
Abstract
Purpose
Ipatasertib, a potent and highly selective small-molecule inhibitor of AKT, is currently under investigation for treatment of cancer. Ipatasertib is a substrate and a time-dependent inhibitor of CYP3A4. It exhibits non-linear pharmacokinetics at subclinical doses in the clinical dose escalation study. To assess the DDI risk of ipatasertib at the intended clinical dose of 400 mg with CYP3A4 inhibitors, inducers, and substrates, a fit-for-purpose physiologically based pharmacokinetic (PBPK) model of ipatasertib was developed.
Methods
The PBPK model was constructed in Simcyp using in silico, in vitro, and clinical data and was optimized and verified using clinical data.
Results
The PBPK model described non-linear pharmacokinetics of ipatasertib and captured the magnitude of the observed clinical DDIs. Following repeated doses of 400 mg ipatasertib once daily (QD), the PBPK model predicted a 3.3-fold increase of ipatasertib exposure with itraconazole; a 2–2.5-fold increase with moderate CYP3A4 inhibitors, erythromycin and diltiazem; and no change with a weak CYP3A4 inhibitor, fluvoxamine. Additionally, in the presence of strong or moderate CYP3A4 inducers, rifampicin and efavirenz, ipatasertib exposures were predicted to decrease by 86% and 74%, respectively. As a perpetrator, the model predicted that ipatasertib (400 mg) caused a 1.7-fold increase in midazolam exposure.
Conclusion
This study demonstrates the value of using a fit-for-purpose PBPK model to assess the clinical DDIs for ipatasertib and to provide dosing strategies for the concurrent use of other CYP3A4 perpetrators or victims.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ipatasertib (GDC-0068) is a potent, highly selective small-molecule inhibitor of the three isoforms of the serine–threonine kinase AKT (Akt1, Akt2, and Akt3) or protein kinase B [1,2,3]. It was developed to treat cancers, such as breast and prostate cancers, with a high prevalence of PI3K/AKT pathway activation, promoting tumor survival, proliferation, growth and changes in cellular metabolic pathways [4, 5]. Ipatasertib in combination with abiraterone has showed prolonged radiographic progression-free survival compared to placebo with abiraterone in patients with metastatic prostate cancer in a phase 2 clinical study [6]. Currently, multiple clinical studies are ongoing to evaluate ipatasertib in combination with hormonal agents, targeted agents or chemotherapy for the treatment of solid tumors.
Ipatasertib has high solubility (> 10 mg/mL across the pH range of 1.1–7.0) and moderate permeability. Food does not affect exposures of ipatasertib [7]. In first-in-human dose escalation study (25–800 mg), ipatasertib showed rapid oral absorption with median time to peak concentration (Tmax) ranging from 0.5 to 3 h [7]. The mean terminal half-life of ipatasertib ranged from 31.9 to 53.0 h at doses above 100 mg [7]. During dose escalation stage, ipatasertib exposures increased with increasing doses and were approximately dose-proportional from 200 to 800 mg following a single dose or multiple doses [7]. Exposure at 100 mg was close to dose-proportional after a single dose and more than dose-proportional increase in exposures was observed at the low doses of 25–50 mg [7]. The absolute bioavailability of ipatasertib was estimated to be 34%. Following a single 200 mg oral dose, approximately 24% unchanged drug was eliminated in feces and 8% in urine, indicating that ipatasertib was primarily eliminated by metabolism (ClinicalTrials.gov Identifier: NCT02390492).
In vitro studies indicated that ipatasertib is a substrate and a competitive and time-dependent inhibitor (TDI) of CYP3A4. The potential drug–drug interaction (DDI) risk of ipatasertib with inhibitors and substrates of CYP3A4 was therefore assessed in clinical studies. Itraconazole, a potent CYP3A4 inhibitor, increased ipatasertib (100 mg single oral dose) area under the curve (AUC) and maximum concentration (Cmax) by 5.45-fold and 2.26-fold, respectively [8]. In the presence of ipatasertib (600 mg QD), the AUC and Cmax of midazolam, a sensitive CYP3A4 substrate, increased by 2.22-fold and 1.29-fold, respectively [7]. Given the observed effects of CYP3A4 modulations on the pharmacokinetics (PK) of ipatasertib, additional evaluation of DDI potentials for ipatasertib is crucial to inform on concomitant medication use during ipatasertib treatment at clinically intended dose of 400 mg.
As recommended by guidance from the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for investigating drug interactions [9, 10], the physiologically based pharmacokinetic (PBPK) modeling and simulation approach has been commonly used to predict the PK and enzyme-mediated DDI, providing dosing recommendations in drug applications [11,12,13]. The aim of this study was to develop a fit-for-purpose PBPK model for ipatasertib to predict additional untested drug interactions for ipatasertib.
Methods
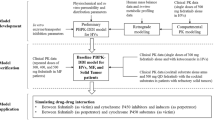
A PBPK model of ipatasertib was developed using Simcyp population-based absorption, distribution, metabolism, and excretion (ADME) simulator v.18 (Certara, Sheffield, UK) and the model was optimized, verified and applied to predict untested DDIs according to the workflow shown in Fig. 1. The initial model development utilized in silico, in vitro, and clinical PK data. Parameter optimization and verification were performed using clinical PK and DDI data. The verified PBPK model was then used to predict untested DDIs of ipatasertib at the clinically intended dose of 400 mg as the victim or perpetrator of CYP3A4. The ipatasertib PK was comparable between patients with cancer and healthy subjects and the Simcyp healthy volunteer population (“Sim-Healthy Volunteers”) was used in all model development and application steps. For each simulation, the age range, proportion of females, and dosing regimen were set to match the observed ranges in the relevant clinical studies.

Workflow of ipatasertib PBPK model development, verification and application. Fa fraction absorbed, Ka first-order absorption rate constant, Qgut a nominal flow in gut model, Vss volume of distribution at steady state, CLR renal clearance, Ki concentration of inhibitor that supports half-maximum inhibition, Kapp concentration of mechanism-based inhibitor associated with half-maximal inactivation rate, Kinact inactivation rate, CLadditional additional systemic clearance, MD multiple dose, SD single dose
Initial model development
The physiochemical and ADME parameters used for the development of the initial PBPK model are summarized Table S1. A first-order absorption model was used to describe the ipatasertib absorption. The fraction absorbed (fa) was estimated based on the human mass balance study following administration of a single oral dose of 200 mg ipatasertib (ClinicalTrials.gov Identifier: NCT02390492), in which unchanged ipatasertib in feces accounted for 24% of the administered dose. Fa was assumed to be dose-independent since ipatasertib has high solubility (> 10 mg/mL across the pH range of 1.1–7.0) and moderate permeability. The Ka and a nominal flow in gut model (Qgut) were predicted within Simcyp using ipatasertib MDCK permeability data. For ipatasertib distribution, a full PBPK model was used. The volume of distribution at steady state (Vss) was obtained from the clinical absolute bioavailability study (ClinicalTrials.gov Identifier: NCT02390492). A tissue to plasma partition coefficient (Kp) scalar was applied to match the predicted Vss (predicted with in Simcyp using Method 3, Rodgers et al. + ion membrane permeability) to the observed value. The IV clearance determined in the clinical absolute bioavailability study was used (ClinicalTrials.gov Identifier: NCT02390492). CYP3A4 was assigned to account for 100% of hepatic clearance (CLH), as in vitro study suggested that ipatasertib was primarily metabolized by CYP3A4 [8]. The renal clearance (CLR) was calculated using Eq. 1:
in which Ae is the amount of unchanged drug excreted in urine following a single oral dose of 200 mg ipatasertib in healthy subjects and AUC0–∞ is the area under the concentration–time curve extrapolated to infinity from the same clinical study (ClinicalTrials.gov Identifier: NCT02390492). The CYP3A4 inhibition parameters for competitive inhibition and TDI determined using in vitro assays (Data on file) were incorporated into the model.
To assess the initial model, the PK of ipatasertib following a single intravenous (IV) dose of 0.08 mg or oral dose of 200 mg was simulated separately with 10 trials containing 8 subjects per trial over 3 days and compared with the clinical PK data in the absolute bioavailability study. The age range of 21–41 and the proportion of females of 0 were used in the simulation.
The performance of the PBPK model that incorporated TDI of CYP3A4 was also evaluated for its ability to explain the nonlinearity in exposures at the lower doses. In this case, ipatasertib PK was simulated following single and multiple oral doses over a range of 25–800 mg and was compared to the observed clinical data in the dose escalation study [7]. The ipatasertib PK (25–800 mg ipatasertib QD) was simulated with 10 trials containing 29 subjects per trial over 8 days. The age range of 32–73 and the proportion of females of 0.55 were used in the simulation.
Optimization and verification of model parameters
In the clinical dose escalation study (25–800 mg), more than a dose-proportional increase of ipatasertib exposure was observed at lower doses (25–50 mg) after single and multiple doses. The initial model with consideration of TDI using the observed IV clearance failed to describe the observed nonlinearity of ipatasertib PK. Therefore, parameters related to ipatasertib metabolism were optimized to capture nonlinear PK as well as clinical DDI data in a stepwise manner (Fig. 1). Briefly, Km and Vmax of CYP3A4 were first optimized by capturing the clinical DDI between ipatasertib and itraconazole without incorporating CYP3A4 TDI parameters in the model. Three models with different Km values were developed in parallel to ensure the observed nonlinear PK of ipatasertib could be properly described in the later step. Then, to ensure the model could properly describe the inhibitory impact of ipatasertib on CYP3A4, the inactivation rate (Kinact) of CYP3A4 was optimized in each model by capturing the clinical DDI between midazolam and ipatasertib. Lastly, a model incorporating the optimized Km, Vmax and Kinact, and capturing the nonlinear PK of ipatasertib at lower doses was selected from three developed models as the final model. Details are described in the subsequent sections.
Simulation of DDI between ipatasertib and itraconazole
To determine the fraction metabolized by CYP3A4, Km and Vmax of CYP3A4 and additional hepatic clearance (CLadditional) were optimized by capturing the observed DDI between ipatasertib and itraconazole.
Briefly, starting from the in vitro measured Km,CYP3A4 of 19.47 μM, a sensitivity analysis was conducted with 10- and 100-fold lower values of Km,CYP3A4 (1.95 μM and 0.195 μM) used in the simulations. The Simcyp retrograde calculator in the recombinant CYP module was then used to calculate the intrinsic clearance of CYP3A4 (CLint,CYP3A4) (assuming CYP3A4 contributes to 100% Hep met CL) from the observed IV clearance and CLR and assigned CLaddtional, and from there, Vmax,CYP3A4 was calculated using Eq. 2:
A total of three models (three sets of Km,CYP3A4, Vmax,CYP3A4, and CLadditional) that can capture the observed DDI between ipatasertib and itraconazole, were used to optimize inhibitory parameters in the subsequent step. Since ipatasertib was given as a low single dose (100 mg) in this DDI study, the impact of auto-inhibition caused by TDI was expected to be minimal, and parameters related to TDI were not included in this step. The Simcyp default compound file of itraconazole (fasted, solution) and OH-itraconazole was used for the simulation. The interaction between ipatasertib (100 mg, single dose, starting on Day 5) and itraconazole (200 mg QD of 9 doses, starting on Day 1) was simulated with 10 trials containing 15 subjects per trial over 12 days. The age range of 22–52 and the proportion of females of 0.67 were used in the simulation.
Simulation of DDI between ipatasertib and midazolam
DDI between ipatasertib and midazolam was simulated using the Simcyp default compound file of midazolam and compared with observed data from a clinical study conducted in patients with cancer [7]. The in vitro measured CYP3A4 inhibitory parameters of ipatasertib, Ki of 4.4 μM, Kapp of 9.66 μM and Kinact of 2.6 1/h, were used in the ipatasertib model. Briefly, the interaction between midazolam (2 mg, single oral dose, starting on Day 8) and ipatasertib (600 mg QD of 8 doses, starting on Day 1) was simulated with 10 trials containing 13 subjects per trial over 8 days. The age range of 37–76 and the proportion of females of 0.69 were used in the simulation.
Initially, the DDI between midazolam and ipatasertib was consistently over-predicted. Therefore, sensitivity analyses of inhibitory parameters, where only one parameter (Ki or Kinact) varied at a time, were performed for all three models described above. Based on results of sensitivity analyses, Kinact was optimized in each model to capture the clinical DDI between midazolam and ipatasertib.
Simulation of ipatasertib PK following single and multiple oral doses
Three models with optimized enzyme kinetic (Vmax and Km) and inhibitory (Kinact) parameters of CYP3A4 and CLadditional were established as described above with the rest of the input parameters remaining the same, and each model predicted the clinical DDI between ipatasertib and itraconazole or midazolam. Lastly, to estimate the value of Km,CYP3A4 while capturing the nonlinearity of ipatasertib PK, the ipatasertib PK following single and multiple doses at the dose range of 25–800 mg was simulated using each model and compared to the PK profile of observed clinical studies. The parameters used in the final model that described the nonlinearity of ipatasertib PK are listed in Table 1.
Verification of ipatasertib PBPK model
To verify the performance of the final PBPK model, ipatasertib PK was simulated and compared with observations from two clinical studies that were not used for model development and optimization. In the first clinical study, AUC and Cmax of ipatasertib decreased by approximately 50% and 53% in the presence of enzalutamide (a strong CYP3A4 inducer) in patients with prostate cancer [14, 15]. The interaction between ipatasertib (400 mg QD of 43 doses, starting on Day 1) and enzalutamide (160 mg QD of 35 doses, starting on Day 9) was simulated with 10 trials containing 23 subjects per trial over 44 days. The age range of 56–83 with the proportion of females of 0 was used in the simulation. A published enzalutamide PBPK model [16] was used and the input parameters of the enzalutamide PBPK model are listed in Table S2.
The second study used for model verification is the study of ipatasertib co-administered with palbociclib (a substrate and TDI of CYP3A4) [17], in which ipatasertib AUC and Cmax increased by approximately 63% and 44% in the presence of palbociclib in patients with breast cancer (ClinicalTrials.gov Identifier: NCT04060862). Briefly, the interaction between ipatasertib (300 mg QD of 22 doses, starting on Day 1) and palbociclib (125 mg QD of 15 doses, starting on Day 8) was simulated with 10 trials containing 9 subjects per trial over 23 days. A published palbociclib PBPK model [18] and the age range of 49–67 with the proportion of females of 1 were used in the simulation. The input parameters of the palbociclib PBPK model are summarized in Table S3.
Application of the ipatasertib PBPK model
The verified ipatasertib PBPK model was used to assess CYP3A4-mediated DDIs between ipatasertib at the intended therapeutic dose of 400 mg with strong, moderate or weak CYP3A4 inhibitors or inducers and a sensitive CYP3A4 substrate. In addition, DDIs between 200 mg ipatasertib and moderate CYP3A4 inhibitors were simulated and compared with 400 mg ipatasertib alone to assess the adequacy of dose reduction when concurrent use of moderate CYP3A4 inhibitors cannot be avoided. An age range of 20–95, the age range for the default cancer population, and a proportion of females of 0.5 were used in the Simcyp default healthy volunteer population for the simulation.
Effect of CYP3A4 inhibitors on ipatasertib PK
To evaluate the effects of strong (itraconazole), moderate (erythromycin and diltiazem) and weak (fluvoxamine) CYP3A4 inhibitors on ipatasertib PK, Simcyp default compound files of itraconazole, erythromycin, diltiazem, and fluvoxamine were used in the simulation with the ipatasertib PBPK model. Briefly, the interaction between ipatasertib (400 mg QD of 21 doses starting on Day 1) and CYP3A4 inhibitor [itraconazole 200 mg QD of 21 doses; erythromycin 500 mg three times daily (TID) of 63 doses; diltiazem 120 mg twice daily (BID) of 42 doses; fluvoxamine 100 mg QD of 21 doses, starting on Day 1] was simulated with 10 trials containing 10 subjects per trial over 21 days.
To evaluate the impact of reducing the ipatasertib dose from 400 to 200 mg when co-administered with moderate inhibitors, simulations were conducted to evaluate interactions between ipatasertib (200 mg) and erythromycin or diltiazem with the remaining trial parameters maintained as described above.
Effect of CYP3A4 inducers on ipatasertib PK
To access the effect of CYP3A4 inducers on ipatasertib exposures, the PK of ipatasertib was simulated in the presence of rifampicin and efavirenz, strong and moderate inducers of CYP3A4, respectively. Simcyp default compound files of rifampicin and efavirenz were used in the simulation. Briefly, the interaction between ipatasertib (400 mg QD of 21 doses starting on Day 1) and CYP3A4 inducer (rifampin 600 mg QD of 21 doses; efavirenz 600 mg QD of 21 doses, starting on Day 1) was simulated with 10 trials containing 10 subjects per trial for 21 days.
Effect of ipatasertib on PK of a sensitive CYP3A4 substrate
To evaluate the effect of ipatasertib (at the clinically intended dose of 400 mg) on the PK of a sensitive CYP3A4 substrate, the DDI between midazolam and ipatasertib was simulated. The Simcyp default compound file of midazolam was used in the simulation. Briefly, the interaction between midazolam (2 mg, single dose, starting on Day 8) and ipatasertib (400 mg QD of 8 doses starting on Day 1) was simulated with 10 trials containing 10 subjects per trial over 8 days.
Results
Initial ipatasertib PBPK model development
The initial PBPK model was able to describe the observed ipatasertib PK following a single IV dose of 0.08 mg (Figure S1a). However, the PBPK model did not adequately capture the observed PK after a single oral dose of 200 mg ipatasertib in the absolute bioavailability study or the observed nonlinear PK following single and multiple oral doses at the dose range of 25–800 mg in the dose escalation study (Figures S1b and c). At steady state, the observed oral clearance of ipatasertib (Dose/AUC0–∞) decreased at the dose range of 25–50 mg, approached linearity at 100 mg and became linear at 200–800 mg after single and multiple doses. In contrast, the model predicted no change in oral clearance across all dose levels after the first dose and at steady state (Figures S1b and c), even though the model incorporated TDI using in vitro parameters.
Optimization and verification of model parameters
The initial model incorporating CYP3A4 inhibitory parameters obtained from in vitro studies did not predict the nonlinear PK of ipatasertib well. Since ipatasertib is primarily metabolized by CYP3A4, the observed nonlinearity could likely be mainly caused by the saturation of CYP3A4 in the gut and liver. To capture the saturation of CYP3A4 and the DDI between ipatasertib and itraconazole, enzyme kinetic parameters (Km and Vmax) of CYP3A4 were used instead of CLiv. The stepwise optimization strategy is illustrated in Fig. 1.
Simulation of DDI between ipatasertib and itraconazole
To capture the observed DDI between ipatasertib and itraconazole, CLadditional and Vmax were optimized with fixed Km values of 0.195, 1.95, or 19.47 μM. Three models were developed in parallel (TDI parameters were not included) and simulated with the optimized CLadditional (L/h) of 2.7, 11.8 and 13.7 and Vmax (pmol/min/pmol) of 0.135, 1 and 9.37 corresponding to Km of 0.195, 1.95, and 19.47 μM, respectively. All three models predicted ipatasertib PK and the DDI between ipatasertib and itraconazole (data not shown).
Simulation of DDI between ipatasertib and midazolam
The model using in vitro measured competitive inhibition and TDI parameters of CYP3A4 over-predicted the DDI between ipatasertib and midazolam. As shown in Figure S2a, changes in competitive inhibition (Ki) had a minor effect while changes in TDI (Kinact) had a substantial impact on the predicted magnitude of DDI (Figure S2b). Therefore, TDI parameters were included and Kinact was optimized to capture the observed DDI between midazolam and ipatasertib. The optimized values for Kinact (1/h) were 0.17, 0.33 and 0.41 corresponding to Km (μM) values of 0.195, 1.95 and 19.47, respectively. All three models predicted the magnitude of the change in midazolam exposure in the presence of ipatasertib (data not shown for models with Km of 19.47 μM, and 1.95 μM; the model with Km of 0.195 µM is shown in Table 2).
Simulation of ipatasertib PK following single and multiple oral doses
The PK profiles of ipatasertib following single and multiple doses (25–800 mg) were simulated using the three optimized models. Among the models that described the DDIs mentioned above, the model with Km of 0.195 µM best described the observed trend of nonlinear PK of ipatasertib at the dose range of 25–800 mg (Fig. 2). The simulated PK of ipatasertib using the model with Km of 0.195 µM matched the observed PK reasonably well, especially around the clinically intended dose of 400 mg (Fig. 3 and Table S4). Therefore, the model with Km of 0.195 µM was considered to be the final model.

Observed and predicted changes of ipatasertib oral clearance (mean ± SD) following a a single dose and b at steady-state

Observed and predicted plasma concentration–time profiles of ipatasertib following single and multiple oral doses. Solid black line—observed mean plasma concentrations; circles—observed individual plasma concentrations; dashed red lines—predicted mean, 95th and 5th percentile of plasma concentrations
Using the final model, the DDI between itraconazole and ipatasertib was simulated in an iterative learn-and-confirm approach. The simulated magnitude of DDI was consistent with the observed magnitude, and the simulated ipatasertib PK profile was in good agreement with the observed PK profile of 100 mg ipatasertib in the absence and presence of itraconazole (Fig. 4 and Table 2). Additionally, the simulated itraconazole PK profile was in agreement with the clinical observation (Figure S3a). The final model also predicted the observed PK of ipatasertib following a single IV or oral dose in the study that was used for initial model development (Figure S3b and c).

Simulated and observed plasma concentration–time profiles of ipatasertib following a 100 mg single oral dose in the a absence and b presence of 200 mg itraconazole. Solid line—observed mean plasma concentrations; circles—observed individual plasma concentrations; dashed line—predicted mean plasma concentrations, 95th and 5th percentile. Shown here is the simulation of the finalized model
Verification of ipatasertib PBPK model
The verification of the ipatasertib PBPK model was performed using clinical data from two independent studies. In the first study, the model predicted 42% and 62% decrease in Cmax and AUC of ipatasertib in the presence of enzalutamide (a strong CYP3A4 inducer), which was comparable to the observed 53% and 50% decrease in Cmax and AUC (Table 2). In the second study, in the presence of palbociclib (a substrate and TDI of CYP3A4), Cmax and AUC of ipatasertib were predicted to increase by 23% and 48%, respectively (Table 2). The predicted changes in ipatasertib exposures were slightly lower but still comparable to the observed 44% and 63% increase in ipatasertib exposures in the presence of palbociclib (Table 2). Moreover, the simulated exposures of enzalutamide and palbociclib were in a good agreement with the clinical observations as described in Table S5.
Application of the ipatasertib PBPK model
Effect of CYP3A4 inhibitors on ipatasertib PK
The impact of strong (itraconazole), moderate (erythromycin and diltiazem), and weak (fluvoxamine) inhibitors of CYP3A4 on the PK of ipatasertib (400 mg) was assessed using the developed PBPK model. The simulation showed that the fold-change in ipataserib AUC was 3.34, 2.51, 2.04, and 1.06 for itraconazole, erythromycin, diltiazem, and fluvoxamine, respectively. The fold-change in ipataserib Cmax was predicted to be 2.01, 1.66, 1.47, and 1.03 for itraconazole, erythromycin, diltiazem, fluvoxamine, respectively (Table 3). The observed and simulated ipatasertib AUC, Cmax ratios and 90% confidence intervals (CI) with various CYP3A4 inhibitors are shown in Fig. 5a.

Observed and simulated ipatasertib AUC and Cmax ratios with various CYP3A4 inhibitors and inducers
Additionally, simulations showed that the steady-state AUC of ipatasertib at a reduced dose of 200 mg administered concurrently with moderate CYP3A4 inhibitors erythromycin or diltiazem was comparable to the predicted steady-state AUC of ipatasertib at the clinically intended dose of 400 mg without inhibitors (Table S6).
Effect of CYP3A4 inducers on ipatasertib PK
The impact of strong (rifampin) and moderate (efavirenz) inducers of CYP3A4 on the PK of ipatasertib (400 mg) was assessed using the PBPK model. The simulation showed the fold-change in ipatasertib AUC was 0.14 and 0.26 for rifampin and efavirenz, respectively, while the fold-change in ipatasertib Cmax was 0.32 and 0.49 for rifampin and efavirenz, respectively (Table 3 and Fig. 5b).
Effect of ipatasertib on PK of a sensitive CYP3A4 substrate
The potential impact of 400 mg ipatasertib on the PK of midazolam, a sensitive CYP3A4 substrate, was evaluated using the ipatasertib PBPK model. The model predicted a fold-change in midazolam AUC of 1.69 (90% CI 1.63–1.75) and a fold-change in Cmax of 1.49 (90% CI 1.45–1.52) when co-administered with 400 mg ipatasertib (Table 3).
Discussion
A PBPK model of ipatasertib was developed using both in vitro and clinical data to assist with DDI predictions. The key clinical studies that helped in a top–down approach for model development and optimization of in vitro parameters included ADME studies (absolute bioavailability and mass balance studies), a dose escalation study showing nonlinear PK, and DDI studies with midazolam and itraconazole. The clinical data were used in a learn-and-confirm iterative process to optimize in vitro parameters of CYP3A4-mediated metabolism (Km and Vmax) and TDI (Kinact). The stepwise parameter optimization process is described in Fig. 1. After initial model development, models with three different Km,CYP3A4 values were optimized in parallel for CLadditional and Vmax,CYP3A4 using clinical PK data from the itraconazole DDI study. The reason for using this clinical DDI data as the initial step of optimization was that the effect of TDI was expected to be minimal after a single dose of 100 mg ipatasertib, thereby making a separate optimization of the CYP3A4 enzyme kinetic parameters feasible. Further, Kinact,CYP3A4 was optimized with the goal of replicating results from the DDI study with midazolam. This optimization approach is not uncommon as the magnitude of DDI is often overestimated using in vitro TDI parameters obtained from human liver microsomes [19].
There were several discrepancies between the optimized parameter values and in vitro values. The model with Km,CYP3A4 that was lower than the measured in vitro value was selected as the final model to capture the nonlinear PK and the clinical DDI. This optimized Km could be considered as a value combining the effect of fraction of unbound drug in the in vitro microsomal incubation and an extrapolation factor that accounting for in vitro to in vivo system translation. Similar explanations could be also applied to the optimized Kinact,CYP3A4. In fact, the extrapolation factor has been used to explain the difference between the in vitro and in vivo system to match the clinical observations for CYP-mediated clearance in multiple studies [20,21,22]. Moreover, based on the current clinical observations and the simulation results, the nonlinear PK observed at low dose levels was likely due to the CYP3A4 saturation rather than TDI of CYP3A4. However, it is possible that other mechanistic factors could also contribute to the nonlinear PK and the optimized enzyme kinetic and inhibitory parameters could be the net values accounting for those factors. In vitro studies showed that ipatasertib was also a substrate of P-glycoprotein (P-gp) and the saturation of P-gp in liver and intestine may contribute to the observed nonlinearity. However, the available clinical data cannot distinguish the contribution of P-gp from CYP3A4 to the observed nonlinearity. Only 2.9–5.8% of unchanged ipataseritb was found in the bile of rats and monkeys dosed with 10 mg/kg and 5 mg/kg ipatasertib, indicating the biliary elimination may be negligible and the likelihood of the P-gp saturation in liver is low. In addition to these points, although ipatasertib is a weak CYP3A4 inducer in vitro, the induction was not incorporated into the model. This is due to the difficulty in differentiating between the inhibition and induction from the clinical data and that the PBPK model parameters were optimized to represent a net inhibition effect.
This PBPK model was verified with two independent clinical studies, in which the ipatasertib PK profiles changed in the presence of enzalutamide, a strong CYP3A4 inducer or palbociclib, a substrate and TDI of CYP3A4. The simulation of the DDIs was comparable to the clinical observations, indicating the capability of the ipatasertib PBPK model in predicting the interaction with CYP3A4 inducers and inhibitors. Notably, in the presence of enzalutamide, ipatasertib exposures decreased approximately 50%, while the predicted decreases of exposures in the presence of rifampin and efavirenz were 86% and 74%, respectively. This could be explained by the fact that enzalutamide may be also a substrate and an inhibitor of CYP3A4 as suggested by the in vitro and clinical data [14, 23]. Moreover, in the presence of ipatasertib, a competitive and TDI of CYP3A4, the simulated palbociclib exposures matched the observations well, indicating the ipatasertib PBPK model reasonably described the inhibitory effect of ipatasertib. This fit-for-purpose PBPK model of ipatasertib was considered adequate for DDI prediction as several clinical studies were used in calibrating and verifying the model and key DDIs were captured.
As a substrate of CYP3A4, a 5.45-fold increase in AUC was observed with 100 mg ipatasertib in the presence of itraconazole. However, the DDI magnitude was expected to be lower at the clinically intended dose of 400 mg as the competition for CYP3A4 is typically concentration-dependent, which was confirmed using PBPK modeling with a predicted 3.34-fold increase in AUC. The exposure of 100 mg ipatasertib in the presence of the strong CYP3A4 inhibitor itraconazole was comparable to the exposure of ipatasertib alone at the clinically intended dose of 400 mg. Additionally, the AUC was predicted to increase by 2–2.5-fold in the presence of moderate inhibitors. The simulation showed that 200 mg ipatasertib in the presence of moderate CYP3A4 inhibitors had comparable exposures as 400 mg ipatasertib taken alone, suggesting if the concurrent use of moderate CYP3A4 inhibitors is unavoidable, ipatasertib could be reduced to 200 mg. Based on the simulation, weak inhibitors did not appear to affect exposures of ipatasertib appreciably, suggesting dose adjustment is not needed when ipatasertib is administered with weak CYP3A4 inhibitors. The simulation also showed that strong and moderate inducers of CYP3A4 could decrease ipatasertib AUC by 86% and 74% and Cmax by 68% and 51%, suggesting co-medications that are strong and moderate CYP3A4 inducers should be avoided because they may reduce the efficacious exposures of ipatasertib. Together, these simulations provided a model-based dosing strategy to maintain safe and efficacious plasma concentrations of ipatasertib when co-administration with CYP3A4 inhibitors or inducers is needed. In the clinical DDI study with midazolam, ipatasertib administered at 600 mg caused a 2.22-fold increase in midazolam AUC. Using the PBPK model, simulations of midazolam DDI with 400 mg ipatasertib confirmed that ipatasertib is a weak CYP3A4 inhibitor at 400 mg dose with a < 2-fold increase of AUC predicted for midazolam.
In summary, in vitro and clinical PK and DDI data were used to develop, optimize and verify a fit-for-purpose ipatasertib PBPK model. Based on the model prediction, at the clinically intended dose of 400 mg, ipatasertib is a weak CYP3A4 inhibitor, dose adjustment may be needed when chronic co-administration with moderate and strong inhibitors of CYP3A4 is needed and the concurrent use of strong CYP3A4 inducers may need to be avoided. Together with observed clinical studies, this work will provide support for safe and effective use of ipatasertib when given in combination with various modulators and substrates of CYP3A4. The PBPK model helps reduce the number of clinical DDI studies needed to characterize CYP3A4-mediated DDI with ipatasertib and provides an ethical benefit for patients with cancer.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Blake JF, Xu R, Bencsik JR et al (2012) Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J Med Chem 55(18):8110–8127. https://doi.org/10.1021/jm301024w
Yan Y, Serra V, Prudkin L et al (2013) Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res 19:6976–6986. https://doi.org/10.1158/1078-0432.CCR-13-0978
Lin J, Sampath D, Nannini MA et al (2013) Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res 19:1760–1772. https://doi.org/10.1158/1078-0432.CCR-12-3072
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169(3):381–405. https://doi.org/10.1016/j.cell.2017.04.001
Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y, Maehara Y (2008) Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets 8(1):27–36. https://doi.org/10.2174/156800908783497140
de Bono JS, De Giorgi U, Rodrigues DN et al (2019) Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res 25(3):928–936. https://doi.org/10.1158/1078-0432.CCR-18-0981
Malhi K, Budha N, Sane R et al (2021) Single- and multiple-dose pharmacokinetics, potential for CYP3A inhibition, and food effect in patients with cancer and healthy subjects receiving ipatasertib. Cancer Chemother Pharmacol 88(6):921–930. https://doi.org/10.1007/s00280-021-04344-9
Sane R, Cheung KWK, Cho E et al (2021) Evaluation of ipatasertib interactions with itraconazole and coproporphyrin I and III in a single drug interaction study in healthy subjects. J Pharmacol Exp Ther. https://doi.org/10.1124/jpet.121.000620
US Food and Drug Administration (2020) clinical drug interaction studies—cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. https://www.fda.gov/media/134581/download. Accessed 08 Oct 2021
European Medicines Agency (2012) Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. Accessed 08 Oct 2021
Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, Wang Y (2019) Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci 108(1):21–25. https://doi.org/10.1016/j.xphs.2018.10.033
Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang SM, Zhu H, Wang Y (2020) Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol 60(Suppl 1):S160–S178. https://doi.org/10.1002/jcph.1767
Yoshida K, Budha N, Jin JY (2017) Impact of physiologically based pharmacokinetic models on regulatory reviews and product labels: frequent utilization in the field of oncology. Clin Pharmacol Ther 101(5):597–602. https://doi.org/10.1002/cpt.622
Gibbons JA, Vries M, Krauwinkel W, Ohtsu Y, Noukens J, Walt JS, Mol R, Mordenti J, Ouatas T (2015) pharmacokinetic drug interaction studies with enzalutamide. Clin Pharmacokinet 54(10):1057–1069. https://doi.org/10.1007/s40262-015-0283-1
Isakoff SJ, Tabernero J, Molife LR et al (2020) Antitumor activity of ipatasertib combined with chemotherapy: results from a phase Ib study in solid tumors. Ann Oncol 31(5):626–633. https://doi.org/10.1016/j.annonc.2020.02.007
Narayanan R, Hoffmann M, Kumar G, Surapaneni S (2016) Application of a “fit for purpose” PBPK model to investigate the CYP3A4 induction potential of enzalutamide. Drug Metab Lett 10(3):172–179. https://doi.org/10.2174/1872312810666160729124745
Dhillon S (2015) Palbociclib: first global approval. Drugs 75(5):543–551. https://doi.org/10.1007/s40265-015-0379-9
Yu Y, Loi CM, Hoffman J, Wang D (2017) physiologically based pharmacokinetic modeling of palbociclib. J Clin Pharmacol 57(2):173–184. https://doi.org/10.1002/jcph.792
Mao J, Tay S, Khojasteh CS, Chen Y, Hop CE, Kenny JR (2016) Evaluation of time dependent inhibition assays for marketed oncology drugs: comparison of human hepatocytes and liver microsomes in the presence and absence of human plasma. Pharm Res 33(5):1204–1219. https://doi.org/10.1007/s11095-016-1865-9
Zhang M, You X, Ke M, Jiao Z, Wu H, Huang P, Lin C (2019) Prediction of ticagrelor and its active metabolite in liver cirrhosis populations using a physiologically based pharmacokinetic model involving pharmacodynamics. J Pharm Sci 108(8):2781–2790. https://doi.org/10.1016/j.xphs.2019.03.028
Siccardi M, Olagunju A, Seden K, Ebrahimjee F, Rannard S, Back D, Owen A (2013) Use of a physiologically-based pharmacokinetic model to simulate artemether dose adjustment for overcoming the drug-drug interaction with efavirenz. In Silico Pharmacol 1:4. https://doi.org/10.1186/2193-9616-1-4
Jing J, Nelson C, Shirasaka Y, Paik J, Amory J, Isoherranen N (2017) Physiologically based pharmacokinetic model of all-trans retinoic acid with application to cancer populations and drug interactions. J Pharmacol Exp Ther 361(2):246–258. https://doi.org/10.1124/jpet.117.240523
XTANDI® (enzalutamide) U.S. Package Insert (2012). https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203415lbl.pdf. Accessed 05 Oct 2021
Acknowledgements
The authors would like to thank Anshin BioSolutions, Inc., for editorial support of the manuscript.
Funding
Funding was provided by Genentech. No external funding was received for this work.
Author information
Authors and Affiliations
Contributions
JJ, YC, KY and RS wrote the manuscript. JJ, YC, LM, JYJ, KY and RS designed the research. JJ, YC, LM, JYJ, KWKC, KY and RS performed research. JJ, YC, KWKC, KY and RS analyzed data.
Corresponding authors
Ethics declarations
Conflict of interest
This study was funded by Genentech (a member of the Roche group). All authors (J.J., Y.C., L.M., J.Y.J., K.W.K.C., K.Y. and R.S.) were employees of Genentech when this work was carried out. All authors are past or present employees of Genentech.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jing, J., Chen, Y., Musib, L. et al. Assessment of cytochrome P450 3A4-mediated drug–drug interactions for ipatasertib using a fit-for-purpose physiologically based pharmacokinetic model. Cancer Chemother Pharmacol 89, 707–720 (2022). https://doi.org/10.1007/s00280-022-04434-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-022-04434-2