
Abstract
Multiple sclerosis (MS) is a chronic autoimmune disease that affects the central nervous system (CNS). Compared to other types of self-limiting myelin disorders, MS compartmentalizes and maintains chronic inflammation in the CNS. Even though the exact cause of MS is unclear, it is assumed that genetic and environmental factors play an important role in susceptibility to this disease. The progression of MS is triggered by certain environmental factors, such as viral infections. The most important viruses that affect MS are Epstein-Barr virus (EBV), human herpes virus 6 (HHV-6), human endogenous retrovirus (HERV), cytomegalovirus (CMV), and varicella zoster virus (VZV). These viruses all have latent stages that allow them to escape immune detection and reactivate after exposure to various stimuli. Furthermore, their tropism for CNS and immune system cells explains their possible deleterious function in neuroinflammation. In this study, the effect of viral infections on MS disease focuses on the details of viruses that can change the risk of the disease. Paying attention to the most recent articles on the role of SARS-CoV-2 in MS disease, laboratory indicators show the interaction of the immune system with the virus. Also, strategies to prevent viruses that play a role in triggering MS are discussed, such as EBV, which is one of the most important.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is a Central nervous system (CNS) autoimmune disease [1, 2] in which nerve cells are demyelinated, causing inflammation and damage in the CNS [3,4,5,6,7]. Although the etiology of MS remains unclear [2, 8,9,10], colossal advancement has been accomplished in distinctive hazard factors connected to MS [11]. Both genetic and environmental variables are discovered in the epidemiologic analysis with a vital role in the progression of MS [11]. The role of infectious and viral agents is still controversial in MS, but there is increasing evidence that some viruses play a role in disease development [2, 12]. The most outstanding proof comes from identifying viral nucleic acids or antigens and antiviral antibody responses in patients with MS [12]. Viral infections can influence MS in different ways and combinations. These pathways include molecular mimicry, direct toxicity, bystander activation, dual T-cell receptors, and epitope spreading [12, 13]. Direct toxicity: It is based on direct damage to the cell without the intermediation of inflammation or autoimmunity. It has been observed in some of the MS plaques that the dystrophy of the oligodendrocyte cells and the precursors (without the mediation of IgG and complement) has happened, which causes a defect in the function of the blood–brain barrier (BBB) and the continuation of the disease [12]. Molecular mimicry: During this phenomenon, cross-reaction occurs, wherein myelin compounds are presented by class II major histocompatibility complex (MHC) on APCs to autoreactive CD4 + T-cell lymphocytes due to the similarity of myelin to some viral structures [12, 14].
Dual T-cell receptors: It is thought that some T lymphocytes express different TCRs with various functions, such as recognizing viral antigens or myelin antigens, and these lymphocytes activate both types of antigen responses [12]. Bystander activation: viral infections cause extensive inflammation. With this inflammation, the surrounding cells are damaged and cause the discovery and presentation of autoantigens presented by Antigen-presenting cells (APC). As a result, T and B lymphocytes became reactive [12]. Epitope expansion: damage to myelogenous cells causes myelin fragmentation in the inflammatory environment, which leads to additional epitopes with self-perpetuating destruction of myelin [12, 14].
Viruses can cause brain damage by directly infecting the CNS or through the inflammatory response that follows. Epstein-Barr virus (EBV), human herpes virus 6 (HHV-6), human endogenous retrovirus (HERV), cytomegalovirus (CMV), SARS-CoV-2 (COVID-19), and varicella zoster virus (VZV) can all enter the CNS, cause acute cellular damage and dysfunction, and remain quiescent or latent in infected cells for long periods [4, 15, 16]. They can stimulate the activation of lymphocytes and the production of pro-inflammatory cytokines that lead to neuron destruction and disrupt cellular activity in other indirect ways. These inflammatory pathways may be triggered by sensitization of brain neurons, which may result from genetic changes in MS. Although these processes related to viral roles in classical neurological disorders remain hypothetical and are still being studied. The association between infection or viral components and the onset or recurrence of symptoms in MS has long been recognized [17]. The persistent latent infections that have concealed, silent or latent phases escape from detection by the immune system and revive when exposed to multiple triggers. Their tendency for CNS and immune system cells explains their potential destructive property in neuroinflammation and neurodegeneration of CNS [18]. Also, other viruses that have been connected to the onset of MS but are not addressed in the article include Herpes simplex virus-1 (HSV-1), HSV-2, and John Cunningham Virus (JCV). For HSV-1, the seroprevalence of IgG against the virus has been found to be expanded in pediatric MS/Clinically isolated syndrome (CIS) but not adult MS compared with controls [19]. For HSV-2 the seroprevalence has been found significantly increased in MS compared with controls [19]. JCV is a non-enveloped double-stranded DNA (dsDNA) virus, which may cause progressive multifocal leukoencephalopathy (PML) characterised by infection of oligodendrocytes and astrocytes in the CNS [20,21,22].
In this review, we address the existing evidence linking particular viruses to MS development or aggravation. Finally, controlled clinical studies utilizing preventative or therapeutic techniques that precisely interfere with any virus in MS disease are the only way to confirm that agent’s participation in the disease effectively.
Viral Triggers in MS
Epstein–Barr Virus (EBV)
EBV, a member of the human Herpesviridae family, has a dsDNA genome of 120 kb encoding approximately 85 proteins, and a large number of non-coding RNAs (ncRNAs) [23, 24]. A history of chronic infectious mononucleosis caused by EBV increases the risk of MS by approximately 40-fold [25]. This is one of the most critical factors in developing MS [1, 9, 26, 27]. EBV is activated, replicates, and latently exists in B lymphocytes during the lifetime of an infected individual [1, 9, 28].
The Immunological Mechanism of EBV in MS
The EBV virus affects epithelial and lymphoid cells in the oropharyngeal Waldeyer’s loop. However, the virus continuously transforms B cells that express CD21, EBV’s primary cellular receptor [25, 28]. To produce the phenotypic (IgD-CD27 + and IgD + CD27 +) memory cells, where the virus causes permanent infection and alters the development of resting B cells like germinal center responses [9, 25, 28], the EBV and cellular genomes experience variable degrees of epigenetic alteration throughout the B cell transformation process, including silencing of severalviral genes required to create a sustained latent infection [1, 25, 28, 29]. Immune control is essential for this crucial process because when a virus released from each ruptured plasma cell infects an epithelial cell, virus production is increased in the tonsils, and the epithelial cell generates enough virions to infect 10,000 native B cells, implying that the harm is gradual. This cycle can have a considerable negative impact on the infected blast population [9, 28, 29].
Deficiency in cytotoxic CD8 + T-cell removal of EBV-infected B cells may increase the risk of developing MS by allowing the buildup of EBV-infected autoreactive B cells in the CNS [9, 28, 30]. There is a widespread lack of CD8 + T-cells in MS, namely the CD62L effector memory (EM/EMRA) fraction, which performs CNS immunosurveillance and defends against viral infection [28, 31]. At all MS levels, CD8 + T- cell lymphocytes do not respond to lytic phase EBV antigens, which indicates an impaired control of EBV reactivation. Contrarily, CD8 + T-cells that target EBV latent antigens are more abundant but less active, meaning an exhausted response to the more significant number of latently infected cells due to diminished CD8 + T-cell regulation of EBV reactivation [9, 28, 30, 32]. There is evidence that the course of MS is associated with cellular exhaustion of EBV-specific CD4 + and CD8 + T lymphocytes [30, 33]. However, other factors, such as a decline in EBV reactivation related to age, may also contribute to these outcomes [9, 28]. A study showed that lysis-specific CD8 + T-cells declined in peripheral blood mononuclear cells (PBMCs) at all stages of multiple sclerosis, besides clinical attacks, as well as CD8 + EMRA T-cells and CD8 + EM, the frequency of lytic-specific CD8 + T-cells was also decreased. EM/EMRA T-cells were reduced compared to ordinary cases. MS patients were significantly less likely to have lytic-specific T-cells in their CD8 + and T-cell phenotypes than normal individuals [28, 34]. The number of EBV-lytic CD8 + T-cells was reduced, and the number of latent-specific CD8 + T-cells increased in MS [28, 30, 34]. The results revealed that the numbers of latent-specific cells in the CD8 + T-cells, CD8 + EM T-cells, CD8 + CM T-cells, and CD8 + EM/EMRA T-cells substantially increased in patients with MS compared to healthy individuals with EBV-positive blood [28, 34].
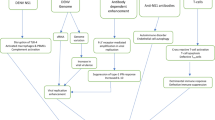
A possible mechanism for CD8 + T-cell memory loss in MS is the decrease in IFN-I production because the number of EBV-specific CD4 + T-cells correlates severely with the number of EBV-specific CD8 + T-cells in MS as opposed to EBV-infected people with no previous records of MS. This could be because IFN-α or IFN-β is critical in developing CD8 + memory T-cells [28, 35]. Memory B cells can trigger the auto proliferation of Th1 cell-homing CD4 + T-cells in MS, detecting autoantigens in both B cells and MS lesions [36]. According to reports, these cells go to the brain and interact with RAS guanyl-releasing protein 2 (RASGPR2) and HLA-DR to cause inflammation [37]. CNS autoreactive T-cells may get activated in lymphoid tissue. They could, by contact with EBV-infected B cells, move into the CNS, where they receive co-stimulatory and survival cues from EBV-infected B cells. Increased B cell-mediated antigen presentation to autoreactive T-cells and prevention of their apoptosis may contribute to local inflammation persistence, attracting more inflammatory cells and causing antigen-directed harm and bystander injury to the CNS [37]. (Fig. 1).

The immunological mechanism involved in EBV can cause MS. 1. EBV infects naive B cells, so these cells proliferate in the germinal centers. Then the receptor of B cells or BCR and proteins of EBV are latent in the self-reactive memory cells. B: blood circulation: 2. Memory cells infected with EBV leave the lymph node and enter the bloodstream. C: Brain: 3. Memory cells that are EBV-infected will enter the brain cells (neurons) and stay there. 4. In neurons, autoreactive T-cells that have already entered the brain cause damage to neurons in two ways. 5. Infected memory cells signal B7 stimulus to CD28 receptors on the surface of active autoreactive T-cells. 6. These co-stimulus signals activate T-cells by producing interferon-gamma, interferon-beta, and IL-2. 7. Autoreactive T-cells detect apoptotic myelin fragments by microglia (brain macrophages) through MCH, eventually causing further apoptosis of the myelin sheath around neurons. 8. Autoantibodies produced by B cells infected with EBV sit on the myelin sheath, releasing myelin and oligodendrocyte fragments. 9. Released myelin fragments by MHC memory cells that are EBV-infected are given to the autoreactive T-cells, and this degradation process by the T-cells continues
MS is associated with high levels of EBV-induced G protein-coupled receptor 2 (EBI2) expression, which mediates CNS autoimmunity, lymphocyte migration, and MS lesions [38]. The EBI2 receptors play a critical role in myelin formation and mediate the halt of demyelination induced by lysophosphatidylcholine-induced demyelination (LPC, lysolecithin). The oxysterol-EBI2 direction is involved in immunoregulatory responses, and the unique expression of this receptor is involved in the antigen-specific B-dependent antibody responses, and T-dependent antibody responses [39, 40].
EBV survival is elevated by EB nuclear antigen-2 (EBNA-2) modulation of gene expression, which leads to a higher incidence of lymphoma and autoimmune diseases, such as MS [26]. Therefore, a recent study has suggested that mutations in EBNA-2 might influence host responses to EBV and MS sensitivity [26, 30, 41]. As revealed here, EBNA-2 connects to five of six genes associated with MS with allele imbalance [42], and suppressing EBNA-2 changes the expression of five of these genes. There is still significant uncertainty concerning how EBNA-2 affects MS susceptibility by promoting the expression of (mutated) Single-nucleotide polymorphisms (SNPs) in lymphoblastoid cell lines, thus affecting EBV’s ability to evade immune responses and exposure to MS. Studies should be conducted to determine whether inhibiting EBNA-2 could offer an excellent therapeutic way of treating MS [26, 30, 43]. It has been suggested that astrocyte EBV infections could activate human endogenous retrovirus-W (HERVW)/MS-associated retrovirus (MSRV)/syncytin1 in humans.
Laboratory Diagnostic Markers of EBV in MS
Although almost all MS patients are seropositive, whereas EBV seropositivity is present in approximately 95% of the world’s population [9]. A record of infectious mononucleosis significantly accelerates the risk of multiple sclerosis. In addition, EBV-specific oligoclonal bands have been identified in cerebrospinal fluid (CSF) in a subgroup of patients with MS [12, 44,45,46]. Recent studies have shown that the level of serum components differs in MS patients compared to healthy people. Antibodies that cross-react with myelin essential protein (MBP) can recognize a unique epitope of EBNA-1 411–426 [12, 28]. This has led to the hypothesis that EBV can reintroduce “forbidden” memory B-lymphocytes to CNS epitopes. EBV memory B cells may lose EBV recognition DNA during replication, which explains why EBV is not always found in MS lesions [9]. Still, authentication of the “forbidden” epitope would be preserved, potentially triggering molecular mimicry. In addition, a “two-hit concept” can explain the relationship between MS and EBV infection. In the initial condition, EBV alters the penetrance of the BBB, which allows activated immune cells to enter the CNS, triggering a cascade of events that leads to inflammation of the CNS [9, 12]. Increased antibodies against the EBV latency-associated nuclear antigen 1 (EBNA-1) can be noted before the onset of neurological symptoms [25, 28]. It is proposed that the raised titer of anti-EBNA-1 IgG reflects an elevated amount of latent EBV antigen load due to faulty control of EBV by T-cells [9, 28]. Moreover, EBV infection evidence in brain-infiltrating B cells in brain lesions of patients with MS has been noted [47, 48]. A novel study about the transcriptome of B cells received from CSF, and MS patients’ peripheral blood was bare of human viral transcripts, which included EBV [9, 25, 47]. It has also been suggested that EBV boosts the survival of oligoclonal antibodies-producing autoreactive B cells in the CSF of patients suffering from MS, and boosts the survival and infiltration of these infected cells toward the CNS, leading to the pathogenesis of MS [25].
As a study showed, the anti-EBNA-1 IgG and anti-VCA IgG titers were elevated in MS patients. The negative correlation between the anti-EBNA-1 IgG titer and the LCL-specific CD8 + T-cell frequency in MS validates their former finding, which noted an inverse correlation between this titer and the LCL-specific T-cell frequency [28, 49]. The immune response to the infection of EBV in people with MS (PwMS) is different than in healthy people. For instance, levels of EBNA-1 IgG are higher in both children and adults suffering from MS [50]. There is a positive relation between anti-EBNA1, but not anti-VCA titers, but anti-EBNA-1 titers of IgG, and blood EBV DNA load in both patients with MS and healthy individuals, as well as noted for patients with Hodgkin lymphoma and healthy relatives [28, 51].
Medication Control of EBV
Effective control of EBV infections has been suggested as a remedy for preventing and curing autoimmune diseases. In patients with MS, controlling infection of EBV can be done by depleting B cells, enhancing immunity, antiviral drugs, and ameliorating immune surveillance [9]. Specific agents Mechanisms of action (MOA), inactivation through viral kinase, narrowed the study duration, single-agent versus a combination of antiviral drugs (e.g., efficacy against HIV). Readers should remember that one of the top five options for the treatment for MS remains a potent antiviral protein, human IFN-beta [52]. IFN-beta, a significant contributor to MS, inhibits the ability of EBV, CMV, and other viruses to replicate, affects T-cell proliferation in response to EBNA-1, and decreases the compartment of B cell memory through a subset of cellular pathogens. [9]. The hypothesis that IFN-beta and other disease-modifying treatments (DMTs) have overlapping antiviral and anti-inflammatory mechanisms supports the concurrent testing of MS medications with various modes of action (MOAs) in the future [53].
Currently, there is not any vaccine that protects against infection of EBV. One viable approach to developing such a vaccine is targeting gp350 or other viral proteins [54]. New progressions in immunization by vaccines, focusing on DNA or RNA vaccines, which are capable of furnishing sequences coding multiple proteins, could facilitate advanced immunization investigations about MS. The idea of producing a prophylactic vaccine for averting acute infection of EBV as a strategy for preventing MS progression remains intriguing. Recent exploration of other herpesvirus vaccines has supplied encouraging results that support the view that designing a vaccine that could stop disease instead of infection might be feasible [55, 56]. The importance of B cells in the pathology of MS was highlighted by the potent efficacy of anti-CD20 B cell abatement treatments like ofatumumab, rituximab, and ocrelizumab. A recent study using adaptive delivery of autologous EBV-specific T-cells that demonstrated encouraging clinical results supports an EBV + B cell pathogenic role in the pathogenesis of MS [7, 9, 25, 57].
HHV-6
Since 1993, a relationship between HHV-6 and MS has been hypothesized, and numerous studies have been conducted. HHV-6 is a neurotrophic virus linked to various nervous disorders, including MS and neuromyelitis opticus. Numerous clinical studies have found a link between MS and HHV-6 infection [18, 58, 59]. HHV-6 A and HHV-6 B [12, 58] are two HHV-6 species with 95% homology [12, 58, 60]. HHV-6A and HHV-6B are large enveloped (Env) beta herpesviruses with a dsDNA genome [59]. Several studies have shown that the incidence of HHV-6A is higher than HHV-6B in samples from MS patients [58, 61].
Activation of the Immune System by HHV-6 in MS
HHV-6 can activate the immune system to generate a “fertile field” for the proliferation of autoreactive T-cells induced by other environmental stimuli. Evidence also suggests that HHV-6 has a role in nervous system disorders by infecting microglia cells in the CNS and causing them to produce a pro-inflammatory response [12, 58]. Adult oligodendrocytes, astrocytes, and microglia cells exhibit the complement system receptor CD46, which is used by HHV-6. These characteristics make them ideal candidates for modulating MS pathogenic pathways. HHV-6A is thought to use this receptor more frequently [4]. HHV-6B’s primary receptor is the CD134 protein on the surface of T-cells [62].
HHV-6A is responsible for the development of demyelination and can cause latent infection in oligodendrocytes, which are thought to be a target of autoimmune responses in MS pathogenesis [58]. Compared to control tissue, HHV-6 DNA and protein were found in MS plaques, particularly oligodendrocytes [4]. In MS, HHV-6A activates latent EBV in B cells. Latent EBV-infected B cells are imported into the CNS after infection with neurotrophic HHV-6A [63]. In MS patients, this virus can operate as an originator or potentiator of inflammatory plaques. CD8 + T lymphocytes responses to HHV-6-infected CNS cells can cause tissue injury and the release of sequestered antigens, which activate self-reactive lymphocytes and boost autoreactive immune reactions. Improvement of the activation of the complementary systems can occur by the CD46 used by HHV-6A as a cellular receptor [4].
Both kinds of HHV-6 may cause Th2 migration in T-helper cell balance by blocking IL-12 production by DCs and macrophages. Other studies have found that HHV-6 infection increases the production of inflammatory cytokines like IL1, TNF, and IFN in PBMCs [58]. It increases the production of IL-18 and IFN-γ receptors in T lymphocytes while decreasing the expression of IL-10 and IL-14, changing the balance of T-helper cells towards the Th2 phenotype. HHV-6A has also been demonstrated to aggravate disease progression and induce the production of IL-15 in NK cells [58]. Infected astrocytes had a reduced ability to ingest glutamate, which was linked to lower expression of the glial glutamate transporter EAAT-2 [64]. Over-activation of AMPA and kainate receptors can cause cytotoxic death of oligodendrocytes and oligodendroglial death when dysregulation of glutamate levels [4].
Laboratory Diagnostic Markers of HHV-6 in MS
Innumerable clinical studies have shown an association between infection of HHV-6 and MS. For instance, serum samples showing HHV-6 DNA levels representing active infection are significantly increased in patients suffering from MS compared to healthy individuals or patients with different neurological-related diseases [58]. In addition, in the CSF and PBMCs of patients with MS, HHV-6 DNA has been identified at a higher level [12, 58]. Another study showed that in brain autopsy samples from patients with MS compared to healthy individuals, genes exclusively expressed in HHV-6 were elevated [48, 58]. These investigations found an extremely high proportion of patients with MS, who were seropositive for anti-HHV-6 IgG, which was remarkably higher than that of controls [48]. A novel multiplex serological assay was used to measure IgG reactivity against the immediate-early protein 1 of HHV-6A (IE1A) and HHV-6B (IE1B) in cohorts of MS. Anti-IE1A IgG responses were positively related to MS, and there was an interaction between IE1A and EBV antibody responses on the risk of MS [61].
Human Endogenous Retrovirus
MS has been linked to the existence and activation of three HERVs, including HERV H, HERV K, and HERV W, which were inserted into the human genome many years ago [12, 65]. Gag (matrix and retroviral nucleus), Pol (reverse transcriptase), Pro (integrase), and Env are the four viral proteins encoded by a HERV’s genome, which is identical to those of exogenous retroviruses’ genomes [66]. Most HERVs are found in heterochromatin and are silenced through epigenetic processes such as germinal centers methylation, histone changes, and RNA silencing. HERV expression has been linked to some physiological functions thus far [8]. The presence of HERV-W, HERV-K, and HERV-H in the embryonic brain suggests that HERV may play an essential function in brain evolution and the progression of brain injury [8, 67]. In addition, high-level expression of the HERV family such as HERV-W and HERV-K has been indicated in many diseases, such as cancers and autoimmune diseases [8]. A link between HERVs and MS has been established [66] as HERV-K18 Env expression is higher in MS patients [8]. The pathogenic coat of the HERV-W Env protein originally termed “MSRV- Env,” has been detected in the brain, serum, perivascular infiltration, and infiltrating macrophages of MS diseases [8, 66, 68], Proposing a role of HERVW/MSRV as a biomarker for the course and treatment outcome of MS [68]. Microglia activated by recombinant HERV-W Env protein can cause myelinated axon injury, implying that pHERV-W Env may function in MS neuron destructions [8, 68]. Although it is unknown whether HERV-W is to blame for the onset of multiple sclerosis, evidence suggests that it may affect the immune system. HERV-W Env has been linked to inflammatory processes and is related to active demyelination locations, and is primarily expressed in macrophages and microglia [8, 68].
Activation of the Immune System by HERV in MS
In MS, some HERV products are overexpressed, which triggers an innate immune response by stimulating IFN-I and III responses. The severity of MS appears to be associated with the p40 subunits of IL-12 and IL-6 [8]. The expression of human inducible nitric oxide synthase (hiNOS) and the promoter activity of hiNOS can be increased by HERV-W Env. Nitric oxide (NO) encompasses a dual function in the CNS. By some mechanisms, NO can demyelinate or destroy oligodendrocytes, disrupting the structure of the BBB and increasing its permeability, boosting neuronal apoptosis or necrosis, and effects on damaging the axons [8, 68]. Dendritic cells’ ability to develop Th1-like effector T lymphocytes and their functional or phenotypic maturation can be induced by HERV-W Env. Expressing the HERV-W Env epitope by active B cells and monocytes in MS patients can demonstrate cross-reactivity toward myelin proteins via molecular mimicry events [8, 68].
The MSRV Env’s potential immunopathogenic and pro-inflammatory effects appear to be associated with the activation of TLR4 and its co-receptor CD14, which is expressed on endothelial and monocyte cells and lead to the formation of inflammatory cytokines, IL-6, IL1β, and TNF-α [69]. The expression induction of ICAM-1 happens by TLR4 activation on endothelial cells, which recruits T-cells from the bloodstream toward the CNS. The Env protein interacts with regions of the T-cell receptor-independent antigen-binding site. It can activate many clones regardless of the antigen dedicated after T-cells enter the CNS. The Env protein, a superantigen, can be a mediator in a cycle and leads to out-of-control autoreactive cell expansion and major secreting of pro-inflammatory cytokines in the CNS with its pro-inflammatory effects on microglia [66]. In addition, in MS patients, the Env protein can be co-localized with normal-appearing white matter oligodendrocyte progenitor cells (OPCs). The production of inflammatory cytokines and inducible nitric oxide synthase (iNOS), which in turn affects the expression of myelin proteins and also causes groups of the nitrotyrosine (superoxide) to form, is determined by the activation of TLR4 expressed in OPC. These adverse effects of the HERV-W env protein on OPC may obstruct myelin repair, result in remyelination abnormalities, and development of MS [70].
Laboratory Diagnostic Markers of HERV in MS
The perceived association between HERV and autoimmune disease hangs primarily on detecting retroviral antigens at the disease site or the presence of the examination case’s serum antiretroviral antibodies [8]. In an investigation, expressing several gag genes of HERV-K was significantly higher in PBMCs and brain cells from patients suffering from MS. In addition to HERV-K and HERV-W, HERV-H Env and gag increased expression in PBMCs and serum of patients with MS [8]. Elevated expression of HERV-H Env protein was found in monocytes and B cells of patients with active MS compared with inactive MS patients or healthy controls [8]. EBV can activate HERV-W in infectious mononucleosis patients and healthy individuals with an elevated titer of EBNA-1 [8].
Medication Control of HERV
These findings indicate that HERV activation may come up with the progression of MS triggering the demyelination process. HERV may be activated by several simulations, consisting of viruses such as VZV, HSV-1, EBV, and HHV-6 [8, 12]. It has been reported that treatment may also affect the expression of HERV. Rituximab can play a role in downregulating the expression of HERV by depleting B cells that co-express proteins of EBV and HERV [8, 68]. Several findings have shown that a considerable decrease may happen in viremia in interferon-beta, natalizumab, and fingolimod-treated patients [68].
Stimulation of MS by Cytomegalovirus
CMV is another human herpes virus implicated in several autoimmune diseases. In most cases, immune-competent individuals infected with CMV have few or no symptoms [1]. In experimental autoimmune encephalomyelitis (EAE), the proportion of peripheral CD4 + CD28 null T-cells is correlated with the severity of the illness [6]. This suggests that peripherally enlarged CD4 + CD28 null T-cells can affect the CNS by migrating to the CNS. In summary, it has been demonstrated that CMV can increase the proliferation of CD4 + CD28 null T-cells, which in turn encourages the escalation of autoimmune-mediated inflammation, demyelination, and activation of disease-specific CD4 + T-cells [6]. CD28 is seen on the surface of naive T-cells, but the loss of CD28 expression can be caused by repeated antigen stimulations. Within chronic activation of the immune system in a subgroup of healthy control subjects and MS patients, CD4 + CD28 null memory T-cells can occur [6]. Memory T-cells specific to CMV may accumulate significantly (on average 10% of total T memory cell compartments) due to CMV persistent quiddity. Due to this high percentage of CMV-specific T cells, immunological surveillance may become noticeably weaker, impairing normal immunity [71]. A linear relationship with the severity of disease in MS has not yet been defined. However, indirect evidence, such as the ability to penetrate target tissues and cytotoxic activity on oligodendrocytes, suggests this hypothesis [6].
Laboratory Diagnostic Markers of CMV in MS
A study exposed that the mean value of anti-cytomegalovirus IgG antibody in MS patients’ blood was not only increased but also statistically significant. The mean value of the anti-cytomegalovirus IgM antibody in the MS patients’ blood increased but was not statistically meaningful [72]. They found that antibody levels to CMV were higher in MS patients (98%) compared to controls (52%) and were statistically significant [72].
Our results also demonstrated the role of CMV in the elevation of autoimmune symptoms in MS patients. The acceleration in IgG and IgM antibody titers against CMV in the MS patients’ sera was statistically significant [72], suggesting a role that this virus may affect autoimmune diseases [72]. The enrichment of CMV-specific antibodies in MS is the most crucial indicator of the disease promoting state. Within those patients with MS where antibodies had been found, this became related to a reduced relapse time, a boom within the wide variety of relapses, and more advantageous mind atrophy [6]. When comparing the serosensitivity of two important antigens, VZV-IgG and CMV-IgG, between controls and patients with MS, we noted that the control group was remarkably more likely to be less favorable for both antibodies compared with the patients suffering from MS [11].
Stimulation of MS by VZV
VZV has been shown to be the most common component of multi-specific humoral responses in the spinal cavity of patients who suffer from MS, which helps diagnose MS. Additionally, a higher risk of MS diagnosis can be associated with VZV infection [12]. After primary infection, VZV can remain dormant in the sensory ganglia. The virus can be reactivated during immunosuppression [48]. The mechanisms by which CMV or VZV may affect the risk of MS are unclear but may arise from an immune response to proteins of the virus or alter the local cytokine milieu by a nonspecific third-party immune response [11].
Laboratory Diagnostic Markers of VZV in MS
In an investigation, anti-VZV IgG seropositivity in patients with MS was slightly elevated than in controls, demonstrating VZV DNA’s short-term presence in mononuclear cells during relapse. A higher risk of developing MS a year after reactivation of VZV was found in the Taiwanese population than in the control group [48]. Some studies have shown that VZV load increases during relapse and decreases during remission of the disease [11].
SARS-CoV-2 Role in MS
SARS-CoV-2 is the seventh human coronavirus known as a positive sense non-segmented RNA virus [73,74,75]. Detecting coronaviruses in the CNS of patients with Alzheimer’s disease (AD), Parkinson’s disease (PD), and MS is prominent [14, 76,77,78]. A possible explanation is that infection occurs as spikes in viral glycoproteins bind to the angiotensin-converting enzyme 2 (ACE2) receptor, which is widespread in the brain [76, 79].
PARP9 (poly ADP-ribose polymerase 9) and PARP14 (poly ADP-ribose polymerase 14) play vital roles in eukaryotic physiology and are actively involved in developing COVID-19. Under normal physiological conditions, the function of the PARP family proteins is mainly unclear. However, both proteins play essential roles in IFN-mediated host antiviral advocacy and DNA repair [80,81,82]. Notably, PARP9 and PARP14 have two opposing positions in IFN-γ-induced macrophage activation, where PARP9 promotes the IFN-γ response and PARP14 inhibits it by preventing STAT1 phosphorylation. On the Open Targets Platform, PARP14 and PARP9 have shown powerful associations with a wide range of human diseases spanning multiple organ systems, many of which have an autoimmune component in their etiology (e.g., MS) [80]. In contrast to PARP14 and PARP9 proteins, lymphocytes can express two TCRs that promiscuously interact with two or more molecular mimics, thereby increasing the potential for self-oriented immunopathology and further epitope diffusion [80, 83].
MS is an interesting disease for several reasons. First, the disease itself has an immunological nature. And then, disease-modifying therapy (DMT) clinical management may alter immune function and increase susceptibility to COVID-19 [76]. People with MS treated with DMT usually have a higher risk of infection, with rituximab having the most significant severe infection incidence [84]. However, COVID-19 infection severity was not associated with the presence of DMT, which was related to a lower risk of hospitalization in univariate analysis [85, 86].
Additionally, neurologists worldwide face the daunting task of stratifying the viral infection risk, especially in MS patients receiving immunosuppressive or immunomodulatory therapies. Although it has been documented that people with MS may, in theory, have a higher risk of infection than the general population, it is still being discussed whether patients with MS are at a higher risk of contracting COVID-19 from infection with SARS-CoV-2 [84]. In particular, some thoughts are required to examine the most recent data showing a lack of relationship with DMT exposure and a significant association between the Extended Disability Status Scale (EDSS) and the severity of COVID-19 and age [85]. The highest degree of heterogeneity in severe COVID-19 outcomes has been linked to EDSS, according to reports [14, 76].
The statement that morbidity and death in COVID-19 may be caused by an overlapping immunological response generated by the virus and the subject’s immune condition can be a sensible explanation. Both adaptive and innate immune responses are critical to preventing infection by the virus. Viral infections can be prevented by innate immunity with inhibition of natural killer (NK) cells and IFN-I as resistance is achieved within an adaptive immune response to immunity induced by T lymphocytes and antibodies, mainly CD8 + T-cells [14, 87, 88]. Therefore, the infection of COVID-19 triggering further amplification of immunity pathways in patients with pre-existing immunocompromised paths like MS can be speculated. Second, COVID-19 infection and age. Age has been reputed to be related to the highest variation in severe COVID-19 outcomes [85]. Third, DMT and the disease of COVID-19. Are these drugs detrimental or protective? In theory, DMT limits the immune response, allowing more replication of the virus and potentially more severe infections. On the other hand, with the aid of limiting the exaggerated immune reaction and cytokine storm due to illness by SARS-CoV-2, those drugs can also have a few defensive and valuable results in opposition to this new virus. Additionally, most of the DMTs don’t specifically interact with the innate immune system, and few of them have serious, long-lasting effects on CD8 + T-cells, which limits protection against COVID-19 [89]. The long-lasting or acute effects of COVID-19 on disease phases in patients with MS should be the subject of future research. This can also be a resilience time; This catastrophic pandemic may be an extraordinary chance for databases to ease collaboration and investigation in the MS field [90]. Although there is an association between neurodegeneration and neuroinflammation in the MS brain, there is currently insufficient evidence that SARS-CoV-2 may have a possible role in these patients’ future neurodegeneration [76].
Conclusions
The significance of viral infections in patients suffering from MS is yet unknown, and the potential for many viruses to be involved in the pathogenesis of MS has to be considered. Furthermore, because of MS heterogeneity, the interaction between viruses and other illnesses, as well as environmental and genetic variables, may vary significantly. The presence of viral components in lesions of MS or an antiviral immune response in patients suffering from MS with clinical relapses strongly suggests that viruses are involved in disease progression, potentially as stimuli or co-factors. The majority of licensed MS disease-modifying medications currently have an indirect or direct effect on memory B cells and also memory T-cells, whose cooperation is expected to be seriously involved in disease pathogenesis. Clinical trials utilizing a B cell-depleting antibody have recently backed up the B cell role in MS. Ocrelizumab is a game-changing therapy for MS, and its ability to reduce disease activity and CNS damage as long as it preferentially targets B cells is consistent with the pathophysiologic role(s) of EBV-infected B cells in MS patients. Cell-based treatments that target specific groups of B cells, such as EBV-infected ones, might be a novel way to do this, with a suggestion for treating relapsing and also progressive types of MS disease, as well as perhaps hindering the disease from developing. The frequency and specificity of these viruses for humans, however, make studying plausible pathways challenging. As a result, the development of novel MS models in infected animals is exceptionally encouraging, and it enables an essential tool for defining the viral involvement in MS.
Data Availability
Not applicable.
References
Hussein HM, Rahal EA (2019) The role of viral infections in the development of autoimmune diseases. Taylor & Francis, NY, pp 394–412
Olsson T, Barcellos LF, Alfredsson L (2017) Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 13(1):25–36
Leitzen E et al (2019) Comparison of reported spinal cord lesions in progressive multiple sclerosis with theiler’s murine encephalomyelitis virus induced demyelinating disease. Int J Mol Sci 20(4):989
Mechelli R et al (2021) Viruses and neuroinflammation in multiple sclerosis. Neuroimmunology and Neuroinflammation 8:269–283
Houen G, Trier NH, Frederiksen JL (2020) Epstein-Barr virus and multiple sclerosis. Front Immunol 2020:3315
Vanheusden M et al (2017) Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci Rep 7(1):1–11
Cheng Y, Skinner DD, Lane TE (2018) Innate immune responses and Viral-Induced neurologic disease. J Clin Med 8(1):3
Latifi T, Zebardast A, Marashi SM (2021) The role of human endogenous retroviruses (HERVs) in multiple sclerosis and the plausible interplay between HERVs, Epstein-Barr virus infection, and vitamin D. Mult Scler Relat Disorders 57:103318–103318
Bar-Or A et al (2020) Epstein-Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med 26(3):296–310
Marrodan M et al (2019) The role of infections in multiple sclerosis. Mult Scler J 25(7):891–901
Karampoor S et al (2017) Cytomegalovirus and varicella zoster virus seropositivity of Iranian patients with multiple sclerosis: a population-based study. J Neuroimmunol 309:4–6
Donati D (2020) Viral infections and multiple sclerosis. Drug Discov Today Dis Model 32:27–33
Gholizadeh O et al (2022) Recent advances in treatment Crimean-Congo hemorrhagic fever virus: a concise overview. Microb Pathog 8:105657
Freire-de-Lima L et al (2021) Autoimmune disorders & COVID-19. Medicines 8(10):55–55
Keikha M et al (2020) HCV genotypes and their determinative role in hepatitis C treatment. Virusdisease 31(3):235–240
Hashemi B et al (2021) Emerging importance of nanotechnology-based approaches to control the COVID-19 pandemic; focus on nanomedicine iterance in diagnosis and treatment of COVID-19 patients. Journal of Drug Delivery Science and Technology 67:102967
Eslami M et al (2021) Dietary pattern, colonic microbiota and immunometabolism interaction: new frontiers for diabetes mellitus and related disorders. Diabet Med 38(2):e14415
Leibovitch EC, Jacobson S (2018) Viruses in chronic progressive neurologic disease. Mult Scler J 24(1):48–52
Xu L et al (2021) Positive association of herpes simplex virus-IgG with multiple sclerosis: A systematic review and meta-analysis. Mult Scler Relat Disorders 47:102633
Del Valle L, White MK, Khalili K (2008) Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J Neuropathol Exp Neurol 67(8):729–740
Meneguzzi G et al (1978) Minichromosome from BK virus as a template for transcription in vitro. Proc Natl Acad Sci 75(3):1126–1130
Saribaş AS et al (2010) JC virus-induced progressive multifocal leukoencephalopathy. Futur Virol 5(3):313–323
Jouanguy E et al (2020) Human inborn errors of immunity to herpes viruses. Curr Opin Immunol 62:106–122
Majerciak V et al (2019) A genome-wide Epstein-Barr virus polyadenylation map and its antisense RNA to EBNA. J Virol 93(2):e01593-e1618
Soldan SS et al (2021) Epigenetic plasticity enables CNS-trafficking of EBV-infected B lymphocytes. PLoS Pathog 17(6):e1009618
Keane JT et al (2021) The interaction of Epstein-Barr virus encoded transcription factor EBNA2 with multiple sclerosis risk loci is dependent on the risk genotype. EBioMedicine 71:103572–103572
Endriz J, Ho PP, Steinman L (2017) Time correlation between mononucleosis and initial symptoms of MS. Neurol-Neuroimmunol Neuroinflam 4(3):e308
Pender MP et al (2017) Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Trans Immunol 6(1):e126
Hammerschmidt W (2015) The epigenetic life cycle of Epstein-Barr virus. Epstein Barr Virus 1:103–117
Cencioni MT et al (2017) Programmed death 1 is highly expressed on CD 8+ CD 57+ T cells in patients with stable multiple sclerosis and inhibits their cytotoxic response to Epstein-Barr virus. Immunology 152(4):660–676
Latour S, Fischer A (2019) Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol Rev 291:174–189
Pender MP et al (2009) Decreased T cell reactivity to Epstein-Barr virus infected lymphoblastoid cell lines in multiple sclerosis. J Neurol Neurosurg Psychiatry 80(5):498–505
Serafini B et al (2019) Epstein-Barr virus-specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: clue for a virus-driven immunopathological mechanism. J Virol 93(24):e00980-e1019
Albanese M et al (2016) Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc Natl Acad Sci 113(42):E6467–E6475
Jangra S et al (2019) Epstein-Barr virus and innate immunity: Friends or foes? Microorganisms 7(6):183
Jelcic I et al (2018) Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 175(1):85–100
Winter S et al (2018) Loss of RASGRP 1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol Med 10(2):188–199
Wanke F et al (2017) EBI2 is highly expressed in multiple sclerosis lesions and promotes early CNS migration of encephalitogenic CD4 T cells. Cell Rep 18(5):1270–1284
Rutkowska A et al (2018) EBI2 regulates pro-inflammatory signalling and cytokine release in astrocytes. Neuropharmacology 133:121–128
Clottu AS et al (2017) EBI2 expression and function: robust in memory lymphocytes and increased by natalizumab in multiple sclerosis. Cell Rep 18(1):213–224
Liu X et al (2020) Human virus transcriptional regulators. Cell 182:24–37
Afrasiabi A et al (2019) Evidence from genome wide association studies implicates reduced control of Epstein-Barr virus infection in multiple sclerosis susceptibility. Genome medicine 11(1):1–13
Glaser LV et al (2017) EBF1 binds to EBNA2 and promotes the assembly of EBNA2 chromatin complexes in B cells. PLoS Pathog 13(10):e1006664
Simonsen CS et al (2020) The diagnostic value of IgG index versus oligoclonal bands in cerebrospinal fluid of patients with multiple sclerosis. Mult Scler J-Exp Trans Clin 6(1):2055217319901291–2055217319901291
Makhani N et al (2019) Oligoclonal bands increase the specificity of MRI criteria to predict multiple sclerosis in children with radiologically isolated syndrome. Mult Scler J-Exp Trans Clin 5(1):2055217319836664
Arrambide G et al (2018) The value of oligoclonal bands in the multiple sclerosis diagnostic criteria. Brain 141(4):1075–1084
Hassani A et al (2018) Epstein-Barr virus is present in the brain of most cases of multiple sclerosis and may engage more than just B cells. PLoS ONE 13(2):e0192109
Czarnowska A et al (2018) Herpesviridae seropositivity in patients with multiple sclerosis: first polish study. Eur Neurol 80(5–6):229–235
Moreno MA et al (2018) Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol-Neuroimmunol Neuroinflamm 5(4):e466
Maple PA et al (2020) A different response to cytomegalovirus (CMV) and Epstein-Barr virus (EBV) infection in UK people with multiple sclerosis (PwMS) compared to controls. J Infect 80(3):320–325
Veroni C et al (2018) Transcriptional profile and Epstein-Barr virus infection status of laser-cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflammation 15(1):1–19
Jakimovski D et al (2018) Interferon β for multiple sclerosis. Cold Spring Harb Perspect Med 8(11):a032003
Zivadinov R et al (2019) Teriflunomide’s effect on humoral response to Epstein-Barr virus and development of cortical gray matter pathology in multiple sclerosis. Mult Scler Relat Disorders 36:101388
Ogembo JG et al (2015) A chimeric EBV gp350/220-based VLP replicates the virion B-cell attachment mechanism and elicits long-lasting neutralizing antibodies in mice. J Transl Med 13(1):1–15
Smith C, Khanna R (2015) The development of prophylactic and therapeutic EBV vaccines. Epstein Barr Virus 2:455–473
Eslami M et al (2019) Mycobacterium avium paratuberculosis and Mycobacterium avium complex and related subspecies as causative agents of zoonotic and occupational diseases. J Cell Physiol 234(8):12415–12421
Mulero P, Midaglia L, Montalban X (2018) Ocrelizumab: a new milestone in multiple sclerosis therapy. Ther Adv Neurol Disord 11:1756286418773025
Keyvani H et al (2020) The role of human herpesvirus-6 and inflammatory markers in the pathogenesis of multiple sclerosis. J Neuroimmunol 346:577313
Dunn N, Kharlamova N, Fogdell-Hahn A (2020) The role of herpesvirus 6A and 6B in multiple sclerosis and epilepsy. Scand J Immunol 92(6):e12984
Greninger AL et al (2018) Comparative genomic, transcriptomic, and proteomic reannotation of human herpesvirus 6. BMC Genomics 19(1):1–17
Engdahl E et al (2019) Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol. https://doi.org/10.3389/fimmu.2019.02715
Tang H et al (2013) CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc Natl Acad Sci 110(22):9096–9099
Fierz W (2017) Multiple sclerosis: an example of pathogenic viral interaction? Virology journal 14(1):1–5
Fotheringham J et al (2008) Human herpesvirus 6 (HHV-6) induces dysregulation of glutamate uptake and transporter expression in astrocytes. J Neuroimmune Pharmacol 3(2):105–116
Morandi E et al (2017) Human endogenous retroviruses and multiple sclerosis: causation, association, or after-effect? Mult Scler J 23(8):1050–1055
Küry P et al (2018) Human endogenous retroviruses in neurological diseases. Trends Mol Med 24(4):379–394
Wang X, Huang J, Zhu F (2018) Human endogenous retroviral envelope protein syncytin-1 and inflammatory abnormalities in neuropsychological diseases. Front Psych 9:422
Dolei A (2018) The aliens inside us: HERV-W endogenous retroviruses and multiple sclerosis. Mult Scler J 24(1):42–47
Duperray A et al (2015) Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int Immunol 27(11):545–553
Madeira A et al (2016) MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll-like receptor 4: relevance of GNbAC1 in multiple sclerosis treatment. J Neuroimmunol 291:29–38
Hanley PJ, Bollard CM (2014) Controlling cytomegalovirus: helping the immune system take the lead. Viruses 6(6):2242–2258
Salim MA et al (2017) Determining the IgM and IgG antibody titer against CMV and helicobacter pylori in the serum of multiple sclerosis patients comparing to the control group in Hamadan. Hum Antibodies 26(1):23–28
Lippi A et al (2020) SARS-CoV-2: at the crossroad between aging and neurodegeneration. Mov Disord 35(5):716
Yasamineh S et al (2022) A state-of-the-art review on the recent advances of niosomes as a targeted drug delivery system. Int J Pharm 624:121878
Yousefi B et al (2020) A global treatments for coronaviruses including COVID-19. J Cell Physiol 235(12):9133–9142
Ferini-Strambi L, Salsone M (2021) COVID-19 and neurological disorders: are neurodegenerative or neuroimmunological diseases more vulnerable? J Neurol 268(2):409–419
Matías-Guiu J et al (2020) Should we expect neurological symptoms in the SARS-CoV-2 epidemic? Neurología (English Edition) 35(3):170–175
Yasamineh S et al (2022) Spotlight on therapeutic efficiency of mesenchymal stem cells in viral infections with a focus on COVID-19. Stem Cell Res Ther 13(1):257
Rodriguez-Perez AI et al (2020) Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav Immun 87:256–271
Kovačić D, Jotanović J, Laković J (2021) The possible role of molecular mimicry in SARS-CoV-2-mediated autoimmunity: an immunobiochemical basis. J Med Sci 90(3):e560–e560
Yousefi B, Eslami M (2022) Genetic and structure of novel coronavirus COVID-19 and molecular mechanisms in the pathogenicity of coronaviruses. Rev Med Microb 33(1):e180–e188
Yousefi B et al (2022) Potential therapeutic effect of oxygen-ozone in controlling of COVID-19 disease. Med Gas Res 12(2):33
Dardalhon V et al (2008) Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun 31(3):252–256
Willis M, Robertson N (2020) Multiple sclerosis and the risk of infection: considerations in the threat of the novel coronavirus, COVID-19/SARS-CoV-2. J Neurol 267(5):1567–1569
Louapre C et al (2020) Clinical characteristics and outcomes in patients with coronavirus disease 2019 and multiple sclerosis. JAMA Neurol 77(9):1079–1088
Yasamineh S et al (2022) An overview on nanoparticle-based strategies to fight viral infections with a focus on COVID-19. Journal of Nanobiotechnology 20(1):440
Berger JR, Brandstadter R, Bar-Or A (2020) COVID-19 and MS disease-modifying therapies. Neurol-Neuroimmunol Neuroinflamm 7(4):e761
Fard NG et al (2021) COVID-19 coinfection in patients with active tuberculosis: first case-report in Iran. Authorea Preprints
Baker D et al (2020) The underpinning biology relating to multiple sclerosis disease modifying treatments during the COVID-19 pandemic. Mult Scler Relat Disorders 43:102174
Racke MK, Newsome SD (2020) Multiple sclerosis and COVID-19: a great opportunity for databases promoting research and collaboration. J Neuroimmunol 345:577283
Acknowledgements
The authors would like to thank the Department of Medical Biotechnology, Semnan University of Medical Sciences for supporting this project.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors.
Author information
Authors and Affiliations
Contributions
SS, OG, SY, SA, PA, PF: Writing—original draft, HA, AF-A, DP, ME, BY: Writing—review & editing. VP and MD: Conceptualization, Supervision, Writing—review & editing. All authors participated in the manuscript in the critical review process of the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sedighi, S., Gholizadeh, O., Yasamineh, S. et al. Comprehensive Investigations Relationship Between Viral Infections and Multiple Sclerosis Pathogenesis. Curr Microbiol 80, 15 (2023). https://doi.org/10.1007/s00284-022-03112-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-022-03112-z