcium modulates interactions between bacteria and hydroxyapatite. J Dent Res 85(12):1124–1128
Mocanu A, Cadar O, Frangopol PT, Petean I, Tomoaia G, Paltinean GA, Racz CP, Horovitz O, Tomoaia-Cotisel M (2021) Ion release from hydroxyapatite and substituted hydroxyapatites in different immersion liquids: in vitro experiments and theoretical modelling study R. Soc Open Sci 8(1):201785
Alshali RZ, Silikas N, Satterthwaite JD (2013) Degree of conversion of bulk-fill compared to conventional resin-composites at two time intervals. Dent Mater 29:213–217
Shortall AC, Palin WM, Burtscher P (2008) Refractive index mismatch and monomer reactivity influence composite curing depth. J Dent Res 87:84–88
Fujita K, Ikemi T, Nishiyama N (2011) Effects of particle size of silica filler on polymerization conversion in a light-curing resin composite. Dent Mater 27:1079–1085
Ikejima I, Nomoto R, McCabe JF (2003) Shear punch strength and flexural strength of model composites with varying filler volume fraction, particle size and silanization. Dent Mater 19:206–211
Chen ZF, Darvell BW, Leung VWH (2004) Hydroxyapatite solubility in simple inorganic solutions. Arch Oral Biol 49:359–367
Acknowledgements
This work was supported by the Ministry of Education and Science.
Funding
This work was supported by the Ministry of Education and Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Buchwald, Z., Sandomierski, M., Smułek, W. et al. Physical–chemical and biological properties of novel resin-based composites for dental applications. Polym. Bull. 80, 11249–11272 (2023). https://doi.org/10.1007/s00289-022-04606-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-022-04606-3