
Abstract
Acute Myeloid Leukemia (AML) is a life-threatening disease whose induction treatment consists of combination chemotherapy with Idarubicin and Cytarabine for fit patients. Treatment failures are frequent, urging the need for novel treatments for this disease. The DNA Damage Response Mechanism (DDR) comprises numerous molecules and pathways intended to arrest the cell cycle until DNA damage is repaired or else drive the cell to apoptosis. AML-derived cell lines after treatment with Idarubicin and Cytarabine were used for studying the expression profile of 84 DDR genes, through PCR arrays. Utilizing de novo AML patient and control samples we studied the expression of PPP1R15A, CDKN1A, GADD45A, GADD45G, and EXO1. Next, we performed PPP1R15A silencing in AML cell lines in two separate experiments using siRNA and CRISPR-cas9, respectively. Our findings highlight that DDR regulators demonstrate increased expression in patients with high cytogenetic risk possibly reflecting increased genotoxic stress. Especially, PPP1R15A is mainly involved in the recovery of the cells from stress and it was the only DDR gene upregulated in AML patients. The PPP1R15A silencing resulted in decreased viability of Idarubicin and Cytarabine-treated cell lines, in contrast to untreated cells. These findings shed light on new strategies to enhance chemotherapy efficacy and demonstrate that PPP1R15A is an important DDR regulator in AML and its downregulation might be a safe and effective way to increase sensitivity to chemotherapy in this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute Myeloid Leukemia (AML) is a life-threatening disease that is currently treated with intensive chemotherapy in eligible patients of young age with no substantial comorbidities. Even though AML is a highly heterogeneous disease, the backbone of induction chemotherapy remains the combination of Idarubicin (or Doxorubicin) and Cytarabine. Both these agents act by causing DNA damage to the leukemic cells, thereby eliminating them [1, 2]. Although effective, a considerable proportion of patients do not respond to induction therapy or lose initial response, resulting in 5-year survival rates of less than 60% in patients up to 60 years of age [3]. Understanding the pathophysiology of the disease and mechanisms of resistance to induction chemotherapy is crucial for developing new targeted therapies that can improve the response rates and overall survival of AML patients.
DNA Damage Response (DDR) represents an attractive pathway for translational research in cancer therapeutics. It refers to an intricate mechanism of responding to DNA damage once it occurs. Comprising numerous molecules and pathways, its main function is to prevent damage expansion by arresting the cell cycle until the damage is repaired, or else driving cells to apoptosis. DDR can act as a double-edged sword, in the sense that impaired DDR may lead to leukemogenesis, while hyperactivated DDR may reverse chemotherapy-induced DNA damage and result in chemoresistance [4]. Although more extensively studied in solid malignancies, DDR has also emerged as an important mechanism in the pathophysiology of hematopoietic malignancies. DDR inhibition in combination with conventional treatment is also being investigated as a means of inducing synthetic lethality in hematological malignancies [4, 5].
Defective DDR has been involved in AML pathogenesis. Hematopoietic stem cells are prone to the accumulation of genetic lesions owing to their long lifespan and repeated exposure to stressful conditions that force them to enter the cell cycle resulting in replication stress [6]. Specific entities related to DDR impairment such as congenital DDR deficiencies [7], inherited and somatic TP53 mutations [8], and multiple polymorphisms in DDR genes [9] have been associated with increased AML incidence.
In this study, we showed that several DDR genes are upregulated in cell lines following treatment with Idarubicin and Cytarabine. One of them, PPP1R15A, was upregulated in AML patients compared to controls, and its knockdown by two different methods resulted in increased AML chemosensitivity.
Materials and methods
Patient and control samples
Mononuclear cells were isolated from bone marrow samples from 74 patients with de novo AML at diagnosis and 30 lymphoma subjects without evidence of bone marrow involvement - the latter were used as healthy controls. The research was approved by the institutional review board of Attikon University Hospital (ΕΒΔ2421/26-05-2017) and all participants gave written informed consent in agreement with the Declaration of Helsinki. All AML patients had received one to two induction cycles of Cytarabine and Idarubicin and response to treatment was evaluated after the end of induction. Isolation was performed with Lymphoprep (STEM CELL Technologies) by density gradient centrifugation. Isolated cells in RPMI were stored in cryovials at – 80˚C. Mononuclear cells were homogenized using a QIA shredder spin column (Qiagen Ltd., Hilden, Germany).
Cell culture
Four AML cell lines namely MV-4-11, MOLM-13 (MLL-AF9 fusion, FLT3-ITD mutated), KASUMI-1 (core binding factor AML), and TF-1 (erythroleukemia), were cultured based on ATCC® guidelines. The growth conditions were 5% CO2, 95% air, and a temperature of 37℃.
MTT assay
Cell lines KASUMI-1 and MV-4-11 were seeded in a 96-well plate at a concentration of 1 × 105 cells/mL, 16 h after seeding they were treated based on bibliographic data with Idarubicin and Cytarabine in order to conduct the gene expression profiling. MOLM-13 and KASUMI-1 cells were seeded in a 96-well plate at a concentration of 1 × 105 cells/mL. Afterward, they were treated with Idarubicin or Cytarabine at a concentration range of 1nM- 10µΜ (with a 10fold gradual increase) and were further incubated for 24, 48, and 72 h. TF-1 cells were seeded in a 96-well plate at a concentration of 1 × 105 cells/mL. Afterward, they were treated with Idarubicin (0.01, 0.05, 0.1, 0.2, and 0.5µΜ) or Cytarabine (0.1, 0.5, 01, 2, 5µΜ) and were further incubated for 24, 48 and 72 h. The colorimetric 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) assay was used for cell viability determination at each time-point and for each agent concentration. MTT (Sigma, USA) was dissolved in PBS at 5 mg/mL, the MTT solution was added to the 96-well plate at volume 20 µL, and the resulting solution was incubated in 5% CO2 for another 4 h at 37 °C. Formazan crystals were dissolved in 200 µL of SDS-HCL. The plates were then analyzed in a plate reader at 570 nm. All experiments were performed in triplicate.
Trypan blue assay
Trypan Blue assay was also used to determine the number of viable cells after treatment of both cell lines with the different concentrations of Idarubicin or Cytarabine in 24, 48, and 72 h. MOLM-13 and KASUMI-1 cells were seeded in a 6-well plate at a concentration of 1 × 105 cells/mL, then treated and incubated at three-time points. Next, we mixed 1 part of 0.4% trypan blue (Sigma-Aldrich, Merck) and 1 part cell suspension and the total cells were counted within 3 to 5 minutes with a hemacytometer. All experiments were performed in triplicate.
The half-maximal inhibitory concentration (IC50) calculation
The IC50 values 24,48 and 72 h after treatment with different concentrations of Idarubicin or Cytarabine in MOLM-13 and KASUMI-1 cell lines were determined by plotting the MTT assay results of % viability vs. time for each drug concentration. Subsequently, cell viability was further assessed using the trypan blue assay to confirm the results.
Clonogenicity assay
TF-1 cells were counted and re-suspended at a density of 2000 cells/ml in methylcellulose-based media. After 10 to 14 days of incubation at 37 °C in 5% CO2, the recovery of colony-forming units was determined by colony counting under bright-field microscopy. A cell aggregate composed of > 50 cells was defined as a colony.
siRNA transfection
In 6-well plates, MOLM-13 and KASUMI-1 cells were seeded at the specified concentrations. Following the designated incubation period, treatment with Idarubicin (10 nM) or Cytarabine (100 nM) was administered to MOLM-13, while KASUMI-1 received Idarubicin (1 nM) or Cytarabine (100 nM). Untreated wells were also incorporated. After 24 h, 10 pmol of siRNA was transfected into each respective well. In both treated and untreated cells, wells containing 10 pmol of a scrambled sequence were included, as were wells comprising only the transfection reagents (mock control). Each reaction was conducted in triplicates using the Lipofectamine™ RNAiMAX (Invitrogen™, Thermo Fisher Scientific Inc.). The cells were incubated with the transfection reagents for 24 and 48 h. The principles of the designed sequences are shown in Supplemental Table S1.
Total RNA isolation and RNA quality control
Total RNA was extracted from patient samples utilizing the RNeasy Plus Mini Kit (Qiagen Ltd., Hilden, Germany). Additionally, total RNA was isolated from transfected cells employing NucleoZOL (MACHEREY-NAGEL GmbH & Co. KG, Düren, Germany). The concentration of total RNA was determined using the BioSpec-nano Micro-volume UV–vis Spectrophotometer (Shimadju, Kyoto, Japan), and their integrity was assessed via electrophoresis in 1.2% agarose gel.
Reverse transcription and real-time quantitative PCR (qPCR)
200 ng of each RNA extract from patient samples was used as a template for cDNA synthesis. First-strand synthesis was conducted using M-MLV reverse transcriptase (Life Technologies Ltd., Carlsbad, CA, USA) and oligo-dT primer. All reactions were performed according to the manufacturer’s instructions. 100 ng of cDNA of patient samples were diluted in QuantiNova Yellow Template Dilution Buffer were mixed with 10 µl of 2x QuantiNova SYBR Green PCR Master Mix (Qiagen Ltd., Hilden, Germany), 2 µl of 10x QuantiTect primer, and RNase free water. Real-time qPCR assays followed using KAPA™ SYBR® FAST qPCR master mix (2X) (Kapa Biosystems Inc., Woburn, MA, USA). Reactions were performed in a QuantStudio 5 Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific Inc.), according to the manufacturer’s protocol. Gene expression was normalized against GAPDH expression and relative gene expression was calculated by the ΔCt method. We studied gene expression in correlation with disease status, ELN2017 AML cytogenetic risk [10], presence of FMS like Tyrosine kinase 3 mutation Internal Tandem Duplication (FLT3-ITD) and nucleophosmine 1 (NPM1) mutations and patient survival.
To investigate the impact of Idarubicin and Cytarabine at their respective IC50 concentrations on PPP1R15A expression in AML cell lines, gene expression analysis was performed using qRT-PCR as described above. Melt curve analysis was also utilized to verify the specificity of each amplification product. For each pairing of cell line and inhibitor, the normalized RQU or fold change values were expressed as log2 fold change. To elucidate the functional significance of PPP1R15A in AML cell viability and chemotherapy response, experiments involving PPP1R15A silencing using siRNA in MOLM-13 and KASUMI-1 cell lines were conducted. Initially, the efficiency of PPP1R15A silencing by qPCR was assessed by comparing the expression levels to cells transfected with a scramble sequence. Furthermore, cell viability comparisons between PPP1R15A-silenced and mock control cells were made after treatment with Cytarabine and Idarubicin at IC50 concentrations for 24 and 48 h. For this purpose, GADPH, HRPT1, and B2M were used as reference genes.
Gene expression profiling through PCR arrays analysis (Qiagen RT² Profiler PCR Array (96-Well Format and 384-Well [4 × 96] Format) Human DNA Damage Signaling Pathway) was performed in triplicate after RNA extraction from untreated, chemotherapy-treated, and live cells following chemotherapy exposure. Human DNA damage signaling pathway-related gene expression was evaluated and analyzed through the RT2 Profiler PCR Array data analysis tool (Rotor-Gene_2_3_5_1) and Fold Change values per condition were calculated using the respective untreated sample as control. All the primer sequences that were used from the above experiments are shown in Supplemental Tables S2 and S3.
CRISPR-Cas9 mediated PPP1R15A deletion
PLKO.1-puro lentiviral vectors expressing CRISPR-associated endonuclease 9 (Cas9) and two different single guide RNAs (sgRNAs) targeting PPP1R15A (sgRNA1: AGGTCCTGGGAGTATCGTTC, sgRNA2: GGACAACACTCCCGGTGTGA) or scramble RNA (GTGTAGTTCGACCATTCGTG) as control purchased from VectorBuilder were transfected into HEK-293FT cells [11]. TF-1 cells were transduced with lentiviruses and selected with puromycin (2 µg/ml). Subsequently, single-cell cloning was done, and clones were screened by performing qPCR to select PPP1R15A deleted clones.
Flow cytometry
KASUMI-1 and MV-4-11 cell sorting was performed using a flow-cytometry-based approach with Annexin V staining. In brief, cultured cells after treatment with either Idarubicin or Cytarabine, washed with ice-cold phosphate-buffered saline (PBS) and resuspended in binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4). Subsequently, cells were stained with Annexin V-FITC (fluorescein isothiocyanate) according to the manufacturer’s instructions (BD Biosciences Annexin V-FITC Apoptosis Detection Kit). Sorting gates were established based on the Annexin V-FITC signal intensity and FSC/SSC characteristics to isolate the viable cell population. The entire sorting process was conducted at 4 °C. TF-1 cells were fixed in 4% formaldehyde for 15 min and permeabilized in 100% methanol for 30 min. Subsequently, cells were labeled with the phycoerythrin (PE) conjugated PPP1R15A antibody purchased from Santa Cruz (sc-373,815). Gating was based on clearly distinguishable populations, or in the absence of such, the negative antibody control. Median fluorescence intensity (MFI) was determined for each marker using FlowJo analysis software version 10.0.8 (FlowJo, Ashland, CO, USA).
Western blotting
Cell pellets were homogenized on ice in lysis buffer Nonident P-40 (NP-40) (Sigma-Aldrich, St. Louis, MI, USA), that containing protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA) and centrifuged for 10 min (4 °C) at 20,000× g. Samples were subjected to SDS-PAGE and immunoblotting analysis after the protein content was calibrated for each sample using the Bradford assay (Bio-Rad, Hercules, CA, USA). Using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), 10 micrograms of protein were separated and then transferred to a nitrocellulose membrane (MACHEREY-NAGEL GmbH & Co. KG, Düren, Germany). Following blocking with 5% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO, USA), the membranes were incubated with primary antibody, GADD34 (E3S6N) Rabbit mAb and β-Actin (13E5) Rabbit mAb (Cell Signaling Technology, Danvers, Massachusetts, USA) overnight at 4 °C, followed by the respective secondary antibodies. Immunoblots were developed using Clarity Western ECL Blotting Substrate (Bio-Rad, Hercules CA, USA). Blot quantification was performed using ImageJ 1.53k (NIH).
Statistical analysis
To visualize the relative response profile of each cell treatment in the KASUMI-1 and MV4-11 cells, Principal Component analysis (PCA) was performed using the gene expression (2−ΔCt) of all 84 DDR genes from the Qiagen’s RT2 Profiler PCR Array. For differential gene expression in patient and control samples, statistical analysis was performed using the Kruskal-Wallis test and Dunn’s test for pairwise multiple comparisons, with a significance level of p < 0.05. Correlation analysis was performed using Spearman’s Rho and survival analysis was performed using the Kaplan-Meier method and the log-rank test. Statistical analysis was performed using STATA 17 software.
Results
Gene expression profiling
PCA of the 84 genes involved in DNA Damage Response (DDR), revealed distinct patterns of DDR gene expression in MV4-11 and KASUMI-1 cell lines before and after treatment with Idarubicin and Cytarabine. DDR gene expression following treatment was measured in both the entire population of treated cells and the live cells, as seen in Supplemental Figure S1.
The following genes were over two-fold up-regulated in live cells from leukemic cell lines after treatment with both agents: PPP1R15A, CDKN1A, and GADD45G genes were up-regulated in both live cell lines, GADD45A in live MV4-11 cells and EXO1 in live KASUMI-1 cells (Fig. 1).

Heatmap of differentially expressed genes with at least one statistically significant fold change (p-value < 0.05) either in KASUMI-1 or MV4-11 live cells treated with Idarubicin or Cytarabine. Marked in bold are the selected genes whose expression was subsequently studied in bone marrow samples of AML patients
These genes were selected for relative quantification using the ΔCt method.
Patient and control qPCR results
To further characterize the identified set of DDR genes we analysed their expression levels in AML patients and healthy controls and correlated them with the AML patients’ cytogenetic risk. The patients’ and controls’ characteristics are depicted in Table 1.
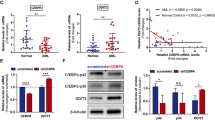
The analysis of the expression level of the DDR genes demonstrated a marked increase in the expression of PPP1R15A as well as a decrease in the expression of GADD45G in AML patients compared to controls (p < 0.01 respectively). GADD45A and CDKN1A were also decreased in AML patients compared to controls, although not significantly (p = 0.076 and p = 0.088 respectively). EXO1 expression was not different between AML patients and controls (Fig. 2a). To confirm our findings in an independent publicly available database we compared the PPP1R15A RNA levels between AML samples and healthy bone marrow mononuclear cell samples using data from the BEAT AML database [12]. We found that PPP1R15A is upregulated in AML samples compared to healthy bone marrow mononuclear cell samples (Fig. 2b). The correlation of expression of various genes showed a significant positive correlation between PPP1R15A and CDKN1A in AML patients (Fig. 2d). The analysis of DDR gene expression based on cytogenetic risk showed that all genes are upregulated in AML patients with higher cytogenetic risk. More specifically, PPP1R15A was upregulated in intermediate risk compared to favorable risk karyotype, CDKN1A was upregulated in intermediate and adverse karyotype compared to favorable risk karyotype, GADD45A, and EXO1 were upregulated in adverse karyotype compared to favorable and intermediate risk karyotype and GADD45G was upregulated in adverse risk compared to favorable risk karyotype (Fig. 2c).

(a) Differential expression of DDR genes in AML patients and controls. Brackets denote p < 0.05 (b) PPP1R15A expression in healthy BM mononuclear cell samples and AML samples based on data from the BEAT AML dataset. (c) Differential expression of DDR genes in different cytogenetic AML risk groups. Brackets denote p < 0.05 (d) Correlation between PPP1R15A and CDKN1A in AML patients
Our analysis showed no difference in the DDR gene expression levels between AML patients who responded to induction treatment and those who did not. No difference was found in the relative gene expression according to FLT3-ITD or NPM1 mutational status either. There was finally no significant effect of gene expression on patients’ overall survival and event-free survival.
Overall, our analysis of human samples supports that PPP1R15A is the only gene out of the DDR genes upregulated in AML cells following Idarubicin and Cytarabine treatment that is also upregulated in AML patients and is associated with worse cytogenetic risk.
IC50 values after treatment with idarubicin or cytarabine on AML cell lines
Our analysis based on the MTT assay showed that Idarubicin has an IC50 of 10 nM in the MOLM-13 cell line and 1 nM KASUMI-1 cells. Cytarabine, on the other hand, has an IC50 of 100 nM in both MOLM-13 and KASUMI-1 cells. All IC50 concentrations were recorded at 72 h post-treatment (Supplemental Figure S2). These results suggest that Idarubicin possesses a slightly stronger effect on cell viability than Cytarabine.
Differential expression of PPP1R15A in MOLM-13 and KASUMI-1 cells after treatment with Idarubicin and cytarabine
After 72 h of treatment, we observed a significant increase in the relative expression of PPP1R15A in both MOLM-13 and KASUMI-1 cells compared to untreated cells. Interestingly, the extent of PPP1R15A upregulation differed between the two cell lines. In MOLM-13 cells, treatment with the IC50 concentration of Idarubicin resulted in a moderate increase in PPP1R15A expression compared to untreated cells. Likewise, Cytarabine treatment induced a similar upward increase of PPP1R15A levels over the control. In the case of KASUMI-1 cells, both Idarubicin and Cytarabine treatments led to a substantial upregulation of PPP1R15A expression. Specifically, Idarubicin treatment resulted in a 5-fold increase in PPP1R15A levels compared to untreated cells. In contrast, Cytarabine treatment exhibited an even more remarkable effect, with PPP1R15A expression showing a robust 12-fold increase over the control. These findings indicate that both Idarubicin and Cytarabine modulate PPP1R15A expression in AML cells, with the KASUMI-1 cell line being more sensitive to the effect of the two chemotherapeutics (Fig. 3).

Differential Expression of PPP1R15A in MOLM-13 and KASUMI-1 cells after treatment with Idarubicin and Cytarabine
Functional impact of PPP1R15A silencing on AML cell viability and response to Idarubicin or Cytarabine
To elucidate the functional significance of PPP1R15A in AML cell viability and chemotherapy response, we conducted PPP1R15A silencing experiments using siRNA in MOLM-13 and KASUMI-1 cell lines. Initially, we assessed the efficiency of PPP1R15A silencing by qPCR and western blot comparing the expression levels to cells transfected with scramble sequence.
In both MOLM-13 and KASUMI-1 cells, siRNA-mediated silencing resulted in a significant reduction in PPP1R15A expression levels compared to the scramble sequence (Supplemental Figure S3). After treatment of PPP1R15A silenced cells with Idarubicin or Cytarabine at their respective IC50 concentrations, we observed a marked decrease in cell viability in PPP1R15A-silenced cells following drug treatment compared to untreated cells. This reduction was evident in both MOLM-13 and KASUMI-1 cells (Fig. 4).

Cell viability after 24 h and 48 h of PPP1R15A silenced and scrambled cells. a: Kasumi-1, Idarubicin treated cell line, b: Kasumi-1, Cytarabine treated cell line, c: MOLM-13, Idarubicin treated cell line d: MOLM-13, Cytarabine treated cell line
It is worth mentioning that cell viability did not significantly change in cell lines that had the PPP1R15A silenced compared to untransfected cells without treatment with Idarubicin or Cytarabine (Supplemental Figure S4).
PPP1R15A knockdown by CRISPR-Cas9 increases the sensitivity of TF-1 cells to Idarubicin and Cytarabine
To further evaluate the implication of PPP1R15A expression in the sensitivity of AML cells to chemotherapy, CRIRPS-Cas9 mediated deletion of PPP1R15A was performed in the chemo-resistant TF-1 cells using two independent sgRNAs. Following puromycin selection, a single-cell clone was performed, and growing clones were screened by using qPCR for PPP1R15A (Supplemental Figure S5a). The PPP1R15A suppression at the protein level was confirmed by flow cytometry and western blot assay (Supplemental Figure S5b, c ).
TF-1 cells counting with trypan blue staining revealed no effect of PPP1R15A knockdown in cell growth (Supplemental Figure S6a) and methylcellulose-based clonogenicity assay showed no effect of PPP1R15A knockdown on cells colony formation (Supplemental Figure S6b).
Control, clone 1 (sgRNA1), and clone 2 (sgRNA2) TF-1 cells were treated with 0–5 µM Cytarabine for 72 h and 0-0.5 Idarubicin µM for 24 h. PPP1R15A knockdown clones (clones 1 and 2) were significantly more sensitive to the chemotherapy agents compared to the control clone (Fig. 5a, b). Consistently, clones 1 and 2 had significantly lower IC50 for both Cytarabine and Idarubicin (Fig. 5c-d).

(a) TF-1 cells with PPP1R15A CRISPR-Cas9-mediated knockdown (clones 1 and 2) have a higher sensitivity to Cytarabine compared to control cells.(b) TF-1 cells with PPP1R15A CRISPR-Cas9-mediated knockdown (clones 1 and 2) have a higher sensitivity to Idarubicin compared to control cells. (c) TF-1 cells with PPP1R15A CRISPR-Cas9-mediated knockdown (clones 1 and 2) have lower IC50 dose for Cytarabine compared to control cells. (d) TF-1 cells with PPP1R15A CRISPR-Cas9-mediated knockdown (clones 1 and 2) have lower IC50 doses for Idarubicin compared to control cells
Discussion
In this study, we examined the effect of Idarubicin and Cytarabine in the expression of a panel of DDR genes in AML cell lines. Genes that exhibited significant changes in their expression were chosen to be studied in bone marrow samples of AML patients and controls. Finally, we studied the effect of PPP1R15A downregulation, as this was the only DDR gene upregulated in AML patients compared to healthy controls and overexpressed in those with higher cytogenetic risk. We showed for the first time that its downregulation significantly increases AML cells’ chemosensitivity.
Using PCR arrays we observed an upregulation of of several DDR genes (CDKN1A, GADD45A, GADD45G, EXO1, and PPP1R15A) in KASUMI-1 and MV4-11 cell lines that survived following treatment with Idarubicin and Cytarabine. In malignancies genotoxic stress can be caused both by excessive DNA replication itself as well as by chemotherapy used to treat them [5]. This is in agreement with a positive association of these five DDR genes’ expression with increasing cytogenetic risk in AML patients in our study given that more complex karyotype has been linked with increased DNA damage. Furthermore, a previous study by Cavelier et al. has demonstrated an association of complex karyotype with increased DNA damage sensing through Histone 2X phosphorylation that resulted in checkpoint inhibition [13]. Overall, these findings suggest a link between genomic instability and upregulated DDR mechanisms. Of note, a higher level of genomic complexity can be detected in AML when genomic and transcriptomic data are used [14]. The association of genomic instability at high resolution with DDR would be very interesting to investigate.
GADD45A, GADD45G, and GADD34 (also known as PPP1R15A) are part of the growth arrest and DNA damage–induced family of genes and they are involved in cell cycle arrest, DNA repair, cell survival, senescence, and apoptosis in response to oncogenic stress [15]. In our study, we found significant downregulation of GADD45G in AML patients compared to controls, as previously reported in a study by Guo et al. [16]. In keeping with their findings, we observed a tendency for shorter event-free survival in our patients who exhibited low GADD45G levels when we used a cut-off value of 45% GADD45G expression (p = 0.084, data not shown). While all 5 genes were overexpressed in samples from AML patients with intermediate and high cytogenetic risk compared to low cytogenetic risk, only PPP1R15A was found to be overexpressed in samples from AML patients compared to healthy controls highlighting its importance as a DDR mediator in this disease.
Our study reports for the first time the upregulation of PPP1R15A in AML patients compared to controls. Our results are further confirmed by data from the BEAT AML dataset. We also report enhanced chemosensitivity of AML cell lines after PPP1R15A downregulation. PPP1R15A is a holophosphatase comprised of two subunits, namely PPP1 and R15A. PPP1R15A is induced after treatment with DNA-damaging agents [17] and it enhances p53 independent apoptosis after treatment with some, but not all DNA damaging agents [18]. Interestingly, it was shown that in 11q23 AML, GADD34-mediated apoptosis is abrogated owing to the inactivating effect of the fusion protein on GADD34 [19]. These reports suggest that PPP1R15A-induced apoptosis is dependent both on the underlying disease as well as the DNA-damaging agent used.
In our study, it was demonstrated that PPP1R15A was overexpressed in AML probably as a result of genotoxic stress. In addition, its downregulation was associated with reduced cell viability in chemotherapy-treated AML cell lines indicating that PPP1R15A may have multiple mechanisms of action in DDR besides induction of apoptosis. This hypothesis is further supported by experimental data in solid tumors associating tumor hypoxia and autophagy with PPP1R15A upregulation, while PPP1R15A downregulation resulted in tumor growth suppression [20,21,22,23]. The mechanisms underlying PPP1R15A upregulation in AML remain to be elucidated.
The main function of PPP1R15A is the inactivation of the subunit of eukaryotic initiation factor 2 (eIF2α). PPP1R15A can bind to catalytic subunit protein phosphatase 1 (PP1c) and promote the dephosphorylation/inactivation of eIF2α. EIF2a is mainly activated under endoplasmic reticulum (ER) stress and it acts by inhibiting protein translation to restore proteostasis. Increased levels of eIF2α induce upregulation of PPP1R15A in order to restore translation and promote cell recovery [24,25,26]. PPP1R15A is currently being investigated for the treatment of protein-misfolding neurodegenerative diseases [27]. Furthermore, we found a significant correlation between PPP1R15A and CDKN1A expression in our patient cohort. CDKN1A is upregulated upon p53 activation after DNA damage, resulting in cell cycle arrest at G1 [28] and it has been reported that ER stress downregulates p21CDKN1A levels, suggesting a link between ER stress and DDR [29].
PPP1R15A silencing studies, performed with two independent methods support that PPP1R15A suppression enhances the sensitivity of AML cells to Idarubicin or Cytarabine treatment, leading to reduced cell viability. Interestingly, untreated cells that were silenced for PPP1R15A did not show a decrease in cell viability suggesting that PPP1R15A silencing alone does not substantially affect cell survival under normal conditions. A previous study has shown that mice homozygous for a functionally inactivating mutation of PP1R15A exhibited normal phenotype [30]. These findings support that PPP1R15A inhibition could be an attractive therapeutic approach to increase the sensitivity of AML to induction chemotherapy.
Conclusions
Overall, this study confirms the findings of previous investigators regarding DDR gene dysregulation in AML, while reporting for the first time the upregulation of PPP1R15A in this disease. Lastly, it highlights the functional relevance of PPP1R15A in AML cell viability and chemotherapy response.
By understanding the role of PPP1R15A in mediating drug sensitivity, we can potentially develop novel strategies to enhance the efficacy of chemotherapy and improve treatment outcomes for AML patients. Further studies are warranted to elucidate the molecular mechanisms underlying the interaction between PPP1R15A and chemotherapy agents, paving the way for the development of targeted therapies in AML.
Data availability
Data will be made available upon request to the corresponding author.
References
Cersosimo RJ (1992) Idarubicin: an anthracycline antineoplastic agent. Clin Pharm 11:152–167
Faruqi A, Tadi P, Cytarabine (2023) StatPearls [Internet]. Treasure Island (FL) ineligible companies
Kantarjian H, Kadia T (2021) Acute myeloid leukemia: current progress and future directions. Blood Cancer J 11:41–66. https://doi.org/10.1038/s41408-021-00425-3
Delia D, Mizutani S (2017) The DNA damage response pathway in normal hematopoiesis and malignancies. Int J Hematol 106:328–334. https://doi.org/10.1007/s12185-017-2300-7
Takagi M (2017) DNA damage response and hematological malignancy. Int J Hematol 106:345–356. https://doi.org/10.1007/s12185-017-2226-0
Walter D, Lier A (2015) Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520:549–552. https://doi.org/10.1038/nature14131
Sharma R, Lewis S (2020) DNA repair syndromes and Cancer: insights into Genetics and phenotype patterns. Front Pediatr 8:1–9. https://doi.org/10.3389/fped.2020.570084
George B, Kantarjian H (2021) TP53 in Acute myeloid leukemia: molecular aspects and patterns of mutation. Int J Mol Sci 22:1–22. https://doi.org/10.3390/ijms221910782
Esposito MT, So CW (2014) DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 123:545–561. https://doi.org/10.1007/s00412-014-0482-9
Dohner H, Estey E (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129:424–447. https://doi.org/10.1182/blood-2016-08-733196
Kitamura T, Tange T (1989) Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol 140:323–334. https://doi.org/10.1002/jcp.1041400219
Burd A, Levine RL (2020) Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the beat AML Master Trial. Nat Med 26:1852–1858. https://doi.org/10.1038/s41591-020-1089-8
Cavelier C, Didier C (2009) Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res 69:8652–8661. https://doi.org/10.1158/0008-5472.CAN-09-0939
Klever MK, Strang E (2023) AML with complex karyotype: extreme genomic complexity revealed by combined long-read sequencing and Hi-C technology. Blood Adv 14:6520–6531. https://doi.org/10.1182/bloodadvances.2023010887
Liebermann DA (2022) Gadd45 in normal hematopoiesis and leukemia. Adv Exp Med Biol 1360:41–54. https://doi.org/10.1007/978-3-030-94804-7_3
Guo D, Zhao Y (2021) GADD45g acts as a novel tumor suppressor, and its activation suggests new combination regimens for the treatment of AML. Blood 138:464–479. https://doi.org/10.1182/blood.2020008229
Hollander MC, Sheikh MS (2001) Activation of Gadd34 by diverse apoptotic signals and suppression of its growth inhibitory effects by apoptotic inhibitors. Int J Cancer 96:22–31. https://doi.org/10.1002/1097-0215(20010220)96:1<22::aid-ijc3>3.0.co;2-k
Hollander MC, Zhan Q (1997) Mammalian GADD34, an apoptosis- and DNA damage-inducible gene. J Biol Chem 272:13731–13737. https://doi.org/10.1074/jbc.272.21.13731
Adler HT, Chinery R (1999) Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol Cell Biol 19:7050–7060. https://doi.org/10.1128/mcb.19.10.7050
Liu L, Ito S (2016) GADD34 promotes Tumor Growth by inducing myeloid-derived suppressor cells. Anticancer Res 36:4623–4628. https://doi.org/10.21873/anticanres.11012
Yang ZJ, Chee CE (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10:1533–1541. https://doi.org/10.1158/1535-7163.MCT-11-0047
Holczer M, Banhegyi G (2016) GADD34 keeps the mTOR pathway inactivated in endoplasmic reticulum stress-related autophagy. PLoS ONE 11:1–20. https://doi.org/10.1371/journal.pone.0168359
Ho KH, Lee YT (2021) Guanabenz sensitizes Glioblastoma Cells to Sunitinib by inhibiting GADD34-Mediated autophagic. Signal Neurother 18:1371–1392. https://doi.org/10.1007/s13311-020-00961-z
Wek RC (2018) Role of eIF2alpha kinases in Translational Control and Adaptation to Cellular stress. Cold Spring Harb Perspect Biol 10:1–16. https://doi.org/10.1101/cshperspect.a032870
Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D (2009) Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci USA 106:1832–1837. https://doi.org/10.1073/pnas.0809632106
Carrara M, Sigurdardottir A (2017) Decoding the selectivity of eIF2alpha holophosphatases and PPP1R15A inhibitors. Nat Struct Mol Biol 24:708–716. https://doi.org/10.1038/nsmb.3443
Das I, Krzyzosiak A (2015) Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348:239–242. https://doi.org/10.1126/science.aaa4484
Engeland K (2022) Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ 29:946–960. https://doi.org/10.1038/s41418-022-00988-z
Mlynarczyk C, Fahraeus R (2014) Endoplasmic reticulum stress sensitizes cells to DNA damage-induced apoptosis through p53-dependent suppression of p21(CDKN1A). Nat Commun 5:1–16. https://doi.org/10.1038/ncomms6067
Marciniak SJ, Yun CY (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18:3066–3077. https://doi.org/10.1101/gad.1250704
Funding
This study received no funding.
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Contributions
A.B. collected and analysed human data, prepared Fig. 2 and wrote the main manuscript text C.D.S. performed PPP1R15A silencing experiments, prepared Figs. 3 and 4 and supplementary Figs. 2-4 and wrote the methods section C.E performed PPP1R15A knockdown experiments, prepared Fig. 5 and suppl Figs. 5-6 and wrote the methods section on PPP1R15A knockdownT.L. analysed PCR array results and prepared Fig. 1 and suppl Fig. 1G.G.D. performed PCR for DDR genes on human samplesS.G.P., M.P., E.H., M.D., S.K., E.A., and T.P.T. provided human samples for the studyC.K.K. supervised the PPP1R15A silencing experimentsT.K. supervised the PPP1R15A knockdown experiments and designed the studyV.P. supervised the PCR arrays and human data experiments and designed the studyAll authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bouchla, A., Sotiropoulou, C.D., Esteb, C. et al. Silencing of the DNA damage repair regulator PPP1R15A sensitizes acute myeloid leukemia cells to chemotherapy. Ann Hematol (2024). https://doi.org/10.1007/s00277-024-05785-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00277-024-05785-x