
Abstract
Aplastic anemia (AA) is an auto-activated T cell–mediated bone marrow failure. Cyclosporine is often used to treat non-severe AA, which demonstrates a more heterogeneous condition than severe AA. The response rate to cyclosporine is only around 50% in non-severe AA. To better predict response to cyclosporine and pinpoint who is the appropriate candidate for cyclosporine, we performed phenotypic and functional T cell immune signature at single cell level by mass cytometry from 30 patients with non-severe AA. Unexpectedly, non-significant differences of T cell subsets were observed between AA and healthy control or cyclosporine-responder and non-responders. Interestingly, when screening the expression of co-inhibitory molecules, T cell trafficking mediators, and cytokines, we found an increase of cytotoxic T lymphocyte antigen 4 (CTLA-4) on T cells in response to cyclosporine and a lower level of CTLA-4 on CD8+ T cells was correlated to hematologic response. Moreover, a decreased expression of sphingosine-1-phosphate receptor 1 (S1P1) on naive T cells and a lower level of interleukin-9 (IL-9) on T helpers also predicted a better response to cyclosporine, respectively. Therefore, the T cell immune signature, especially in CTAL-4, S1P1, and IL-9, has a predictive value for response to cyclosporine. Collectively, our study implies that immune signature analysis of T cell by mass cytometry is a useful tool to make a strategic decision on cyclosporine treatment of AA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aplastic anemia (AA) is a bone marrow failure syndrome attacked by auto-activated T cell on autologous bone marrow hematopoietic progenitors, which is characterized by pancytopenia [1,2,3]. Non-severe AA (non-SAA) with a variably clinical feature accounts for 80% of AA [4, 5]. Up to two-thirds of patients would finally progress to transfusion dependence or severe AA (SAA) without intervention [6,7,8]. Although cyclosporine, which has effect on reducing T cell proliferation and activation, can reverse pancytopenia and alleviate transfusion requirements, there still exist 50% of patients who fail to respond and 35% relapse after responding [9,10,11]. Additional novel therapeutic approaches are required for AA patients who fail to achieve response, and along with this, more robust diagnostic examinations that predict response to cyclosporine are necessary.
Mechanisms of T cell–mediated bone marrow failure include excessive production of inhibitory cytokines conferred by Th1 polarization, Th17 immune response, and direct toxicity to hematopoietic cells [12,13,14]. Previous researches also have revealed a decreased number and impaired function of regulatory T cells (Tregs) in AA [15,16,17]. Furthermore, a wide variety T cell immune checkpoint molecules have been identified to regulate T cell activation, including increased expression of T cell immunoglobulin mucin-3 (TIM-3) and programmed death-1 (PD-1) in AA [18,19,20]. Moreover, the sphingosine-1-phosphate receptor (S1PR) signaling is related to various physiological processes including T lymphocyte recirculation and their biology [21, 22]. Cytokines also play a crucial role in driving immune responses and HSPC growth inhibition and apoptosis [23]. Interleukin-9 (IL-9) is a pleiotropic cytokine secreted by T helpers. This growth factor not only is associated with the pathogenesis of autoimmune diseases but also regulates hematopoiesis [24,25,26]. Altogether, T cell function and activation are critical to regulate the pathophysiology of AA [27, 28]. Thus, we hypothesize that defining T cell immune signature will facilitate the development of predicting cyclosporine response and exploring novel immune-based therapeutics.
In this study, a panel of surface and intracellular markers associated with T cell phenotype and function was designed, and the complexity of T cell subsets was examined by mass cytometry. This study enabled us to identify the T cell signature in AA patients and explore immune biomarkers for predicting the response to cyclosporine treatment.
Materials and methods
Patients’ and healthy donors’ samples
A total of thirty patients with acquired non-SAA from Institute of Hematology and Blood Diseases Hospital were enrolled in the study. Inclusion criteria were predefined as follows: (1) meet the international study group criteria for AA [29]; (2) did not meet the criteria of SAA; and (3) obtained consent to cyclosporine. Exclusion criteria were defined as follows: (1) inherent bone marrow failure; (2) suffer from other hematological disorders or autoimmune diseases; (3) received an immunosuppressive agent before; and (4) suffer fever or infection within 4 weeks before diagnosis. Based on disease severity [30, 31], patients were classified into transfusion dependent (defined as red blood cell or platelet transfusion dependence or neutrophil count < 0.5 × 109/L) and transfusion independent.
Each patient received oral cyclosporine at 3–5 mg/kg daily. Dose adjustment was based on the serum level of cyclosporine which was maintained between 100 and 200 ng/mL. According to established criteria [29], response to cyclosporine was evaluated in the first 6 months after starting therapy. Partial response (PR) included transfusion independent (if previously dependent on transfusion), or at least one cell lineage doubling or reach to the normal level, or increase of hemoglobin concentration above 30 g/L (if initially less than 60 g/L), absolute neutrophil counts > 0.5 × 109/L (if initially less than 0.5 × 109/L), and platelets > 20 × 109/L (if initially less than 20 × 109/L). Complete response (CR) was defined as hemoglobin concentration recovered to be normal, neutrophil count > 1.5 × 109/L, and platelet count > 100 × 109/L. Patients with no increase of blood cell or still transfusion dependent or progressed to SAA were defined as no response (NR).
Four healthy males were enrolled, with the median age of 44.5 years old (range from 16 to 50 years old). Routine blood tests were done before collecting bone marrow samples, and all of them had normal levels of hemoglobin (range from 145 to 160 g/L), platelets (range from 161 to 244 × 109/L), and absolute neutrophil counts (range from 2.1 to 2.75 × 109/L).
Bone marrow specimens were collected from participants including AA patients and healthy volunteers. Bone marrow mononuclear cells were isolated by Ficoll-Hypaque (1.077 g/mL) density gradient centrifugation, frozen in cryoprotectant, and stored at liquid nitrogen until laboratory analysis.
Mass cytometry staining and acquisition
A panel of antibodies based on surface and intracellular markers was designed (see supplemental Table 1). Each antibody was labeled with a rare metal isotope. The CyTOF mass cytometer (Fluidigm) was used for data acquisition. Forty-one bone marrow samples were tested, including thirty of patients without any treatment, seven of post cyclosporine, and four healthy individuals.
After 72-h stimulation with phorbol myristate acetate (50 ng/mL) and ionomycin (1 μg/mL), cells were collected and stained with metal-tagged antibodies. Samples were processed by six batches separately; the same protocol and culture conditions were done to minimize the variety from stimulation and staining. The protocol was referred to previous report, detailed steps are listed in supplementary methods. Cisplatin solution staining was used for identifying dead cells. After stating with metal-coupled surface antibody cocktail, nuclear antigen staining perm working solution was added for fixation and permeabilization. Then, samples were stained with antibody cocktail of cytokines and transcription factors. Next, 1.6% paraformaldehyde was used to fix cells, and 1 mL of intercalation solution was added for each sample by adding cell-ID Intercalator-Ir into fixation and permeabilization buffer to a final concentration of 125 nM and vortex to mix. To prepare cells for acquisition, cells were adjusted to 0.5–2 × 106/mL in double-distilled H2O and filtered by a 35–70-μm mesh. EQ beads were added prior to data acquisition by using a CyTOF mass cytometer. Data was exported in a format of flow cytometry file for further analysis.
Referred to previous report [32], CD45RA+CCR7+ defined naive T, CD45RA−CCR7+ defined central memory T, CD45RA−CCR7− defined effector memory T, CD45RA+CCR7− defined effector T, CD38+HLA-DR+ defined activated T, CD4+CXCR3+CCR6− defined Th1, CD4+CXCR3−CCR6+ defined Th17, CD4+CXCR3−CCR6− defined Th2, and CD4+CCR4+CD25+Foxp3+ defined Tregs.
Statistical analysis
All analyses were performed using Cytobank software and Prism 8.0.1. Data were presented as median or mean ± standard error mean (SEM). Mann–Whitney test and t test were used for assessing the significance of differences. It was considered to be statistically significant with the value of p < 0.05.
Study approval
Prior to inclusion in the study, informed consent was provided by all participants. Biological specimen collection protocols were complied with local guidelines and approved by the ethics committee of the Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences.
Results
T cell subsets and key transcription factor analyses in AA patients
Demographic information of 30 non-SAA patients was obtained from medical records. The cohort demonstrated a median age of 32 years old, and 56.7% was male. Intervals from onset to diagnosis differed largely and the median interval was 8 months (ranging from 1 month to 10 years). Twenty percent of patients were dependent on red blood cell or platelet transfusion; the median concentration of hemoglobin and platelet was 92.5 g/L (ranging from 47 to 149 g/L) and 25 × 109/L (ranging from 13 to 69 × 109/L) respectively. Fifteen patients (50%) respond to 6-month oral cyclosporine treatment (Table 1).
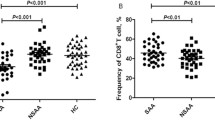
We compared the frequency of sixteen T cell subsets between AA patients and healthy individuals (Supplementary Fig. 1). Non-significant increased ratio of effector or memory CD8+ cytotoxic T cells was observed between patients and healthy individuals (Fig. 1A). Furthermore, the ratio of T helpers between patients and healthy donors was similar (Fig. 1B). Previous studies revealed that the frequency of Tregs in patients with AA was lower than that in healthy controls [15, 17]. Here, we also observed a relatively lower but not statistically significant level of Tregs in AA patients than healthy controls (2.30 ± 1.03% vs. 3.42 ± 1.13%, p = 0.056). Considering that T-box expressed in T cells (T-bet) and Eomesodermin (Eomes) participate in regulating T cell differentiation and phenotype shift [33, 34], we further analyzed the expression of T-bet and Eomes between patients and healthy individuals but found no significant differences due to the variation between individuals (Fig. 1C).

No difference on T cell subset proportion and T-box transcript factor expression between non-SAA patients and healthy donors. A, B Frequencies of T cell subpopulations expressed as frequency of CD8+ T cells (A) and of CD4.+ T cells (B). Data are shown as box plots extending from the 25th to 75th percentiles and the whiskers from the minimum to the maximum point; middle line represents the median. Samples from 30 non-SAA patients and 4 healthy donors were tested. C The UMAP plots showed the expression of T-bet and Eomes on CD3 T cells in patients (left). The violin plots showed frequencies of T-bet and Eomes on CD3 T cells (right). T-bet T-box expressed in T cells, Eomes Eomesodermin, HDs healthy donors. Data was shown as mean ± SEM; p < 0.05
To explore the relationship between T cell subsets and responsiveness to cyclosporine treatment, the frequency of T cell subpopulations between responders and non-responders was analyzed. However, no significant differences were observed, suggesting that the frequency of T cell subpopulations was not correlated with patients’ sensitivity to cyclosporine (Supplementary Fig. 2A–B). Furthermore, to investigate whether calcineurin/nuclear factor of activated T cell (NFAT) pathways are related to cyclosporine response, we further detected the expression levels of NFAT2, NFAT4, and calcineurin on T cells. Unexpectedly, no differences were observed between responders and non-responders (Supplementary Fig. 3).
Taken together, our data suggest that the frequency of T cell subpopulations and the expression level of T cell functional regulatory transcript factors are unrelated to the occurrence and development or the cyclosporine sensitivity of non-SAA which might be due to the heterogeneity of non-SAA. Additionally, the expression level of calcineurin/NFAT pathway could not discriminate the response to cyclosporine.
Cyclosporine treatment restores the expression of immune checkpoint molecule in responder patients
Immune checkpoint molecules were considered to play a critical role in the immune response of AA patients [18, 19, 35]. Thus, we analyzed the expression of these inhibitory molecules on T cell subsets. As compared to healthy individuals, higher frequency of CTLA-4+ CD8+ T cells was found in AA (22.63 ± 2.52% vs. 11.7 ± 2.11%, p < 0.05, Fig. 2A). Furthermore, increased level of CTLA-4 was displayed on naive CD8+ T cells (25.55 ± 2.92% vs. 11.86 ± 2.28%, p < 0.05) and effector memory CD8+ T cells (19.19 ± 2.02% vs. 10.26 ± 1.63%, p < 0.05, Fig. 2B). But no significant difference of CTLA-4 on effector CD8 was observed between healthy controls and AA patients (28.66 ± 2.77% vs. 17.38 ± 2.04%, p = 0.105). Unexpectedly, no statistically significant difference on PD-1+, LAG-3+, TIM3+, or TIGHT+ T cells was observed between the AA cohort and healthy volunteers (Supplementary Fig. 4).

Expression of CTLA-4 is altered upon cyclosporine treatment. A The UMAP figure showed the expression of CTLA-4 on CD8 T cells in patients at diagnosis. Samples from 30 non-SAA patients and 4 healthy donors were tested. B Box plots showed the expression difference of CTLA-4 on naive CD8 and EM CD8 in patients at diagnosis. C The percentage of CTLA-4 on CD8, naive CD8, and EM CD8 was significantly lower in responders. Samples were collected from 15 responders and 15 non-responders. D The heatmap showed increased expression of CTLA-4 on CD8 subsets following cyclosporine treatment in responders (left, n = 3). The alteration was shown in dot plots (right, n = 3). E SPADE clustering of CD4.+ T cells showed significant difference of CTLA-4 on CD4 subsets after cyclosporine treatment in responders (left), and the alteration was also shown in dot plots (right). Prior and post treatment samples were collected from 3 responders. HDs healthy donors, EM CD8 effector memory CD8, CM CD4 central memory CD4, EM CD4 effector memory CD4. Data was shown as mean ± SEM; *p < 0.05
As shown in Fig. 2C, responders displayed a lower frequency of CTLA-4+ CD8+ at diagnosis, compared to non-responders (16.59 ± 1.83% vs. 28.67 ± 4.22%, p < 0.05). Decreased expression of CTLA-4 on naive CD8+ T cells was also observed in responders (18.05 ± 2.18% vs. 33.05 ± 4.75% p < 0.01). Additionally, effector memory CD8+ T cells expressed a lower level of CTLA-4 in responders (14.46 ± 1.55% vs. 23.93 ± 3.36%, p < 0.05).
Following cyclosporine treatment, the expression of CTLA-4 was significantly increased in responders, but the alteration was not observed in non-responders. As shown in Fig. 2D, the frequency of CTLA-4+ CD8+ T cells was significantly increased upon treatment (11.28 ± 2.73% vs. 33.94 ± 6.38%, p < 0.05) in responders. The increased expression of CTLA-4 on effector CD8+ T cells (13.80 ± 3.28% vs. 41.46 ± 6.11%, p < 0.05) and activated CD8+ T cells (15.63 ± 3.57% vs. 44.76 ± 3.82%, p < 0.01) was also observed after cyclosporine (Fig. 2D). Furthermore, the expression of CTLA-4 on CD4+ T subsets in responders, including naive CD4+ T cells (37.20 ± 6.37% vs. 65.70 ± 2.76%, p < 0.05), CD4+ effector memory (29.20 ± 5.31% vs. 56.78 ± 4.77%, p < 0.05), CD4+ central memory (26.09 ± 5.87% vs. 56.74 ± 9.54%, p < 0.05), and CD4+ effector (36.24 ± 6.74% vs. 63.0 ± 3.34%, p < 0.05), was also significantly increased after the intervention of cyclosporine (Fig. 2E).
Collectively, our data demonstrates that the frequency of CTLA-4 on T cells is altered upon cyclosporine in responders, and an increased expression is correlated to cyclosporine response.
T cell trafficking mediator is linked to cyclosporine response in AA
S1P1 receptor signaling is correlated with various physiological processes including lymphocyte recirculation, their biology, and phenotype determination [21]. To explore whether the expression of S1P1 has influence on immune function of T cells in AA, we compared the expression of S1P1 on T cell subsets between healthy donors and AA. As expected, increased expression of S1P1 on naive CD4+ T cells (27.02 ± 2.35% vs. 10.48 ± 6.24%, p < 0.05) and naive CD8+ T cells (27.02 ± 2.35% vs. 10.48 ± 6.24%, p < 0.05) was observed in AA (Fig. 3A). Central memory CD8+ T cells also showed an increased level of S1P1 in AA (24.71 ± 1.96% vs. 10.61 ± 5.55%, p < 0.05). Furthermore, expression difference of S1P1 on T helpers was also observed, and Th2 subset demonstrated a higher level of S1P1 (18.81 ± 1.78% vs. 7.85 ± 3.82%, p < 0.05).

Expression of S1P1 is correlated to clinical response. A The UMAP plots showed higher expression of sphingosine-1-phosphate-receptor 1 (S1P1) on naive T cells (left) and the higher percentage of S1P1 on naive CD4 and naive CD8 (right). Samples from 30 non-SAA patients and 4 healthy donors were tested. B Heatmap showed the difference of S1P1 on CD4 subsets between responders and non-responders (left); the expression of S1P1 was significantly lower in responders (right). Samples were collected from 15 responders and 15 non-responders. Data was shown as mean ± SEM; *p < 0.05
We also found that patients with a lower level of S1P1 on CD4+ T subsets at diagnosis seem to be more prone to achieve response after cyclosporine (Fig. 3B). As compared to non-responder patients, a significantly decreased level of S1P1 on naive CD4 was observed in responders (21.86 ± 2.67% vs. 32.18 ± 3.46%, p < 0.05). Furthermore, a significantly decreased level of S1P1 on CD4+ effector (20.03 ± 2.40% vs. 28.10 ± 3.23%, p < 0.05) and activated CD4+ T cells (23.70 ± 2.49% vs. 34.36 ± 3.49%, p < 0.05) was observed in responder patients. No significant difference of S1P1 expression on CD8 subsets was observed between two groups.
In summary, these results demonstrate that T cells, especially naive T cells, in AA had an increased level of S1P1, which is linked to the response to cyclosporine.
Expression of IL-9 is correlated to cyclosporine response in AA
IL-9/IL-9 receptor plays a dual effect on inflammation and autoimmune disease, including both pro-inflammatory and protective roles in occurrence of disease [36, 37]. We explored whether there was difference on the expression of IL-9 between healthy controls and AA patients. We found that patients displayed a significant higher level of IL-9 on naive CD4+ T cells (18.68 ± 2.62% vs. 7.73 ± 2.41%, p < 0.05, Fig. 4A). Furthermore, an increased frequency of IL-9 on Th2 cells was also found in AA patients (18.78 ± 2.36% vs. 7.64 ± 2.78%, p < 0.05, Fig. 4B). These results indicate that IL-9 may participate in the pathophysiology of AA.

Non-SAA has a higher level of IL-9 at diagnosis and non-responders have even higher IL-9 level. The UMAP plots showed higher frequency of interleukin-9 (IL-9) on naive CD4.+ T and Th2 in patients. Samples from 30 non-SAA patients and 4 healthy donors were tested. B The percentage of IL-9 on naive CD4 and Th2 was significantly higher in patients than HDs. C The UMAP plots showed the difference of IL-9 on Th1, Th2, and Th17 between responders and non-responders (left); columns showed a lower expression of IL-9 in responders (right). Samples were collected from 15 responders and 15 non-responders. Data was shown as mean ± SEM; *p < 0.05
To investigate the correlation between IL-9 expression prior treatment and cyclosporine response, we further analyzed the characteristics of IL-9 on naive T cells and T helper subsets. In responders, naive CD4+ T cells showed a significantly lower expression of IL-9 as compared to non-responder patients (24.52 ± 2.97% vs. 40.42 ± 4.80%, p < 0.05). Of note, the percentage of IL-9+ Th1 cells and IL-9+ Th2 cells in responders was 25.64 ± 2.67% and 13.55 ± 1.64%, respectively, which was significantly lower compared with non-responders (39.35 ± 4.55% and 24.00 ± 4.07%, p < 0.05). Moreover, Th17 clusters expressed lower expression of IL-9 in responders (17.30 ± 2.18% vs. 29.13 ± 3.88%, p < 0.05, Fig. 4C). This encouraging finding suggests a significant association between IL-9 expression and clinical response to cyclosporine.
Discussion
Abnormal T cell subsets and function play a critical role on the occurrence and progression of AA. Kordasti et al. tested 63 patients with AA and have shown that patients had an increased frequency of Th1 and Th2 compared to healthy donors. They also found that the absolute counts of Th2 and Th17 were higher in SAA [16]. Shi et al. detected Tregs in 55 AA patients, which included 27 of SAA, and demonstrated a decreased number and impaired functions of Tregs [17]. It has been reported that frequencies of some T cell subsets were age-related and the disease severity of AA might have a different pathogenesis with different immunological signatures, where very severe or long-lasting diseases might have a Th17 signature while severe or new-onset AA a Th1 profile [23, 38]. Therefore, due to the heterogeneity of group and disease severity of non-SAA, our study did not demonstrate significant difference on T cell subpopulations between patients and healthy individuals. But the frequency of Tregs is in inclined to be decreased in non-SAA. This result provided new evidence that the reduction of Tregs plays a critical role in the pathogenic mechanism of AA.
A variety of T cell immune checkpoint molecules have been identified to regulate T cell activation; increased TIM-3 and PD-1 have been observed in AA [18,19,20]. Although no obvious differences on TIM3 and PD-1 were found, an increased expression of CTLA-4 was shown. CTLA-4 belongs to the CD28 family; the expression and function of this molecule are intrinsically correlated with T cell activation. In autoimmunity, it can be targeted to curb the over-exuberant immune response [20, 39]. It has also been reported that the expression of CTLA-4 on Tregs in SAA was higher than that in healthy controls [40,41,42], but its expression on effector T cells is still unclear. We demonstrated that patients with AA had an increased expression of CTLA-4 on naive CD8+ T and central memory CD8+ T cells, whereas no significant difference was observed on effector T cells. This result indicates that CD8+ T cells in patients were undergoing an abnormal effector function, which may aggravate injury to hematopoietic stem progenitor cells. Furthermore, a higher level of CLTA-4 predicted a worse response to cyclosporine, and responders displayed significant increment in CTLA-4 expression following cyclosporine. Thus, we conclude that patients with lower expression of CTLA-4 are more sensitive to immunosuppression and increased CTLA-4 may play a synergistic effect to inhibitor dysfunctional T cells.
T cell circulation among secondary lymphoid organs was guided by the chemoattractant S1P. The S1P1 receptor regulates the emigration of T cells out of the thymus and lymphoid organs [43]. Furthermore, the S1P signaling pathway maintains the mitochondrial content of naive T cells, supplying cells with energy for their constant egress [44]. We discovered that the expression of S1P1 on naive T cells was higher in AA. It may be related to disrupted immune homeostasis, which would aggravate hematopoiesis injury. Moreover, patients with higher expression of S1P1 on effector and activated CD4+ T cells was resistant to achieve response, which may be due to hyperimmune condition. It has been reported that S1P1 antagonist could inhibit progress of chronic colitis [45]. Considering that, the inhibition of S1P1 may also be considered a promising choice for AA patients.
As the expression of IL-9 was connected with the expansion of Th2 subsets, it was originally identified as a Th2-type cytokine. In recent years, Th9 has been verified to be an important population of Th cells to produce IL-9. This special population is considered to be less exhausted, fully cytolytic, and hyperproliferative [46, 47]. Recent studies have shown that Th9 population and IL-9 are closely correlated with several autoimmune disorders, including systemic sclerosis, inflammatory bowel diseases, and experimental autoimmune encephalitis [25, 48,49,50]. In addition to Th9 and Th2 cells, other cell types are also able to produce IL-9, such as Th17, natural killer T cells, and innate lymphoid cell type 2 [51]. Under the conditions of Th9 differentiation, cytotoxic CD8+ T cells can also polarize into IL-9-producing cytotoxic CD8+T cells [48]. We found a significantly higher level of IL-9 in AA than in healthy individuals, which indicates that IL-9 may play an important role in T cell dysfunction. Although it has been reported that IL-9 could activate STAT3 and STAT5 to promote Treg survival and enhance Treg-mediated suppression [52], no significantly higher expression of IL-9 on Tregs was shown in patients, which may be related to impaired function of Tregs in AA. Furthermore, in our study, patients with a lower level of IL-9 showed better response to cyclosporine, suggesting that IL-9 expression could act as a predictor for overall response. Evidence from animal studies had shown that IL-9 deficiency or inhibition of IL-9 was correlated with decreased inflammatory cytokines and attenuated inflammatory condition [53, 54]. Thus, IL-9 may be a potential therapeutic target for braking T cell dysfunction in AA.
In summary, we showed the differences on T cell immune signature between AA and healthy individuals. More importantly, we observed that cyclosporine restored the expression of CTLA-4 on T cells in responders. A lower level of S1P1 and IL-9 predicts a better response. Evaluation of targeting these T cell markers should be widely taken into consideration for the optimal management of AA taken into the future. Further studies focusing on immune checkpoint molecules, T cell trafficking, and secretion of IL-9 could provide us more information to do decision-making of cyclosporine treatment strategy.
Data availability
The data that support the findings of this study are available on request from the corresponding author [Jun Shi].
References
Brodsky RA, Jones RJ (2005) Aplastic anaemia. The Lancet 365(9471):1647–1656. https://doi.org/10.1016/s0140-6736(05)66515-4
Young NS, Calado RT, Scheinberg P (2006) Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 108(8):2509–2519. https://doi.org/10.1182/blood-2006-03-010777
Young NS, Scheinberg P, Calado RT (2008) Aplastic anemia. Curr Opin Hematol 15(3):162–168. https://doi.org/10.1097/MOH.0b013e3282fa7470
Camitta BM, Rappeport JM, Parkman R, Nathan DG (1975) Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood 45(3):355–363
Bacigalupo A, Hows J, Gluckman E, Nissen C, Marsh J, Van Lint MT, Congiu M, De Planque MM, Ernst P, McCann S et al (1988) Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol 70(2):177–182. https://doi.org/10.1111/j.1365-2141.1988.tb02460.x
Nishio N, Yagasaki H, Takahashi Y, Muramatsu H, Hama A, Yoshida N, Kudo K, Kojima S (2009) Natural history of transfusion-independent non-severe aplastic anemia in children. Int J Hematol 89(4):409–413. https://doi.org/10.1007/s12185-009-0302-9
Kwon JH, Kim I, Lee YG, Koh Y, Park HC, Song EY, Kim HK, Yoon SS, Lee DS, Park SS, Shin HY, Park S, Park MH, Ahn HS, Kim BK (2010) Clinical course of non-severe aplastic anemia in adults. Int J Hematol 91(5):770–775. https://doi.org/10.1007/s12185-010-0601-1
Young NS (2018) Aplastic anemia. N Engl J Med 379(17):1643–1656. https://doi.org/10.1056/NEJMra1413485
Schrezenmeier H, Schlander M, Raghavachar A (1992) Cyclosporin A in aplastic anemia–report of a workshop. Ann Hematol 65(1):33–36. https://doi.org/10.1007/bf01715123
Scheinberg P, Young NS (2012) How I treat acquired aplastic anemia. Blood 120(6):1185–1196. https://doi.org/10.1182/blood-2011-12-274019
Liu C, Sun Y, Shao Z (2019) Current concepts of the pathogenesis of aplastic anemia. Curr Pharm Des 25(3):236–241. https://doi.org/10.2174/1381612825666190313113601
Giannakoulas NC, Karakantza M, Theodorou GL, Pagoni M, Galanopoulos A, Kakagianni T, Kouraklis-Symeonidis A, Matsouka P, Maniatis A, Zoumbos NC (2004) Clinical relevance of balance between type 1 and type 2 immune responses of lymphocyte subpopulations in aplastic anaemia patients. Br J Haematol 124(1):97–105. https://doi.org/10.1046/j.1365-2141.2003.04729.x
Solomou EE, Keyvanfar K, Young NS (2006) T-bet, a Th1 transcription factor, is up-regulated in T cells from patients with aplastic anemia. Blood 107(10):3983–3991. https://doi.org/10.1182/blood-2005-10-4201
de Latour RP, Visconte V, Takaku T, Wu C, Erie AJ, Sarcon AK, Desierto MJ, Scheinberg P, Keyvanfar K, Nunez O, Chen J, Young NS (2010) Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood 116(20):4175–4184. https://doi.org/10.1182/blood-2010-01-266098
Solomou EE, Rezvani K, Mielke S, Malide D, Keyvanfar K, Visconte V, Kajigaya S, Barrett AJ, Young NS (2007) Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood 110(5):1603–1606. https://doi.org/10.1182/blood-2007-01-066258
Kordasti S, Marsh J, Al-Khan S, Jiang J, Smith A, Mohamedali A, Abellan PP, Veen C, Costantini B, Kulasekararaj AG, Benson-Quarm N, Seidl T, Mian SA, Farzaneh F, Mufti GJ (2012) Functional characterization of CD4+ T cells in aplastic anemia. Blood 119(9):2033–2043. https://doi.org/10.1182/blood-2011-08-368308
Shi J, Ge M, Lu S, Li X, Shao Y, Huang J, Huang Z, Zhang J, Nie N, Zheng Y (2012) Intrinsic impairment of CD4(+)CD25(+) regulatory T cells in acquired aplastic anemia. Blood 120(8):1624–1632. https://doi.org/10.1182/blood-2011-11-390708
Zhang J, Gu Y, Xu C, Qu X (2008) Increased T cell immunoglobulin mucin-3 and its ligand in acquired aplastic anemia. Eur J Haematol 81(2):130–139. https://doi.org/10.1111/j.1600-0609.2008.01095.x
Zhao W, Zhang Y, Zhang P, Yang J, Zhang L, He A, Zhang W, Hideto T (2017) High programmed death 1 expression on T cells in aplastic anemia. Immunol Lett 183:44–51. https://doi.org/10.1016/j.imlet.2017.01.016
Salik B, Smyth MJ, Nakamura K (2020) Targeting immune checkpoints in hematological malignancies. J Hematol Oncol 13(1):111. https://doi.org/10.1186/s13045-020-00947-6
Garris CS, Blaho VA, Hla T, Han MH (2014) Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology 142(3):347–353. https://doi.org/10.1111/imm.12272
Li Y, Wang F, Guo R, Zhang Y, Chen D, Li X, Tian W, Xie X, Jiang Z (2019) Exosomal sphingosine 1-phosphate secreted by mesenchymal stem cells regulated Treg/Th17 balance in aplastic anemia. IUBMB Life 71(9):1284–1292. https://doi.org/10.1002/iub.2035
Giudice V, Selleri C (2022) Aplastic anemia: pathophysiology. Semin Hematol 59(1):13–20. https://doi.org/10.1053/j.seminhematol.2021.12.002
Holbrook ST, Ohls RK, Schibler KR, Yang YC, Christensen RD (1991) Effect of interleukin-9 on clonogenic maturation and cell-cycle status of fetal and adult hematopoietic progenitors. Blood 77(10):2129–2134
Goswami R, Kaplan MH (2011) A brief history of IL-9. J Immunol 186(6):3283–3288. https://doi.org/10.4049/jimmunol.1003049
Shao Q, Wang Y, Liu Z, Liu H, Wang Y, Zhao Y, Li L, Fu R (2020) Th9 cells in peripheral blood increased in patients with immune-related pancytopenia. J Immunol Res 2020:6503539. https://doi.org/10.1155/2020/6503539
Javan MR, Saki N, Moghimian-Boroujeni B (2021) Aplastic anemia, cellular and molecular aspects. Cell Biol Int 45(12):2395–2402. https://doi.org/10.1002/cbin.11689
Patel BA, Giudice V, Young NS (2021) Immunologic effects on the haematopoietic stem cell in marrow failure. Best Pract Res Clin Haematol 34(2):101276. https://doi.org/10.1016/j.beha.2021.101276
Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, Hillmen P, Ireland R, Kulasekararaj A, Mufti G, Snowden JA, Samarasinghe S, Wood A, Marsh JC, British Society for Standards in H (2016) Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol 172(2):187–207. https://doi.org/10.1111/bjh.13853
Olnes MJ, Scheinberg P, Calvo KR, Desmond R, Tang Y, Dumitriu B, Parikh AR, Soto S, Biancotto A, Feng X, Lozier J, Wu CO, Young NS, Dunbar CE (2012) Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 367(1):11–19. https://doi.org/10.1056/NEJMoa1200931
Lengline E, Drenou B, Peterlin P, Tournilhac O, Abraham J, Berceanu A, Dupriez B, Guillerm G, Raffoux E, de Fontbrune FS, Ades L, Balsat M, Chaoui D, Coppo P, Corm S, Leblanc T, Maillard N, Terriou L, Socié G, de Latour RP (2018) Nationwide survey on the use of eltrombopag in patients with severe aplastic anemia: a report on behalf of the French Reference Center for Aplastic Anemia. Haematologica 103(2):212–220. https://doi.org/10.3324/haematol.2017.176339
Maecker HT, McCoy JP, Nussenblatt R (2012) Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol 12(3):191–200. https://doi.org/10.1038/nri3158
Li G, Yang Q, Zhu Y, Wang HR, Chen X, Zhang X, Lu B (2013) T-bet and Eomes regulate the balance between the effector/central memory T cells versus memory stem like T cells. PLoS One 8(6):e67401. https://doi.org/10.1371/journal.pone.0067401
McLane LM, Banerjee PP, Cosma GL, Makedonas G, Wherry EJ, Orange JS, Betts MR (2013) Differential localization of T-bet and Eomes in CD8 T cell memory populations. J Immunol 190(7):3207–3215. https://doi.org/10.4049/jimmunol.1201556
Wu H, Miao M, Zhang G, Hu Y, Ming Z, Zhang X (2009) Soluble PD-1 is associated with aberrant regulation of T cells activation in aplastic anemia. Immunol Invest 38(5):408–421. https://doi.org/10.1080/08820130902912332
Chakraborty S, Kubatzky KF, Mitra DK (2019) An update on interleukin-9: from its cellular source and signal transduction to its role in immunopathogenesis. Int J Mol Sci 20(9):2113. https://doi.org/10.3390/ijms20092113
Yazdani R, Shapoori S, Rezaeepoor M, Sanaei R, Ganjalikhani-Hakemi M, Azizi G, Rae W, Aghamohammadi A, Rezaei N (2019) Features and roles of T helper 9 cells and interleukin 9 in immunological diseases. Allergol Immunopathol (Madr) 47(1):90–104. https://doi.org/10.1016/j.aller.2018.02.003
Giudice V, Feng X, Lin Z, Hu W, Zhang F, Qiao W, Ibanez M, Rios O, Young NS (2018) Deep sequencing and flow cytometric characterization of expanded effector memory CD8(+)CD57(+) T cells frequently reveals T-cell receptor Vβ oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica 103(5):759–769. https://doi.org/10.3324/haematol.2017.176701
Edner NM, Carlesso G, Rush JS, Walker LSK (2020) Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov 19(12):860–883. https://doi.org/10.1038/s41573-020-0081-9
Yan L, Fu R, Liu H, Wang H, Liu C, Wang T, Qi W, Guan J, Li L, Shao Z (2015) Abnormal quantity and function of regulatory T cells in peripheral blood of patients with severe aplastic anemia. Cell Immunol 296(2):95–105. https://doi.org/10.1016/j.cellimm.2015.04.001
Liu B, Shao Y, Liang X, Lu D, Yan L, Churov A, Fu R (2020) CTLA-4 and HLA-DQ are key molecules in the regulation of mDC-mediated cellular immunity by Tregs in severe aplastic anemia. J Clin Lab Anal 34(10):e23443. https://doi.org/10.1002/jcla.23443
Huang G, Zhang Y, Wei X, Yu Z, Lai J, Shen Q, Chen X, Tan G, Chen C, Luo W, Li Y, Zhou M, Li Y, Li B (2021) CD8(+)GITR(+) T cells may negatively regulate T cell overactivation in aplastic anemia. Immunol Invest 50(4):406–415. https://doi.org/10.1080/08820139.2020.1770785
Cyster JG, Schwab SR (2012) Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30:69–94. https://doi.org/10.1146/annurev-immunol-020711-075011
Mendoza A, Fang V, Chen C, Serasinghe M, Verma A, Muller J, Chaluvadi VS, Dustin ML, Hla T, Elemento O, Chipuk JE, Schwab SR (2017) Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature 546(7656):158–161. https://doi.org/10.1038/nature22352
Shimano K, Maeda Y, Kataoka H, Murase M, Mochizuki S, Utsumi H, Oshita K, Sugahara K (2019) Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 functional antagonist, inhibits progress of chronic colitis induced by transfer of CD4+CD45RBhigh T cells. PLoS One 14(12):e0226154. https://doi.org/10.1371/journal.pone.0226154
Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, Qian J, Dong C, Yi Q (2018) Th9 cells represent a unique subset of CD4(+) T cells endowed with the ability to eradicate advanced tumors. Cancer Cell 33(6):1048-1060.e1047. https://doi.org/10.1016/j.ccell.2018.05.004
Angkasekwinai P, Dong C (2021) IL-9-producing T cells: potential players in allergy and cancer. Nat Rev Immunol 21(1):37–48. https://doi.org/10.1038/s41577-020-0396-0
Noelle RJ, Nowak EC (2010) Cellular sources and immune functions of interleukin-9. Nat Rev Immunol 10(10):683–687. https://doi.org/10.1038/nri2848
Pan HF, Leng RX, Li XP, Zheng SG, Ye DQ (2013) Targeting T-helper 9 cells and interleukin-9 in autoimmune diseases. Cytokine Growth Factor Rev 24(6):515–522. https://doi.org/10.1016/j.cytogfr.2013.09.001
Kaplan MH, Hufford MM, Olson MR (2015) The development and in vivo function of T helper 9 cells. Nat Rev Immunol 15(5):295–307. https://doi.org/10.1038/nri3824
Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, Coyle AJ, Kasper LH, Noelle RJ (2009) IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med 206(8):1653–1660. https://doi.org/10.1084/jem.20090246
Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, Bettelli E, Oukka M, van Snick J, Renauld JC, Kuchroo VK, Khoury SJ (2009) IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci USA 106(31):12885–12890. https://doi.org/10.1073/pnas.0812530106
Kung TT, Luo B, Crawley Y, Garlisi CG, Devito K, Minnicozzi M, Egan RW, Kreutner W, Chapman RW (2001) Effect of anti-mIL-9 antibody on the development of pulmonary inflammation and airway hyperresponsiveness in allergic mice. Am J Respir Cell Mol Biol 25(5):600–605. https://doi.org/10.1165/ajrcmb.25.5.4533
Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, Lehr HA, Wirtz S, Vieth M, Waisman A, Rosenbauer F, McKenzie AN, Weigmann B, Neurath MF (2014) TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol 15(7):676–686. https://doi.org/10.1038/ni.2920
Funding
This work was supported by the Tianjin Municipal Science and Technology Commission Major Project (18ZXDBSY00070), CAMS Innovation Fund for Medical Sciences (2021-I2M-1–073, 2021-I2M-040), and Ministry of Science and Technology of China (2018YFA0107801, 2020YFE0203000).
Author information
Authors and Affiliations
Contributions
LZ and JM performed the study and wrote the paper; JS and WY designed the study; YL, YC, and WL operated bone marrow samples; QL, HP, ZG, LF, and JZ collected essential clinical data. JS gave an approval of the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
277_2023_5097_MOESM1_ESM.pdf
Supplementary file1 (PDF 1219 KB) Supplementary Fig. 1. Clusters of T cell sub-populations in non-SAA patients (n = 30) and healthy donors (n = 4)
277_2023_5097_MOESM2_ESM.pdf
Supplementary file2 (PDF 344 KB) Supplementary Fig. 2. T cell subsets between responders and non-responders. (A) Frequencies of T cell subpopulations of CD8+ T cells, and (B) of CD4+ T cells. Samples from 30 non-SAA patients and 4 healthy donors were tested. Data are shown as box plots extending from the 25th to 75th percentiles, and the whiskers from the minimum to the maximum point; middle line represents the median
277_2023_5097_MOESM3_ESM.pdf
Supplementary file3 (PDF 1305 KB) Supplementary Fig. 3. No difference on Calcineurin-NFAT signaling was found between responders and non-responders. (A) UMAP plots showed the expression of NFAT2, NFAT4 and Calcineurin on CD3+ T cells in patients. (B) The violin plots showed frequencies of NFAT2, NFAT4 and Calcineurin on CD3+ T. Samples were collected from 30 non-SAA patients and 4 healthy donors
277_2023_5097_MOESM4_ESM.pdf
Supplementary file4 (PDF 236 KB) Supplementary Fig. 4. Expression of checkpoint molecules on T subsets. Higher frequency of CTLA-4 on CD8 T cells in patients was displayed. And no difference of LAG-3, PD-1, TIGHT and TIM3 on T cell subsets was found. Samples from 30 non-SAA patients and 4 healthy donors were tested
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, L., Mao, J., Lian, Y. et al. Mass cytometry analysis identifies T cell immune signature of aplastic anemia and predicts the response to cyclosporine. Ann Hematol 102, 529–539 (2023). https://doi.org/10.1007/s00277-023-05097-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05097-6