
Abstract
Purpose
This phase I study investigated the maximum tolerated dose (MTD), safety, pharmacokinetics, pharmacodynamics, and antitumor activity of the Aurora B kinase inhibitor BI 811283 in patients with advanced solid tumors.
Methods
BI 811283 was administered via 24-h infusion on Days 1 and 15 of a 4-week cycle (schedule A) or Day 1 of a 3-week cycle (schedule B) in a modified 3 + 3 dose-escalation design. Pharmacodynamic assessments included immunohistochemistry for phosphorylated histone H3 (pHH3) on skin biopsies to determine Aurora B kinase inhibition and plasma concentrations of caspase-cleaved CK-18 (apoptosis marker).
Results
A total of 121 patients were treated. The MTDs of BI 811283 were 125 mg (schedule A) and 230 mg (schedule B). Dose-limiting toxicities were primarily hematological (febrile neutropenia and grade 4 neutropenia); the most common drug-related adverse effects included neutropenia, fatigue, leukopenia, nausea, alopecia, diarrhea, and decreased appetite. A trend toward a decrease in pHH3 was observed, with increasing BI 811283 doses, indicating target engagement; there was no consistent trend regarding caspase-cleaved CK-18 levels. No objective response was observed although 19 patients in each schedule achieved clinical benefit (stable disease).
Conclusions
BI 811283 demonstrated a generally manageable safety profile and disease stabilization in some patients.
Trial registration
EudraCT No: 2007-000191-17, ClinicalTrials.gov Identifier: NCT00701324.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
The Aurora kinases belong to a family of three serine/threonine protein kinases (A, B, and C) that have a prominent role in regulating cell division [1, 2]. Aurora B kinase is part of the chromosome passenger complex (CPC), a group of proteins identified by its change in cellular localization over the duration of the cell cycle [3, 4]. Aurora B kinase has an important role in regulating many aspects of mitotic cell division, such as chromosome bi-orientation and sister chromatid cohesion, where Aurora B kinase is associated with the centromeres [5, 6], and spindle disassembly and cytokinesis following re-localization of the CPC to the midbody [7]. Furthermore, Aurora B kinase assists with chromosome condensation via phosphorylated histone H3 (pHH3) [8, 9].
There is considerable evidence linking Aurora B kinase to tumorigenesis. An increased expression of Aurora B kinase promotes chromosome instability and aneuploidy in vitro [10], and cells overexpressing Aurora B kinase have been shown to form aggressive tumors in nude mice [11]. In vivo, overexpression of Aurora B kinase is associated with several different tumor types when compared with matched normal tissue, such as brain, thyroid, breast, lung, colorectal, and prostate cancers, and is linked with characteristics including genetic instability, disease progression, and poor outcome in these patients [12–19]. In vitro, cells lacking Aurora B kinase cannot complete cytokinesis, become tetraploid and subsequently die [20]. This, together with the essential functions of Aurora B kinase during cell replication and its elevated expression in many cancer cell types, suggests that this mitotic enzyme is a valid target for therapeutic intervention.
There are multiple Aurora kinase inhibitors in preclinical studies or clinical trials. These agents are either pan-Aurora kinase inhibitors (targeting Aurora A, B, and occasionally C), or specific for either Aurora A or Aurora B [21].
BI 811283 is an adenosine triphosphate-competitive, reversible, and potent inhibitor of Aurora B kinase (half-maximal inhibitory concentration [IC50], 9 nM) [22, 23]. In a preclinical, in vitro study, BI 811283 exhibited broad antiproliferative activity in 24 tumor cell lines of different tissue origin (half-maximal effective concentration [EC50], <14 nM) [22, 23]. Within 1 h of treatment, pHH3 was reduced, indicating Aurora B kinase inhibition. Polyploidy was observed in up to 80 % of cells 48 h following treatment, with a subset of cells subsequently becoming senescent and going through apoptosis [22, 23]. In studies utilizing in vivo xenograft models of human non-small cell lung cancer, colon carcinoma, and pancreas carcinoma, inhibition of Aurora B kinase with BI 811283 resulted in a dose-dependent inhibition of tumor growth, and tumor regression was observed in some cases. A decrease in pHH3 was also observed, acting as a marker of Aurora B kinase inhibition [24].
Here we report the findings of a phase I, dose-escalation trial of BI 811283 that was performed in patients with advanced solid tumors. Two dosing schedules were examined, with the maximum tolerated dose (MTD) determined for each. The safety, pharmacokinetics, pharmacodynamics, and antitumor activity of BI 811283 are also described.
Methods
Patient selection
Eligible patients were aged 18 years or older presenting with advanced, non-resectable and/or metastatic solid malignant tumors, who were either not amenable to established forms of treatment or for whom no therapy of proven efficacy was available, and with a life expectancy of ≥6 months. Further eligibility criteria included an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤2; recovery from reversible toxicities of previous anticancer therapies; evaluable tumor deposits; secure central venous access; adequate bone marrow, liver, and renal function [absolute neutrophil count ≥1500/mm3, platelet count ≥100 000/mm3, bilirubin ≤1.5 mg/dL (≤26 μmol/L, SI unit equivalent), aspartate amino transferase and/or alanine amino transferase ≤2.5 × upper limit of normal (ULN; if related to liver metastases then ≤5 × ULN), and serum creatinine ≤1.5 mg/dL (≤132 μmol/L, SI unit equivalent)]; no chemo-, radio-, immuno-, hormone-, or investigational therapy within 2 weeks prior to the start of treatment with the trial drug; no known brain metastases or second malignancy requiring therapy, and no serious illness or concomitant disease which could compromise patient safety (including clinically significant cardiovascular disease, left ventricular ejection fraction <50 %, myocardial infarction within the last 6 months prior to inclusion and/or symptomatic coronary artery disease). Written informed consent was obtained from all study participants.
Study design and dose escalation
This was an open-label, first-in-human, phase I, dose-escalation trial of BI 811283 in patients with advanced solid tumors, conducted at two sites in Germany (EudraCT No: 2007-000191-17, ClinicalTrials.gov Identifier: NCT00701324). The primary endpoint was determination of the MTD of BI 811283 administered as a 24-h continuous infusion in a 4- or 3-week schedule. Several secondary endpoints were examined concurrently: the incidence and intensity of adverse events [AEs; determined by the common terminology criteria for AEs (Common Terminology Criteria for Adverse Events (CTCAE) version 3.0)], incidence of dose-limiting toxicity (DLT), objective response rate [according to the Response Evaluation in Solid Tumor (RECIST) criteria version 1.0] [25] and duration of response, pharmacodynamic analysis of skin biopsies for signs of target inhibition, and pharmacokinetic profile.
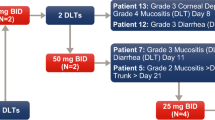
A modified 3 + 3 dose-escalation study design was chosen to evaluate the MTD of BI 811283, which was administered over 24 h by intravenous infusion (via central venous access). BI 811283 was administered on Days 1 and 15 of a 4-week cycle (schedule A) or Day 1 of a 3-week cycle (schedule B). Good laboratory practice-toxicology studies provided the safety data in selecting an intravenous 24 h continuous infusion (c.i.) and the two schedules tested, based on findings that the overall time of the plasma level over threshold was an important parameter for efficacy and was superior when compared with a bolus regimen in an HCT 116 colorectal cancer model (Boehringer Ingelheim; data on file); in detail, weekly and 2-weekly administrations of BI 811283 as 24-h continuous infusion resulted in similarly strong antitumor activity in preclinical models. Thus, this provides the rationale for the two dosing schedules tested in this trial. Cohorts of three patients were enrolled sequentially into escalating dose tiers of BI 811283. The MTD was defined as the highest dose of BI 811283 in which ≤1 of six patients experienced a DLT during the first cycle of treatment. The starting dose level for schedule A was 5 mg estimated according to the Food and Drug Administration (FDA) flow diagram “General guide for starting dose selection for a cytotoxic agent in cancer patients”, with dose escalation in steps of 100 % until the first drug-related AE grade ≥2. Thereafter, escalation steps of BI 811283 were limited to ≤50 %. After the first DLT, escalation steps of no more than 35 % of the previous dose were made. To reduce the overall number of patients in the study, initial patient cohorts were assigned only to the 4-week treatment cycle (schedule A), until the first occurrence in Cycle 1 of a drug-related AE grade ≥2. Thereafter, subsequent patients were randomized between the 4-week cycle (schedule A) and the 3-week cycle (schedule B) to determine the respective MTD in parallel groups. The starting dose level in schedule B was the dose at which a first drug-related grade ≥2 AE occurred in schedule A. After determination of the MTD, up to 9 additional patients were entered at this dose level to obtain further safety data. Patients were treated until disease progression. However, if a patient who experienced disease progression tolerated the drug well and wished to continue with treatment, an intra-patient dose escalation was allowed. Treatment was also terminated if a patient had an intolerable AE or withdrew consent, or if a treatment cycle was delayed for >2 weeks. The trial was conducted in accordance with the principles laid down by the Declaration of Helsinki and approved by the Independent Ethics Committees and/or Institutional Review Boards of the participating centers.
Definition of dose-limiting toxicity
A DLT was recorded if one or more of the following events occurred during the first treatment cycle: (1) drug-related grade ≥3 non-hematological toxicity (except untreated nausea, vomiting, or diarrhea), (2) drug-related grade 4 neutropenia for ≥7 days or febrile neutropenia, or (3) drug-related grade 4 thrombocytopenia or anemia. Additionally, for schedule A only, the following events on Day 15, if drug related, also constituted a DLT: (1) grade ≥2 non-hematological toxicity (excluding alopecia and untreated nausea, vomiting, or diarrhea), or (2) grade ≥3 neutropenia, thrombopenia, or anemia. The MTD was defined on the basis of DLT observed during the first treatment cycle only. However, DLTs observed after that time period were considered along with the type, number, and intensity of AEs to indicate how well BI 811283 was tolerated.
Study assessments
Safety
All treated patients were included in the safety evaluation. Key safety measures included evaluation of the incidence and intensity of AEs using the Medication Dictionary for Regulatory Activities (MedDRA) and classified according to CTCAE version 3.0, assessment of vital signs and laboratory parameters, and 12-lead electrocardiogram (ECG) and echocardiography measurements.
Pharmacokinetics
Blood samples for the evaluation of pharmacokinetic parameters were collected 5 min before drug administration, at 1, 2, 4, 10, and 20 h during the infusion, at 23:59 (just before the end of infusion), and at the following timepoints after the start of infusion: 24:15, 24:30, 25:00, 26:00, 28:00 and 32:00 h (Day 2), 48:00 h (Day 3), 72:00 h (Day 4), and 120 h (Day 6). Further samples were obtained in each subsequent treatment cycle (up to Cycle 6) just prior to the start and at the end of the infusion. Urine samples were also obtained after the first and second infusions of BI 811283 (schedule A: Cycle 1 Days 1 and 15; schedule B: Cycle 1 Days 1 and 22). The concentration of BI 811283 in plasma and urine was determined by a validated high-performance liquid chromatography–tandem mass spectrometry (HPLC–MSMS) assay. Briefly, samples were subjected to protein precipitation with methanol. Then, the samples were analyzed on a PerkinElmer Sciex API 5500 LC-MSMS system using a Zorbax® Eclipse Plus C18 column. An electrospray ion source [atmospheric pressure ionization (API)] was used for ionization. Measurements were performed in the positive ionization mode. Preclinical assessments demonstrated that BI 811283 protein binding is dependent on the plasma concentrations of alpha-1-acid glycoprotein (AGP) (Boehringer Ingelheim, data on file). Therefore, AGP concentrations were determined in clinical plasma samples following BI 811283 dosing and were obtained during the first two treatment cycles at the same time points as those collected for pharmacokinetic analysis, with AGP concentration determined using a validated immunoturbidimetric assay. In the assay, AGP was precipitated using a specific antiserum and the resulting turbidimetric reading at 340 nm was used to determine its concentration. Pharmacokinetic analyses were summarized using descriptive statistics.
Pharmacodynamics
Target inhibition following BI 811283 dosing was assessed. Skin biopsies were analyzed for a reduction of pHH3, a marker of Aurora B kinase inhibition [26]. Three- to four-millimeter punch skin biopsies were obtained during screening and within 6 h after the end of infusion on Day 16 of schedule A or Day 2 of schedule B of the first treatment cycle. The skin biopsies were divided, with one half each for western blotting and immunohistochemistry (IHC). For western blotting, the sample was lysed in Lysing Matrix A (MP Biomedicals #6910-050) and lysis buffer [100 mM NaHCO3 pH 9.6, 1 mM dithiothreitol, Protease inhibitor cocktail (Serva 3910)]. Equivalent amounts of proteins from the cell lysates were applied to 12 % TRIS–glycine gels (Invitrogen NuPAGE Novex NP0341BOX) and blotted with phospho-H3 antibody (Cell Signalling #9701) and H3 antibody (Cell Signalling #9715) to determine the ratio of phospho-H3/total H3 protein. For IHC analysis, the sample was fixed in formalin, embedded into paraffin with 5-µm sections prepared. The slides were probed with phospho-H3 antibody (Cell Signalling #9701) and counterstained with Mayer’s hemalum (Merck 1.09249.1000). Epidermal areas were marked and measured, and positive nuclei counted with the aid of the NIKON image software. Quantification was done in a blinded fashion by two independent researchers.
Serum levels of caspase-cleaved cytokeratin-18 (CK-18) act as a marker of the cell apoptosis anticipated with Aurora B kinase inhibition by BI 811283 [27]. Therefore, plasma samples for quantifying the caspase-cleaved fragment of CK-18 were obtained pre-dose and 48, 72, and 120 h after the first two BI 811283 administrations in each schedule. Samples were also collected pre-dosing and 120 h post-dosing, up to Cycle 6. Plasma concentrations of caspase-cleaved CK-18 were determined using a validated ELISA.
Antitumor activity
Objective tumor response was evaluated using computed tomography (CT) or magnetic resonance imaging (MRI) scans at baseline and at the end of every other treatment cycle, according to RECIST criteria version 1.0 [25].
Results
Patient demographics and disposition
A total of 129 patients were enrolled into the study which was performed between July 2007 and August 2011. Of these, 124 patients were entered to one of the two treatment schedules and 121 were treated with study medication (schedule A: 63 patients; schedule B: 58 patients). Patient demographics and disease characteristics are summarized in Table 1. Median patient age was 58 years (range 23–79 years), and 47.1 % of patients were male. The majority of patients (77.7 %) presented with stage IV disease at screening, with colorectal cancer being the most frequent tumor type (24.8 %). Most patients had undergone previous surgery (90.9 %) or systemic chemotherapy (92.6 %), and many were heavily pre-treated: 57 (47.1 %) had received ≥4 lines of chemotherapy (Table 1).
Dose-limiting toxicities, safety, and tolerability
Patients in the schedule A cohort were treated with BI 811283 at doses from 5 to 140 mg. First-cycle DLTs were primarily hematological events; one patient treated with 10 mg had raised liver function tests (Table 2). DLTs were seen in two of three patients treated with the 140 mg dose. The MTD was determined to be 125 mg; one patient in the initial cohort at this dose experienced grade 4 neutropenia on Day 15. Patients in the schedule B cohort were treated with BI 811283 starting at 13.5 mg and escalating to 300 mg. As with schedule A, DLTs were primarily hematological events; in addition, one patient treated with 105 mg experienced grade 3 fatigue (Table 2). Two out of five patients experienced a DLT at the 270 mg dose. The MTD was determined to be 230 mg. During the data review meeting following the end of the study, five events of grade 4 neutropenia in schedule B were retrospectively identified as possible DLTs (105 mg, n = 2; 125 mg, n = 1; 180 mg, n = 1; 230 mg, n = 1). In all five patients, the initial hematology laboratory tests performed during Cycle 1 showed grade 4 neutropenia. However, there were no laboratory tests performed on Day 7 or 8 after the start of the grade 4 neutropenia to show whether neutropenia was still present to qualify as a DLT (i.e., ≥7 days), although in all of these cases the neutropenia had resolved by Day 8–10 after the start of the grade 4 event. Five additional DLTs [grade 4 febrile neutropenia (n = 2), grade 3 febrile neutropenia (n = 2), and grade 4 neutropenia ≥7 days (n = 1)] were also identified in the 230-mg dose expansion cohort of seven patients. Therefore, although per protocol the MTD was determined as 230 mg for schedule B, based on retrospective DLTs from Cycle 1 and in patients enrolled in the expansion cohort, a lower dose may have been recommended for further study. Expansion cohorts included nine additional patients in schedule A, and seven additional patients in schedule B treated at the respective MTDs (125 and 230 mg). All patients in both treatment schedules had at least one AE during the treatment course, with 100 (82.6 %) patients having AEs that were considered drug related by the investigators [schedule A: n = 49 (77.8 %); schedule B: n = 51 (87.9 %)]. Drug-related AEs that occurred in ≥10 % of all patients across all treatment cycles were neutropenia (n = 40, 33.1 %), fatigue (n = 38, 31.4 %), leukopenia (n = 35, 28.9 %), nausea (n = 29, 24.0 %), alopecia (n = 24, 19.8 %), diarrhea (n = 20, 16.5 %), and decreased appetite (n = 18, 14.9 %). Tables 3 and 4 summarize the most common drug-related AEs in schedules A and B by dose. Serious AEs (SAEs) were reported in 61 (50.4 %) patients [schedule A: n = 33 (52.4 %); schedule B: n = 28 (48.3 %)]. Thirteen of these patients had SAEs regarded as drug related [schedule A: n = 3 (4.8 %); schedule B: n = 10 (17.2 %)]. The most common drug-related SAE was febrile neutropenia (experienced by seven patients); other such events reported in more than one patient were leukopenia, neutropenia, vomiting, and diarrhea. A total of 22 (18.2 %) patients had fatal AEs [schedule A: n = 10 (15.9 %); schedule B: n = 12 (20.7 %)]. None of the deaths were considered related to the study treatment. Overall, 19 (15.7 %) patients had AEs that resulted in study drug discontinuation or a dose reduction [schedule A: n = 9 (14.3 %); schedule B: n = 10 (17.2 %); Supplementary Table 1]. In 10 patients (five patients in each dosing schedule), the AEs resulting in discontinuation or dose reduction were classified as significant according to the International Conference on Harmonization (ICH) E3.
Pharmacokinetics
Plasma concentrations of BI 811283 rose rapidly for several hours after the start of the 24-h infusion, reaching maximum concentrations after 20–24 h (Fig. 1). This was followed by a biphasic decline in plasma BI 811283, which was initially very rapid such that most of the compound was eliminated within the first 6 h following the end of the infusion. The mean terminal half-life of BI 811283 was 11.4–30.5 h for schedule A and 10.1–27.0 h for schedule B. In general, pharmacokinetic parameters were comparable between the two schedules and there were no significant differences between the first and second doses given to patients, regardless of treatment schedule. Although there was high inter-subject variability for both treatment schedules, the maximum measured concentration in plasma (C max) and the area under the concentration–time curve over the time interval from 0 extrapolated to infinity (AUC0–∞) values increased in a dose-dependent manner. The fraction of BI 811283 excreted in urine was low (ranging from 4 to 12 % of the administered dose) and did not differ between the two treatment arms. AGP plasma concentrations did not change significantly with time after BI 811283 dosing and were not dependent on BI 811283 concentrations. There was a trend toward increased exposure to total BI 811283 (bound and unbound) in patients with higher AGP plasma concentrations, although there was high variability.

Geometric mean plasma concentrations of BI 811283 following 24-h intravenous infusion in schedule A (a) and schedule B (b) (linear scale)
Pharmacodynamics
In total, 28 out of 63 patients in schedule A [5, 10, 18, 24, 43, 125, and 140 mg (n = 2 each); 58 mg (n = 3); 78 mg (n = 4); 105 mg (n = 5)], and 28 out of 58 patients in schedule B [32 and 300 mg (n = 1 each); 13.5, 18, 43, 58, 105, 125 (n = 2 each); 150 mg (n = 3); 270 mg (n = 4); 230 mg (n = 7)] provided skin biopsies for pharmacodynamic analyses. Two different types of analyses were performed: an IHC determination of cells with nuclei that were positive for pHH3 and a western blot analysis to determine the ratio of pHH3 to total histone H3 in cells. Western blot analyses resulted in a low to undetectable pHH3 band, sometimes in combination with a very dominant globin band, which interfered with the pHH3 signal. IHC analyses using formalin-fixed paraffin-embedded sections revealed a trend for a reduction in the number of phospho-histone H3-positive nuclei in the epidermis with increasing doses of BI 811283 from 58 mg upward in schedule A, and from 43 mg upward in schedule B [Fig. 2 and example images from a patient receiving schedule A treatment (Supplementary Fig 1)], suggesting Aurora B kinase inhibition in this patient population [11, 28]. There was no consistent trend regarding the level of caspase-cleaved CK-18 in plasma after infusion of BI 811283, no consistency between the first and second infusions, and data obtained from the effect curves were extremely variable (Supplementary Fig 2 and 3).

Effect of BI 811283 following 24-h intravenous infusion on histone H3 phosphorylation measured by immunohistochemistry in schedule A (a) and schedule B (b)
Antitumor activity
No patients in either treatment schedule achieved an objective response. However, 19 (30 %) patients in schedule A and 19 (33 %) patients in schedule B achieved clinical benefit from treatment [defined as stable disease (SD; n = 37) or non-evaluable but clinically not progressive disease (n = 1)]. To confirm a status of SD, an overall response assessment that met the SD criteria was to be recorded at least 42 days after study entry. Ten patients with SD received ≥10 cycles of treatment [tumor types: melanoma (n = 3), thyroid or parathyroid (n = 2), sarcoma (n = 2), breast (n = 1), lung (n = 1), and colorectal cancer (n = 1)].
Discussion
Novel treatment strategies for patients with advanced solid tumors are urgently needed. Although antimitotic drugs in the form of microtubule-targeting agents (MTAs), such as the taxanes, are one of the most widely used classes of cancer therapeutics, the broad and often toxic effects of these agents, together with the emergence of MTA resistance, have triggered interest in new strategies that involve selective inhibition of enzymes involved in the regulation of mitosis [29, 30]. Aurora B kinase is one such enzyme that plays several important roles in mitosis [2, 31, 32]. This fact, coupled with its elevated expression across a wide range of tumor types suggests that this enzyme could be an effective therapeutic target in oncology [33].
This phase I clinical trial was designed as an open, parallel group, first-in-human study to define the MTD of BI 811283, an inhibitor of Aurora B kinase, in patients with advanced solid tumors. The MTD of BI 811283 was determined as 125 mg on Days 1 and 15 of a 4-week cycle (schedule A) and 230 mg on Day 1 of a 3-week cycle (schedule B). The most common DLTs observed in both schedules were primarily hematological events, particularly neutropenia. One patient with metastatic soft tissue sarcoma and no liver metastases treated with 10 mg BI 811283 in schedule A experienced a DLT of grade 3 liver enzyme (alanine and aspartate aminotransferase) elevation in Cycle 1 which resolved spontaneously within 5 days. She again developed grade 3/4 liver enzyme elevation following treatment on Cycle 2 Day 15 which did not improve sufficiently for her to receive subsequent treatment, and she discontinued from the study. Given the recurrence of this AE upon rechallenge of the drug, it was deemed almost certainly drug related. This was the only occurrence of dose-limiting liver enzyme elevation in the study. Such DLTs do not appear to be typical of this class of compound, although DLTs of liver enzyme elevations were observed with the Aurora A kinase inhibitor MLN8054, and development of this compound was ceased in favor of alisertib (MLN8237) [34]. The retrospective identification of additional AEs in schedule B which possibly fulfilled the criteria for DLTs and the additional DLTs identified in the expansion cohort of 230 mg in schedule B suggests that the MTD and recommended phase II dose for further study may have been less than 230 mg with schedule B. However, due to the lack of promising efficacy data in this study and the development of an oral Aurora kinase inhibitor [35–37] offering improved convenience to the patient compared with BI 811283 (which requires 24-h continuous infusion through a central line), the clinical development of BI 811283 was halted.
No patients achieved an objective response with BI 811283 in this study. The best overall response was SD in both treatment schedules, observed in approximately 30 % of patients, all of whom had previously been heavily pre-treated. This is not unexpected; however, as this outcome is consistent with the cytostatic mechanism of action of this class of drugs and comparable with the efficacy data observed in phase I trials of other Aurora kinase inhibitors [35, 36, 38–41].
The overall safety profile was as to be expected in a population of patients with advanced cancer. In addition to the AEs typically associated with the underlying disease, the most common toxicities were hematological. BI 811283 is known to cause a transient inhibition of the proliferation of normal dividing cells in the bone marrow, resulting in a temporary decrease of blood cells and platelets. Inhibition of mucosal proliferation can also occur with BI 811283, leading to gastrointestinal symptoms such as nausea, diarrhea, and abdominal pain. These AEs are not considered unusual following treatment with a compound targeting rapidly dividing cells, and similar toxicities were observed in phase I trials of other Aurora kinase inhibitors [35, 36, 41].
Pharmacokinetic analyses showed that plasma concentrations of BI 811283 rose rapidly in patients during the first few hours of the 24-h infusion, with maximum concentrations generally reached 20–24 h after the start of the infusion. This was followed by a rapid, biphasic decline. Pharmacokinetic parameters did not differ between the two treatment arms and there were no meaningful differences in pharmacokinetic parameters of BI 811283 between the first and second doses given to patients, regardless of treatment schedule. The fraction of BI 811283 excreted in urine was low and did not differ between the two treatment arms. Although there was high variability, exposure of total BI 811283 appeared to increase in patients with higher AGP concentrations. However, unbound BI 811283 was not determined in this study, so the relationship to AGP exposure in this case is not known.
During mitosis, histone H3 is phosphorylated by Aurora kinase B on serine 10 [11, 28]. Aberrant pHH3 is associated with chromosome instability and carcinogenesis [11, 26]. A reduction in phosphorylation on serine 10 indicates Aurora kinase B inhibition. Indeed in preclinical studies, BI 811283 reduced histone H3 phosphorylation in multiple cancer cell lines [22, 24]. In this exploratory study, pHH3 was measured using both western blotting and IHC. Results from the western blot analyses were not considered meaningful due to a low to undetectable pHH3 band, sometimes in combination with a very dominant globin band, which interfered with the pHH3 signal. In contrast, IHC, which relies on single cell analysis, had a signal that was highly concentrated in the rare mitotic cells resulting in a signal-to-noise ratio that was much improved. The IHC analyses revealed a trend for a reduction in the ratio of cellular pHH3 to total histone H3 with increasing doses of BI 811283 in the skin as surrogate tissue, suggesting a dose-dependent inhibition of Aurora B kinase in this patient population. Among the 10 patients who achieved SD for ≥10 cycles, skin biopsy samples from only four patients were evaluable (two patients from each treatment arm). Of these four patients, the biopsy samples from only two patients showed a pharmacodynamics effect; therefore, it is difficult to conclude whether the pharmacodynamics effect had any correlation with clinical outcome. Overall, these results were not unexpected, due to differences in molecular and physiological characteristics between tumor versus skin tissue, and these analyses provide encouraging clinical data regarding BI 811283 target engagement. There was no consistent increase seen in caspase-cleaved CK-18, a marker of tumor cell death [27] in plasma samples of patients treated with BI 811283. Therefore, caspase-cleaved CK-18 was not an informative clinical biomarker of Aurora B kinase inhibition by BI 811283 in this study.
There are several Aurora kinase inhibitors currently in clinical development, which either specifically target Aurora A or Aurora B kinase, or demonstrate some activity toward both kinases [42]. Preliminary evidence from a phase II study indicates that alisertib (MLN8237), an oral Aurora A kinase inhibitor, has a generally manageable safety profile and results in durable disease control in multiple cancer types [37]. The Aurora A kinase inhibitor danusertib has shown an acceptable safety profile and promising clinical activity in patients with advanced hematological malignancies [43], but limited activity in phase I/II studies in advanced solid tumors [44, 45]. The most widely tested Aurora B kinase inhibitor is barasertib (AZD1152), which has been evaluated in both patients with advanced solid tumors and those with hematological cancers. While barasertib demonstrated limited activity in solid tumors [38, 46], greater activity has been observed in patients with acute myeloid leukemia (AML) [47–50]. In these patients, barasertib has demonstrated clinical activity in several phase I/II trials as monotherapy (hematological response rate of 19–25 %) and in combination with low-dose cytosine arabinoside (LDAC; response rate of 45 %). Grade ≥3 neutropenia/febrile neutropenia was reported as a common AE in these studies. Barasertib was also compared to LDAC in a randomized phase II trial in elderly patients with AML, demonstrating higher complete response rates (35.4 vs 11.5 %); however, barasertib was much less tolerable than LDAC in these patients (71 vs 15 % stomatitis; 67 vs 19 % febrile neutropenia) [47]. Another Aurora B kinase inhibitor, BI 831266, has also been recently studied in a phase I trial of patients with advanced solid tumors. Similar to the agent investigated in the current study, BI 831266 treatment resulted in objective response in only one patient (cervical cancer), with 16 % of patients experiencing SD [39]. The BI 831266 trial was discontinued based on these data, as well as the limited activity displayed by BI 811283 in this population.
Although all Aurora kinases are involved in cell division, only Aurora A regulates centrosome maturation/separation and bipolar spindle assembly, whereas Aurora B and C are involved in the regulation of mitotic chromosome dynamics [43]. Based on these different intracellular functions, it has been hypothesized that inhibition of multiple Aurora kinases may provide better antitumor activity. Agents that target both Aurora A and B kinases are currently in early-stage clinical development (e.g., PF-03814735, AT9283) in both solid tumors and hematological malignancies; however, these agents have shown limited antitumor activity thus far in phase I trials [40, 43].
Conclusions
In summary, this phase I trial demonstrated an acceptable safety profile with BI 811283 administered on two different treatment schedules. MTDs were determined as 125 mg (Days 1 and 15 of a 4-week cycle) and 230 mg (Day 1 of a 3-week cycle); however, the retrospective identification of additional potential AEs in schedule B during a data review at the end of the study suggests that the MTD may have been less than 230 mg. The limited antitumor activity observed with BI 811283 does not support its continued development in solid tumors. Further biomarker studies would be beneficial to better understand the role of Aurora kinase inhibition in tumor development and anticancer therapy.
References
Glover DM, Leibowitz MH, McLean DA, Parry H (1995) Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81(1):95–105
Hochegger H, Hegarat N, Pereira-Leal JB (2013) Aurora at the pole and equator: overlapping functions of Aurora kinases in the mitotic spindle. Open Biol 3(3):120185
Adams RR, Wheatley SP, Gouldsworthy AM, Kandels-Lewis SE, Carmena M, Smythe C, Gerloff DL, Earnshaw WC (2000) INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol 10(17):1075–1078
Terada Y (2001) Role of chromosomal passenger complex in chromosome segregation and cytokinesis. Cell Struct Funct 26(6):653–657
Losada A, Hirano M, Hirano T (2002) Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev 16(23):3004–3016
Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K (2002) Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell 108(3):317–329
Buvelot S, Tatsutani SY, Vermaak D, Biggins S (2003) The budding yeast Ipl1/Aurora protein kinase regulates mitotic spindle disassembly. J Cell Biol 160(3):329–339
Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P (2002) Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 22(3):874–885
Monier K, Mouradian S, Sullivan KF (2007) DNA methylation promotes Aurora-B-driven phosphorylation of histone H3 in chromosomal subdomains. J Cell Sci 120(Pt 1):101–114
Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, Suzuki F, Terada Y (1998) Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res 58(21):4811–4816
Ota T, Suto S, Katayama H, Han ZB, Suzuki F, Maeda M, Tanino M, Terada Y, Tatsuka M (2002) Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res 62(18):5168–5177
Araki K, Nozaki K, Ueba T, Tatsuka M, Hashimoto N (2004) High expression of Aurora-B/Aurora and Ipll-like midbody-associated protein (AIM-1) in astrocytomas. J Neurooncol 67(1–2):53–64
Chieffi P, Cozzolino L, Kisslinger A, Libertini S, Staibano S, Mansueto G, de Rosa G, Villacci A, Vitale M, Linardopoulos S, Portella G, Tramontano D (2006) Aurora B expression directly correlates with prostate cancer malignancy and influence prostate cell proliferation. Prostate 66(3):326–333
Smith SL, Bowers NL, Betticher DC, Gautschi O, Ratschiller D, Hoban PR, Booton R, Santibanez-Koref MF, Heighway J (2005) Overexpression of aurora B kinase (AURKB) in primary non-small cell lung carcinoma is frequent, generally driven from one allele, and correlates with the level of genetic instability. Br J Cancer 93(6):719–729
Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, Spalletti-Cernia D, Laccetti P, Linardopoulos S, Chieffi P, Fusco A, Portella G (2005) Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab 90(2):928–935
Tchatchou S, Wirtenberger M, Hemminki K, Sutter C, Meindl A, Wappenschmidt B, Kiechle M, Bugert P, Schmutzler RK, Bartram CR, Burwinkel B (2007) Aurora kinases A and B and familial breast cancer risk. Cancer Lett 247(2):266–272
Tuncel H, Shimamoto F, Kaneko Guangying QH, Aoki E, Jikihara H, Nakai S, Takata T, Tatsuka M (2012) Nuclear Aurora B and cytoplasmic survivin expression is involved in lymph node metastasis of colorectal cancer. Oncol Lett 3(5):1109–1114
Vischioni B, Oudejans JJ, Vos W, Rodriguez JA, Giaccone G (2006) Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol Cancer Ther 5(11):2905–2913
Zeng WF, Navaratne K, Prayson RA, Weil RJ (2007) Aurora B expression correlates with aggressive behaviour in glioblastoma multiforme. J Clin Pathol 60(2):218–221
Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM (2003) The small molecule hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161(2):281–294
Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M (2012) Aurora kinase inhibitors: progress towards the clinic. Investig New Drugs 30(6):2411–2432
Gürtler U, Tontsch-Grunt U, Jarvis M, Zahn SK, Boehmelt G, Quant J, Adolf GR, Solca F (2010) Effect of BI 811283, a novel inhibitor of Aurora B kinase, on tumor senescence and apoptosis. J Clin Oncol 28(suppl):e13632 (abstr)
Tontsch-Grunt U, Ulrich G, Zahn SK, Boehmelt G, Jarvis M, Adolf GR, Solca F (2010) Molecular and cellular pharmacology of BI 811283, a potent inhibitor of Aurora B kinase. Cancer Res 70(8, suppl 1):1080 (abstr)
Gürtler U, Tontsch-Grunt U, Zahn S, Gürtler U, Quant J, Guenther R, Solca F (2010) BI 811283, a potent inhibitor of the mitotic kinase Aurora B, show dose- and schedule-dependent efficacy in human cancer xenograft models. Cancer Res 70(8, suppl 1):1082 (abstr)
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M (1999) Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem 274(36):25543–25549
Linder S, Havelka AM, Ueno T, Shoshan MC (2004) Determining tumor apoptosis and necrosis in patient serum using cytokeratin 18 as a biomarker. Cancer Lett 214(1):1–9
Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, Storici P, Zugnoni P, Pesenti E, Croci V, Ceruti R, Giorgini ML, Cappella P, Ballinari D, Sola F, Varasi M, Bravo R, Moll J (2006) PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res 12(13):4080–4089
Chan KS, Koh CG, Li HY (2012) Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis 3:e411
Morris PG, Fornier MN (2008) Microtubule active agents: beyond the taxane frontier. Clin Cancer Res 14(22):7167–7172
Lampson MA, Cheeseman IM (2011) Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol 21(3):133–140
Salimian KJ, Ballister ER, Smoak EM, Wood S, Panchenko T, Lampson MA, Black BE (2011) Feedback control in sensing chromosome biorientation by the Aurora B kinase. Curr Biol 21(13):1158–1165
Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PNJ, Gandara DR (2008) Aurora kinases as anticancer drug targets. Clin Cancer Res 14(6):1639–1648
Macarulla T, Cervantes A, Elez E, Rodríguez-Braun E, Baselga J, Roselló S, Sala G, Blasco I, Danaee H, Lee Y, Ecsedy J, Shinde V, Chakravarty A, Bowman D, Liu H, Eton O, Fingert H, Tabernero J (2010) Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther 9(10):2844–2852
Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Rosello S, Andreu J, Jung J, Sanchis-Garcia JM, Piera A, Blasco I, Manos L, Perez-Fidalgo JA, Fingert H, Baselga J, Tabernero J (2012) Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective Aurora A kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 18(17):4764–4774
Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, Manfredi M, Fingert H, Burris HA III, Infante JR (2012) Phase I study of Aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res 18(17):4775–4784
Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X, Danaee H, Schilder RJ (2012) Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol 127(1):63–69
Boss DS, Witteveen PO, van der Sar J, Lolkema MP, Voest EE, Stockman PK, Ataman O, Wilson D, Das S, Schellens JH (2011) Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann Oncol 22(2):431–437
Dittrich C, Fridrik MA, Koenigsberg R, Lee C, Goeldner R-G, Hilbert J, Greil R (2015) A phase I dose escalation study of BI 831266, an inhibitor of Aurora kinase B, in patients with advanced solid tumors. Investig New Drugs 33(2):409–422
Schöffski P, Jones SF, Dumez H, Infante JR, van Mieghem E, Fowst C, Gerletti P, Xu H, Jakubczak JL, English PA, Pierce KJ, Burris HA (2011) Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur J Cancer 47(15):2256–2264
Traynor AM, Hewitt M, Liu G, Flaherty KT, Clark J, Freedman SJ, Scott BB, Leighton AM, Watson PA, Zhao B, O’Dwyer PJ, Wilding G (2011) Phase I dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother Pharmacol 67(2):305–314
Bavetsias V, Linardopoulos S (2015) Aurora Kinase inhibitors: current status and outlook. Front Oncol 5:278
Borthakur G, Dombret H, Schafhausen P, Brummendorf TH, Boissel N, Jabbour E, Mariani M, Capolongo L, Carpinelli P, Davite C, Kantarjian H, Cortes JE (2015) A phase I study of danusertib (PHA-739358) in adult patients with accelerated or blastic phase chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant or intolerant to imatinib and/or other second generation c-ABL therapy. Haematologica 100(7):898–904
Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, Dogliotti L, Berardi R, Cappuzzo F, Tagawa ST, Sternberg CN, Jannuzzo MG, Mariani M, Petroccione A, de Wit R (2013) Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int 111(1):44–52
Schöffski P, Besse B, Gauler T, de Jonge MJ, Scambia G, Santoro A, Davite C, Jannuzzo MG, Petroccione A, Delord JP (2015) Efficacy and safety of biweekly i.v. administrations of the Aurora kinase inhibitor danusertib hydrochloride in independent cohorts of patients with advanced or metastatic breast, ovarian, colorectal, pancreatic, small-cell and non-small-cell lung cancer: a multi-tumour, multi-institutional phase II study. Ann Oncol 26(3):598–607
Schwartz GK, Carvajal RD, Midgley R, Rodig SJ, Stockman PK, Ataman O, Wilson D, Das S, Shapiro GI (2013) Phase I study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Investig New Drugs 31(2):370–380
Kantarjian HM, Martinelli G, Jabbour EJ, Quintas-Cardama A, Ando K, Bay JO, Wei A, Gropper S, Papayannidis C, Owen K, Pike L, Schmitt N, Stockman PK, Giagounidis A (2013) Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer 119(14):2611–2619
Kantarjian HM, Sekeres MA, Ribrag V, Rousselot P, Garcia-Manero G, Jabbour EJ, Owen K, Stockman PK, Oliver SD (2013) Phase I study assessing the safety and tolerability of barasertib (AZD1152) with low-dose cytosine arabinoside in elderly patients with AML. Clin Lymphoma Myeloma Leuk 13(5):559–567
Lowenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY, Ifrah N, Martinelli G, Amadori S, Berman E, Sonneveld P, Jongen-Lavrencic M, Rigaudeau S, Stockman P, Goudie A, Faderl S, Jabbour E, Kantarjian H (2011) Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 118(23):6030–6036
Tsuboi K, Yokozawa T, Sakura T, Watanabe T, Fujisawa S, Yamauchi T, Uike N, Ando K, Kihara R, Tobinai K, Asou H, Hotta T, Miyawaki S (2011) A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk Res 35(10):1384–1389
Acknowledgments
The authors were fully responsible for all content and editorial decisions, and were involved at all stages of manuscript development and have approved the final version. Helen Wilkinson of GeoMed, an Ashfield Company, part of UDG Healthcare plc, provided medical writing assistance during the preparation of this manuscript, supported financially by Boehringer Ingelheim. This study was sponsored by Boehringer Ingelheim. Clinical trial registration: ClinicalTrials.gov registration number, NCT00701324.
Author information
Authors and Affiliations
Corresponding author
Additional information
K. Mross and H. Richly have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
IHC analyses of phospho-histone H3 using formalin-fixed paraffin-embedded sections from a patient receiving 78 mg BI 811283 Day 1 and Day 15 A) Baseline, B) Day 16 (TIFF 3887 kb)
Supplementary Fig. 2
Mean percentage change from baseline of caspase-cleaved CK-18 concentrations following 24-h infusion of BI 811283 for schedule A for dose levels at which sufficient samples were available for mean calculations (TIFF 336 kb)
Supplementary Fig. 3
Mean percentage change from baseline of casepase-cleaved CK-18 concentrations following 24-h infusion of BI 811283 for schedule B for dose levels at which sufficient samples were available for mean calculations (TIFF 347 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mross, K., Richly, H., Frost, A. et al. A phase I study of BI 811283, an Aurora B kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol 78, 405–417 (2016). https://doi.org/10.1007/s00280-016-3095-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3095-6