
Abstract
In this work, the results of the research on the properties of hydroxyalkyl derivatives of bisoxamidoester have been presented. Special attention was paid for the assessment of their potential application for obtaining foamed polyurethane materials. Using these derivatives as polyol components and 4,4′-diphenylmethane diisocyanate, rigid foamed polyurethane materials of increased thermal stability have been obtained. The new polyurethane foams obtained using hydroxyalkyl derivatives with bisoxamidoester groups show good dimension stability and better heat-insulating and strength properties as compared to classic foams. These foams can be used as heat-insulating materials of permanent operating temperature 150 °C and temporarily even approaching 200 °C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
From 1 year to the next increasing demand for polyurethane materials is observed. Depending on the kind of the foam, the materials are exploited in many fields, and that is why it is so important to improve their properties and the way of their production.
It should be emphasized that the typical polyurethane foams loose their properties most commonly above the temperature 100 °C and they are not resistant to higher temperatures [1]. It considerably limits their application. In addition, typical polyurethane foams are combustible. The high content of carbon and hydrogen, large specific surface area and cellular structure make polyurethane foams very low resistant to burning. To reduce the risk of the inflammability foams, flame retardants are used. Flame retardants are substances which delay polymer ignition by increasing their ignition temperature, slowing the spread of flame and products of combustion and reducing the formation of smoke. Halogen-containing hydrocarbons, organophosphates, organohalogenophosphates, compounds of nitrogen atoms, and organoboron compounds and metal hydroxides are used as the flame retardants [2].
Introducing additive flame retardants for the foam compositions often causes deterioration of the polyurethane foams’ properties, therefore, more effective and safe is the use of the reactive flame retardants.
Therefore, new polyols are sought after which will play double role and will improve the properties of polyurethane foams.
In recent years, much research has been devoted to the synthesis of the so-called polyols from renewable raw materials, i.e., derivatives of vegetable oils (rapeseed oil, sunflower oil, palm oil, coconut oil, soybean oil [3–5]). They are esters of glycerol and higher unsaturated fatty acids. Oligoesterols are most commonly prepared from them by epoxidation or other oxidation of double bonds, etc. [6].
Foams prepared from oligoesterols are more thermally stable [7].
Furthermore, the glycolysis products of PUR waste [8–10] and other materials may also be used as polyols. In industrial scale, the polyol from recycled poly(ethylene terephthalate) is used in the preparation of full valuable, porous polyurethane materials [11].
Due to the facts I decided to use oxamidoester derivatives as polyol components to improve the properties of the foams.
In polymer chemistry, oxamic acid (oxalic acid monoamide) and its derivatives [12–14] may be used for modification of the structure and properties of materials. According to the literature data, the oxamidoester group was being introduced into polymer chains mainly using diethyl oxalate [15, 16].
Searching for new polyols for obtaining polyurethane foams, it was decided to synthesize N- and O-(hydroxyalkyl)-substituted bisoxamidoesters, and then use them to obtain oligoetherols containing bisoxamidoester group.
Izumi et al. [17] reacted 2-methyl-2-propylpropane-1,3-diol with oxalyl chloride and the product obtained was reacted with ammonia. In this way, it was obtained an oxamidoester with unsubstituted amide groups (I).

Following the model of this synthesis it was decided to obtain N-(hydroxyalkyl)-substituted bisoxamidoesters, and then to subject them to reactions with alkylene carbonates. In this way, the new polyol components for polyurethane foam syntheses have been obtained, and then using 4,4′-diphenylmethane diisocyanate (MDI), polyurethane foams were obtained and their properties were examined.
Experimental
Raw materials
The starting materials: dipropylene glycol p.a., Fluka, diethyl ether p.a., POCH, Poland, oxalyl chloride p.a., Merck, Germany, 2-aminoethanol, p.a., Chempur, Poland; ethylene carbonate, p.a., Fluka, Switzerland; propylene carbonate p.a., Fluka, Switzerland, potassium carbonate, p.a., POCH Gliwice, Poland; diazabicyclo[2.2.2]octane (DABCO), p.a., Avocado, Germany, polymeric 4,4′-diphenylmethane diisocyanate, 98 %, Merck, Germany (wt% NCO = 32, f = 2.5); triethylamine, p.a., Avocado, GB; silicone L6900, Momentive, US were purchased and used without further purification.
Syntheses
Obtaining 2,4-dimethyl-3-oxapentane-1,5-diyl-N,N′-bis(2-hydroxyethyl)-oxamate
In a 100-cm3 three-necked flask, a solution of 7.1 cm3 (7.2 g, 0.05 mol) of dipropylene glycol in 10 cm3 of diethyl ether was placed and cooled down to 0 °C. Next the solution of 8.8 cm3 (12.8 g, 0.1 mol) of oxalyl chloride in 10 cm3 of diethyl ether was dropped in, at such a rate that the reaction temperature was not exceeding 5 °C. Next 6.2 cm3 (6.5 g, 0.1 mol) of 2-aminoethanol was dropped in, maintaining temperature below 5 °C. The post-reaction mixture was left for 24 h, and then ether was evaporated in a rotary evaporator and a precipitate was crystallized from methanol. The product obtained (BOAE) was dried to constant weight in a vacuum dryer at 40 °C and at the pressure of 0.09 MPa.
 : IR (KBr), ν = 3200–3600 (w, O–H, N–H valence), 2952 (w, –CH2–, asym. valence), 2882.5 (w, –CH2–, sym. valence), 1745 and 1668 (s, C=O, valence), 1515 (w, N–(CO), valence), 1480 and 1440 (s, –CH2–, scissor), 1260 and 1195 (m, C–O(CO), asym. and sym. valence), 1114 (s, C–O–C, valence), 1058 (m, C–O–H, valence), (cm−1); 1H-NMR (500 MHz, d6-DMSO), δ = 1.00 (3 H, d, –CH–CH
3, J
7,6 = 5.7 Hz), 2.85 (2H, t, –CH
2–NH, J
1,2 = 5.5 Hz), 3.20 (1 H, m, –CH–CH3, J
6,7 = 5.7 Hz), 3.60 (2 H, t, –CH
2–OH, J
1,2 = 5.5 Hz), 3.80 (2 H, t, –CH
2–O(CO), J
5,6 = 5.9 Hz), 8.15 (1 H, s, –OH), 8.15 (1 H, s, –NH) (ppm), EA: % N
calc.
= 7.69, % N
det.
= 7.57, % C
calc.
= 46.15, % C
det.
= 46.30, % H
calc.
= 6.69, % H
det.
= 7.12.
: IR (KBr), ν = 3200–3600 (w, O–H, N–H valence), 2952 (w, –CH2–, asym. valence), 2882.5 (w, –CH2–, sym. valence), 1745 and 1668 (s, C=O, valence), 1515 (w, N–(CO), valence), 1480 and 1440 (s, –CH2–, scissor), 1260 and 1195 (m, C–O(CO), asym. and sym. valence), 1114 (s, C–O–C, valence), 1058 (m, C–O–H, valence), (cm−1); 1H-NMR (500 MHz, d6-DMSO), δ = 1.00 (3 H, d, –CH–CH
3, J
7,6 = 5.7 Hz), 2.85 (2H, t, –CH
2–NH, J
1,2 = 5.5 Hz), 3.20 (1 H, m, –CH–CH3, J
6,7 = 5.7 Hz), 3.60 (2 H, t, –CH
2–OH, J
1,2 = 5.5 Hz), 3.80 (2 H, t, –CH
2–O(CO), J
5,6 = 5.9 Hz), 8.15 (1 H, s, –OH), 8.15 (1 H, s, –NH) (ppm), EA: % N
calc.
= 7.69, % N
det.
= 7.57, % C
calc.
= 46.15, % C
det.
= 46.30, % H
calc.
= 6.69, % H
det.
= 7.12.
Reactions of bisoxamidoester (BOAE) with alkylene carbonates
BOAE (36 g, 0.1 mol) and such amount of ethylene carbonate, that initial reagent ratio equaled 1:8, 1:14 and 1:20, or 68 cm3 (81.6 g, 0.8 mol) propylene carbonate were placed in a 100-cm3 three-necked flask. The catalyst was: 1.242 g potassium carbonate (12.42 g/mol BOAE, 0.09 mol/mol BOAE)—for reaction with EC or 1.008 g diazabicyclo[2.2.2]octane (DABCO) (1.008 g/mol BOAE, 0.09 mol/mol BOAE)—for reaction with PC. The flask was provided with a reflux condenser, mechanical stirrer and then heated to 160 and 180 °C, for reaction with EC and PC, respectively, under constant mixing. The course of the reaction was followed by determining the percentage of unreacted alkylene carbonate (AC) in the reaction mixture [18]. The reaction was completed if the percentage of unreacted AC was less than 1 wt%.
Obtaining foamed polyurethane materials
10 g of proper hydroxyalkyl oxamidoester derivative, 0.1 g of a surfactant (L-6900 Silicon, Momentive, USA), 0.8–3.2 wt% of triethylamine (TEA, catalyst) and 2 wt% of water (Table 5) were inserted into a 250-cm3 polyethylene cup. Having thoroughly mixed all the components, a properly calculated [19] amount of 4,4′-diphenylmethane diisocyanate (with a triisocyanate additive) was added and later on—experimentally corrected. Next, an isocyanate was added up, it was vigorously agitated by a mechanical stirrer at a constant rotation speed of 2000 rpm (stirrer, Ver. VOS PC S40, Croatia). When the reacting mixture was homogeneous, it was transferred to a cubic mold of 10 × 10 × 15 mm size. The creaming took place only in the mold. The creaming time was measured from commencement of mixing to start of raising. Next, raising and drying time of the foams were measured, too. With the aim of the next tests, after 48 h of foams’ seasoning in room temperature, fittings were cut out of them.
Methods of analysis
Hydroxyl number (HN) of the obtained polyols was determined with the use of acetic anhydride and pyridine [PN-EN ISO 2554].
Thermogravimetric analysis (TG and DTG) of the above-mentioned products was made in a ceramic pan in the following registration conditions: temperature range: 20–600 °C, registration time: 60 min, sample weight: 1–2 mg, atmosphere: nitrogen, with the use of a derivatograph Termowaga TGA/DSC, Mettler.
The polyols’ molar masses [number-average (\( \bar{M}_{\text{n}} \)), weight-average (\( \bar{M}_{\text{w}} \)), z-average (\( \bar{M}_{\text{z}} \))] were determined with the use of a gel chromatograph Viscotec T60A equipped with RI detector (of refractive index). Separation were performed by means of two independent columns: PSS SDV (of 7.8 mm × 300 mm dimensions), accompanied by TSK deposits of 100 and 1000 Å pore diameter and the following recording conditions: 25 ± 0.1 °C, eluent flow rate: 1 cm3/min, injected sample size: 20 μl, polymer concentration in solution: 5 mg/cm3, analysis time: 30 min, eluent: THF (distilled from above sodium); calibration was performed on the basis of typical polystyrene and branched references.
The following physical properties of the obtained polyols were determined: density with the pycnometric method [20], viscosity using Höpler’s viscometer [21] and surface tension with the use of torsion balance [22]. Measurements were carried out in a temperature range of 20–80 °C.
The elemental analysis (C, H, N) of foams was made by means of an element analyzer Vario EL III C, H, N, S and O, manufactured by Elementar.
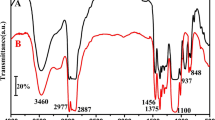
The IR spectra of the foams were recorded using potassium bromide tablets on PARAGON 1000 FTIR spectrometer, produced by Brucker.
The following polyurethane foams’ properties were determined: apparent density [PN-EN ISO 845], water uptake [PN-EN ISO 2896], dimensional stability [PN-EN ISO 2796], glass transition temperature (by means of DSC method), thermal stability measured by weight loss during monthly annealing at 150 and 175 °C (1 month is required to acquire stable weight), compressive strength [PN-93C/89071, ISO 884:1978] with the use of strength testing machine, Instron company, with measuring heads 30 and 100 kN 1 N coupled to optical strain measurement system, Aramis, GOM company, thermal conductivity [PN ISO 4589-2:1999] and oxygen index [PN-EN ISO 11357-1].
The microscopic observation of foams was performed at optical microscope Nikon Eclipse LV100 POL, camera Digital Sight DS-5Mc, objective 2.5 or 5 × L Plan. Microscopy measurements were performed in the Biophysical Laboratory of the Department of Physic of Rzeszow University of Technology. The Laboratory has been constructed in the frame of UE Polish Integrated Regional Operation Program.
Measurements of DSC foams were made with the use of DSC822e calorimeter, manufactured by Mettler Toledo. About 2–3 mg of the foam was put into an aluminum pan. The measurements were carried out from 20 to 200 °C at the heating rate of 10 °C/min, followed by cooling at 10 °C/min in nitrogen atmosphere [PN-EN ISO 13165:2003]. Tg was collected from the second run.
Tests on thermal conductivity and heat capacity coefficients were made at room temperature and by means of Isomet 2114 (Applied Precision, Slovakia), portable measuring apparatus. A rebate probe was used in the experiments as well—it required sample of 40 mm diameter and 80 mm length [PN ISO 4589-2:1999].
Oxygen index experiment, based on minimal oxygen concentration in oxygen and nitrogen mixture, in which a sample of tested material set in measuring column lights a fire longer than 3 min, was performed with the use of a device destined for the oxygen index determination (Fire Testing Technology Limited, Great Britain) [PN-EN ISO 11357-1].
Results and discussion
Synthesis of row material with bisoxamidoester groups
For obtaining polyetherols with bisoxamidoester groups, the initial substrate—a bisoxamidoester derivative—was synthesized in the first stage of the work. With this end in view, oxalyl chloride was reacted with dipropylene glycol and then obtained product (II) was reacted with 2-aminoethanol giving bisoxamidoester (III)—2,4-dimethyl-3-oxapentane-1,5-diyl-N,N′-bis(2-hydroxyethyl)oxamate (BOAE) with the yield of ca. 50 wt%.

This is a white, crystalline substance of melting point ca. 50 °C. The composition and the structure of the substance were confirmed by elemental and spectroscopic (IR, 1H-NMR) analysis, respectively (see “Experimental”).
Polyols’ production and properties
BOAE (III) obtained was further hydroxyalkylated by alkylene carbonates (ACs).

where R = H– or CH3; x + y + z + r ≤ 8, 14 or 20.
Reactions with EC were carried out at a temperature of 160 °C in the presence of potassium carbonate as a catalyst. In turn, in the reactions of BOAE with PC, DABCO was used as a catalyst and the reactions were run at 180 °C (Table 1, item no. 4). It should be emphasized that reactions with PC require higher temperature than reactions with the use of EC because PC is less reactive than EC and to achieve quite short time of reaction higher temperature has to be applied. Because of the same reason, more effective catalysts have to be used in reactions with PC, e.g., DABCO [23].
The composition of the post-reaction mixtures was investigated by MALDI ToF mass spectrometry. For example, the products obtained at 8-molar excess of EC consist of oligomers containing up to 7 mol of oxyethylene units per 1 mol of oxamidoester groups (Fig. 1). In turn, the products obtained with the use of 14-molar excess of EC contain oligomers consisting of maximum 24 oxyethylene units.

MALDI ToF spectrum of the product obtained using initial molar ratio BOAE:EC 1:8 in the presence of 0.09 mole K2CO3/mole BOAE at 160 °C
The content of by-products in the reactions of BOAE with ACs has been investigated. It was noticed, that with the growth in excess of EC (≥14) used for reaction, the content of particular glycols increases. Ethylene glycol (EG) is formed in the least amounts, whereas the products of consecutive reactions of EG with EC are produced in larger quantities (Table 1, item no. 1–3). The reverse is true for the product of the reaction of BOAE with PC, i.e., the amount of propylene glycol (PG) formed is the largest and the consecutive reaction products with PC are decreasingly less abundant (Table 2, item no. 4).
The distribution of molar masses for particular products has been investigated by gel chromatography (Fig. 2). According to the increase in EC excess used for the reaction, the content of higher molecular mass fractions increases; the raise in intensity of the short retention time peak is observed in Fig. 2. By comparison of chromatograms for products of reaction of BOAE with 8-molar excess of EC and PC, it is visible that higher mass oligomers are formed in the reaction with the latter.

Distribution of molar masses of the obtained polyols with retention volumes
The thermal stability of the products obtained was examined by thermogravimetric analysis (Table 3). The 5 % mass loss for the products of the reaction of BOAE with 8- and 14-molar EC excess is observed at the temperature 130 and 150 °C, respectively. The other products show 5 % mass loss at temperature ≤100 °C, what follows from the loss of contained water. Two extrema are present on the DTG curve for the products of the reaction of BOAE with EC. The first is at the temperature ca. 250 °C and the second at 360 °C. In turn, the products of the reaction of BOAE with PC show extrema at 170 and 265 °C. The first peak originates from decomposition of carbonate groups [24] and shows that the content of these groups in the products of reactions of BOAE with PC is larger than that for reactions with EC. On the DTG curve of the products formed in the reaction with EC, the peak with extremum at 170 °C is not present. All the products show an extremum at ca. 250 °C originating from the decomposition of oxamidoester groups.
The physical properties of the products obtained, such us density, viscosity and surface tension have been examined. It was found that the change in these properties with temperature follows the same tendency as the change of physical properties of typical polyols, used for obtaining polyurethane foams (Table 4). The physical properties of the products of reaction of BOAE with EC show higher values than that of the properties of polyols obtained using PC at a given temperature.
Obtaining polyurethane foams
The products containing bisoxamidoester groups were also tested as polyol substrates for polyurethane foam syntheses. The foaming with 4,4′-diphenylmethane diisocyanate (MDI), containing 30 wt% of triisocyanates was carried out in a small laboratory scale. The investigations aimed at determining the influence of the optimum amount of catalyst, isocyanate, foaming agent (water), kind of hydroxyalkyl derivative on the course of the foaming.
It was preliminary found that 2 wt% water content in polyol should be used and such percentage was used for the foaming experiments (Table 5). The amount of TEA (catalyst) depends on the kind of the derivative used. For polyols obtained with the smallest excess of EC, it was not necessary to use TEA. It was found that polyols obtained with higher (14- and 20-molar) excess of EC are not suitable for polyurethane foam syntheses. In turn, the product obtained at 8-molar excess of PC, containing secondary hydroxyl groups, is less reactive toward isocyanate groups and requires the use of TEA in the amount of 2.4/100 g of polyol.
It was observed that the creaming time, particularly rising and drying time, is definitely shorter than that of in case of polyols obtained with the excess of EC (Table 5, composition 1 and 7). The creaming time is usually ca. 23 s for products of EC hydroxyalkylation and is 1.5 times shorter than that for polyols obtained using PC. The differences in the rising time are even bigger. The foams obtained from EC polyols raise 3 times faster than those from PC polyols. The foams obtained from hydroxyethyl derivatives containing bisoxamidoester groups are practically dry directly after the growth, whereas those from PC polyols need to dry for approx. 80 s (Table 5, composition 1 and 7).
Properties of the polyurethane foams
The apparent density of the polyurethane foams obtained is in the range of 51–69 kg/m3 (Table 6) and the foams obtained using hydroxypropyl derivatives exhibit lower density (Table 6, composition 7).
The water uptake of the foams exposed to water for 24 h at room temperature is also lower for foams obtained from PC polyols. During the exposition, the differences in water absorbability of the foams obtained using hydroxyethyl and hydroxypropyl derivatives containing bisoxamidoester group are decreasing. After 5 min, the water absorbability of EC polyols is 3 times greater, after 3 h it increases 2 times and after 24 h the difference is only 25 % (Table 6).
The morphology of the foams, examined by means of optical microscope, shows that they contain closed pores (Fig. 3). The pore sizes of the foams obtained using hydroxypropyl derivatives with oxamidoester groups are in the range of 0.209–0.419 mm, and the thickness of the wall is 23–47 μm (Fig. 3a). These pores are characterized by definitively angular cross-section. In turn, the foams obtained using the products of the reaction of BOAE with excess of EC have rounded shaped pores (of a size of 0.142–0.333 mm) and uniform walls of a thickness of 24–36 μm (Fig. 3b). It was found that pores of the foams obtained using hydroxypropyl derivatives with bisoxamidoester groups have slightly thicker walls and larger dimensions.

The morphology of the foam obtained using the product of the reaction of BOAE with 8-molar excess of: a PC, b EC, magnification 5×
Foams from polyols obtained using PC show better dimension stability than foams synthesized from EC polyols (Table 7). The foams obtained from PC polyols shrink while heating and linear dimension changes do not exceed 1 %. The foams obtained from EC polyols increase in sizes up to 6 % for linear ones.
Polyurethane foams usually decrease their dimensions during the temperature exposure. Nevertheless, the foams, obtained with the use of EC polyols, increase their dimensions and these changes are pretty large and much larger than in the case of foams from PC polyol. Perhaps, it is the result of higher reactivity of EC polyols with primary hydroxyl groups and shorter mixing time and especially the expanding time, which can cause trapping of evolved carbon dioxide in the closed-cell foams. At higher temperature (150 °C) during testing the dimensional stability, carbon dioxide escapes and increases the foam dimensions.
The glass transition temperature (Tg) of polyurethane foams obtained was examined and it was found they show significant differences in Tg values, depending on the kind of hydroxyalkyl groups present in polyol component. The glass transition temperature for foams obtained using hydroxyethyl derivatives is 74 °C lower than that for hydroxypropyl derivatives. A higher value of Tg denotes a more rigid structure and is more advantageous from the point of view of functional properties of the foams in a wider temperature range (Table 8). However, the glass transition temperature of every foam is higher than room temperature, therefore, the obtained foams can be unequivocally classified as rigid ones [25].
Thermogravimetric analysis showed high thermal stability of the polyurethane foams obtained, regardless of the polyol kind. So there is difficultly to observe the influence of thermal stability of polyols (Table 3) on thermal stability of the obtained polyurethane foams (Table 8). The 5 % mass loss of the foams is not observed up to temperature approx. 230–240 °C (Table 8). On the DTG curve of the foams obtained using hydroxypropyl derivatives with oxamidoester group two extrema are observed at 340 and 415 °C. The first extremum originates from decomposition of oxamidoester, urea [26], carbamate and alkoxy groups. The second extremum comes from the decomposition of carbodiimide groups [27, 28], the presence of which was shown by IR spectra of the foams obtained. A band at 2138 cm−1 characteristic of valence vibrations of carbodiimide N=C=N bonds is observed in IR spectra of the foams. They are formed as a consequence of dehydration of N,N′-disubstituted urea derivatives [29]. During the foam production, polyol reacts with MDI, but isocyanate groups also react with water, forming free amine groups. These groups form N,N′-disubstituted urea derivatives in the reaction with isocyanate and these derivatives dehydrate under synthesis conditions giving thermally stable carbodiimide groups.
The heat resistance and thermal stability are the main factors which limit the application of polyurethane foams as thermal insulation at temperatures higher than 110 °C [1, 30]. To check the possibility of higher temperature work of the obtained foams, they were heated for 30 days at 150, 175 and 200 °C (Fig. 4), and the mass losses were examined. As shown in Fig. 4, the mass losses of the foams increase according to the increase in heating temperatures. In spite of growing defects as the heating temperature increases, the foams obtained using hydroxypropyl derivatives of bisoxamidoester do not undergo distortions in their shapes. It was noticed, that foams obtained from polyols with oxypropylene groups show lower mass losses during exposure at each temperature. It follows from the results obtained, that foams synthesized using hydroxypropyl derivatives of bisoxamidoester could work at 150 °C (the mass loss below 10 wt%).

Thermal stability of the obtained foams tested by heating for 30 days at: 150, 175, 200 °C
The foams obtained and their samples heated for 30 days at 150, 175 and 200 °C were tested for compression strength (Fig. 5). The foams after annealing at temperature 150 °C and higher were tested for compression strength for practical reasons. The foams were annealed at temperature 150 °C and higher to check the possibility of the use of foams at these temperatures. From the same reasons, tests of compression strength were made, i.e., to check if the compression strength of foams did not worsen after annealing. During annealing, foams cured but at the same time they could undergo degradation. Of the degradation process predominates, the compression strength of foams decreases.

Compression strength at 10 % strain
The foamed polyurethane materials, obtained using hydroxyethyl and hydroxypropyl derivatives with oxamidoester groups, show increased compression strength (0.5 MPa) as compared to classic foams (σ M = 0.1–0.2 MPa). Nevertheless, their compressive strength is not as high as that of condensation polyurethanes with different hard segments [31]. Generally the foams obtained using EC polyols exhibit slightly higher compression strength before temperature exposition. After temperature exposition at 150 °C, the compression strengths of all foams increase. The foams obtained using hydroxypropyl derivatives containing bisoxamidoester group increase in compression with strength after heating at 175 °C. In turn, further increase in heating temperature up to 200 °C results in decrease in compression strength of the foams. However, considering the initial compression strength of the foams and that after heating at 200 °C, the latter is higher.
Due to projected application of the obtained foams as heat-insulating materials for high temperature work, their insulating properties have been tested (Table 9). The thermal conductivity increase for the foams obtained using hydroxypropyl derivatives of bisoxamidoester, however, is clearly lower than that obtained using hydroxyethyl derivatives. According to increase of the thermal conductivities, the volume heat capacity increases, i.e., the better insulating power, the worse heat storage possibility. Thermal conductivity less than 0.035 (W/m K) [32] is characteristic of typical polyurethane foams obtained as insulating materials. Therefore, the foams obtained using hydroxypropyl derivatives containing bisoxamidoester group as a polyol component can be used as a heat-insulating materials.
Due to application of hydroxyalkyl derivatives containing bisoxamidoester groups and incorporation of additional nitrogen atoms into foam structure by polyol component, the oxygen index was determined for the foams, to illustrate the influence of nitrogen presence on flammability of polyurethane foams (Table 9). IO value is in the range 19.3–19.8 vol%, what denotes that the obtained foams are flammable ones. For comparison, the IO value for classic foam, obtained using Rokopol RF-55 and MDI, is 19.6 vol% [33], what indicates that the obtained foams show OI values, that are typical for non-modified foams.
Summary and conclusions
In the work, the synthesis of the new polyol components for polyurethane foam has been described. To obtain the new polyol, 2,4-dimethyl-3-oxapentane-1,5-diyl-N,N′-bis(2-hydroxyethyl)oxamate was synthesized by reaction of oxalyl chloride, dipropylene glycol and 2-aminoethanol. Next, the conditions of reaction of this oxamate with alkylene carbonates were worked out.
The properties of the products of the reaction of 2,4-dimethyl-3-oxapentane-1,5-diyl-N,N′-bis(2-hydroxyethyl)oxamate with excess of alkylene carbonates were examined, for assessment of their potential application as new polyol components for obtaining polyurethane materials.
Using these derivatives as polyol components, rigid foamed polyurethane materials have been obtained. Incorporation of nitrogen by polyol component did not decrease flammability of the foams. The foams obtained are still flammable and display oxygen index typical for regular polyurethane foams. Nevertheless, the new polyol improved properties of obtained polyurethane foams, especially enhanced their thermal stability.
The polyurethane foams obtained using hydroxypropyl derivatives containing bisoxamidoester groups can be used as heat-insulating materials of operating temperature as high as 150 °C.
References
Lefebvre J, Bastin B, Bras M, Duquesne S, Paleja R, Delobel R (2005) Thermal stability and fire properties of conventional flexible polyurethane foam formulations. Polym Degrad Stab 88:28
Modesti M, Lorenzetti A (2002) Flame retardancy of polyisocyanurate–polyurethane foams: use of different charring agents. Polym Degrad Stab 78:341
Petrović Z (2008) Polyurethanes from vegetable oils. Polym Rev 48:109
Prociak A (2007) Properties of polyurethane foams modified with natural oil-based polyols. Cell Polym J 26:381
Zlatanic A, Lava C, Zhang W, Petrovic Z (2004) Effect of structure on properties of polyols and polyurethanes based on different vegetable oils. J Polym Sci B Polym Phys 42:809
Guo A, Demydov D, Zhang W, Petrovic Z (2002) Polyols and polyurethanes from hydroformylation of soybean oil. J Polym Environ 10:49
Dominguez-Rosado E, Liggat J, Snape C, Eling B, Pichtel J (2002) Thermal degradation of urethane modified polyisocyanurate foams based on aliphatic and aromatic polyester polyol. Polym Degrad Stab 78:1
Datta J, Kacprzyk M (2008) Thermal analysis and static strength of polyurethanes obtained from glycolysates. J Therm Anal Calorim 93:753
Datta J, Janicka M (2007) Synthesis and properties of polyurethanes made of secondary raw materials. Przem Chem 86:624
Datta J, Rohn M (2008) Structure, thermal stability and mechanical properties of polyurethanes, based on glycolysate from polyurethane foam waste, prepared with use of 1,6-hexanediol as a glycol. Polimery 53:871–875
Kacperski M, Spychaj T (1999) Rigid polyurethane foams with poly(ethylene terephthalate)/triethanolamine recycling products. Polym Adv Technol 10:620
Schimmel K, Ward T, Seiner J (1987) Half-amide reaction products of dioxalates and amino group containing materials. US Patent 4,684,710
Shinsuke S, Kazunori K, Mitsuo Y, Satoshi U, Kei A (1989) Epoxy resin containing zwitterionic phosphate group. Europe Patent 371,640
Shinsuke S, Kazunori K, Mitsuo Y (1997) Compound containing an oxamic acid group, a process for producing the compound, and a resin composition containing the compound. US Patent 5,663,262
Dickey J (1950) Polymers and copolymers of unsaturated amides. US Patent 2,496,275
Becke F, Wick K (1961) Improvements in the production of expanded polyamides. US Patent 858,254
Izumi M, Aiko I, Nakanishi M, Saruwatari K, Gono T, Naraki A, Matsushima M (1960) Process for preparing oxamide derivatives. JP 5:628
Kucharski M, Kijowska D (2003) Synthesis of polyetherols from melamine and propylene carbonate. II. J Appl Polym Sci 89:104
Uhlig K (1999) Discovering polyurethanes. Carl Hanser Verlag, Munich, pp 55–82
Kowalski P (2004) Laboratorium chemii organicznej. WNT, Warsaw
Karniewicz J, Sokołowski T (1993) Podstawy fizyki laboratoryjnej, Wyd. Politechniki Łódzkiej, Łódź
Orzechowski Z, Prywer J, Zarzycki R (2001) Mechanika płynów w inżynierii środowiska. WNT, Warsaw
Clements JH. Reactive applications of cyclic alkylene carbonate. http://www.huntsman.com/portal/pls/portal/docs/37717815.PDF
Reed H, White C, Rao V, Bidstrup-Allen S, Henderson C, Kohl P (2001) Fabrication of microchannels using polycarbonates as sacrificial materials. J Micromech Microeng 11:733
Klempner D, Sendijarevic V (2004) Polymeric foams and foam technology, 2nd edn. Hanser, Munich, pp 275–288
Awad W, Wilkie Ch (2010) Investigation of the thermal degradation of polyurea: the effect of ammonium polyphosphate and expandable graphite. Polymer 51:2277
Imashiro Y, Amano S (1990) Process for producing parabanic acid ring-containing polymer. Patent USA 4,939,210
Imai Y, Taguchi Y, Ikeda T, Imai C (1987) Parabanic acid oligomer composition. Patent Japan 62,124,124 ( CA: 87, 199504 y)
Mikołajczyk M, Kiełbasiński P (1981) Recent developments in the carbodiimide chemistry. Tetrahedron 37:231
Singh H, Jain A (2009) Ignition, combustion, toxicity, and fire retardancy of polyurethane foams: a comprehensive review. J Appl Polym Sci 111:1115
Bakar M, Bialkowska A, Szymanska J (2013) Synthesis and evaluation of mechanical and thermal properties of segmented condensation polyurethanes. Plast Rubber Compos 42:203
Garbalińska H, Bochenek M (2011) Izolacyjność termiczna a akumulacyjność cieplna wybranych materiałów ściennych. Architektura, Czasopismo techniczne 108(11):89
Paciorek-Sadowska J, Czupryński B, Liszowska J (2012) Fire-safe polyurethanes modified with new antipyrene. Chemik 4:297
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zarzyka, I., Pacześniak, T. Polyurethane foams modified with bisoxamidoester groups. Polym. Bull. 72, 1321–1338 (2015). https://doi.org/10.1007/s00289-015-1339-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-015-1339-y