
Abstract
Patients (pts) with polycythemia vera (PV) suffer from pruritus, night sweats, and other symptoms, as well as from thromboembolic complications and progression to post-PV myelofibrosis. Ruxolitinib (RUX) is approved for second-line therapy in high-risk PV pts with hydroxyurea intolerance or resistance. The RuxoBEAT trial (NCT02577926, registered on October 1, 2015, at clinicaltrials.gov) is a multicenter, open-label, two-arm phase-IIb trial with a target population of 380 pts with PV or ET, randomized to receive RUX or best available therapy. This pre-specified futility analysis assesses the early clinical benefit and tolerability of RUX in previously untreated PV pts (6-week cytoreduction was allowed). Twenty-eight patients were randomly assigned to receive RUX. Compared to baseline, after 6 months of treatment, there was a significant reduction of median hematocrit (46 to 41%), the median number of phlebotomies per year (4.0 to 0), and median patient-reported pruritus scores (2 to 1), and a trend for reduced night sweat scores (1.5 to 0). JAK2V617F allele burden, as part of the scientific research program, also significantly decreased. One hundred nine adverse events (AEs) occurred in 24/28 patients (all grade 1 to 3), and no pt permanently discontinued treatment because of AEs. Thus, treatment with ruxolitinib in untreated PV pts is feasible, well-tolerated, and efficient regarding the above-mentioned endpoints.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycythemia vera (PV) is a myeloproliferative neoplasm (MPN) that is caused by somatic gain-of-function mutations in the jak2 gene in hematopoietic stem and progenitor cells [1], most of which result in the JAK2V617F oncoprotein [1]. Survival of patients (pts) with PV is decreased compared with age-matched controls [2], and this is mainly due to thromboembolic complications, or, in a smaller proportion of pts, progression to post-PV myelofibrosis, or even acute leukemia [3]. Strikingly, essentially all pts with PV report symptoms when they are systematically evaluated, i.e., using the MPN symptom assessment form (MPN-SAF) [4]. The most common symptoms include fatigue (91.7% of pts), insomnia (68.1%), numbness (66.2%), itching/pruritus (65.0%), sad mood (65.0%), early satiety (62.1%), and concentration problems (61.2%) [4]. Night sweats and bone pain are also present in about half of all PV patients (57.4% and 47.5%, respectively) [4]. Overall, 85.5% of PV pts report decreased quality of life [4]. Importantly, most of these symptoms persist even during complete hematologic remission (CHR) [5].
Treatment of PV is mainly directed at preventing thrombotic/thromboembolic events [6], and, based upon randomized trials [7] [8], currently, ASA and phlebotomies with a target Hct of < 45% are recommended for all PV patients [6]. In addition, cytoreductive therapy with hydroxyurea (HU) or ropeginterferon-alpha (ropegIFNa) is recommended for “high-risk” pts with PV, i.e., those aged > 60 years and/or with a history of thrombosis/thromboembolism [6], based upon a post hoc propensity-matched analysis of HU treatment in the prospective ECLAP cohort [9] and the randomized phase 3 trial [10] of ropegIFNa vs. HU treatment in such high-risk pts.
Two randomized phase 3 trials [11] have evaluated the role of the JAK inhibitor ruxolitinib (RUX) in PV pts who were intolerant or resistant to HU treatment and either had (RESPONSE trial [11]) or did not have splenomegaly (RESPONSE-2 trial [12]). In both studies, RUX was compared with best available therapy (BAT), which consisted of continued HU therapy in most pts. The results showed that RUX was superior to BAT in controlling Hct and improving PV-related symptoms [11], and in reducing spleen volume in those pts with splenomegaly [11]. Of note, although the study was not designed to formally address reductions in thromboembolic events, the RESPONSE trial did find a decreased rate of thromboembolic complications in the RUX- vs. BAT-treated group [13]. In addition, a retrospective real-world analysis of pts with HU-intolerant or HU-resistant PV treated with ruxolitinib or BAT found significantly less arterial thrombotic events in the RUX-treated group [14]. In a double-blinded comparison to hydroxyurea in PV patients who had been treated with HU before but still suffered from symptoms, RUX did not achieve the primary endpoint (> = 50% improvement of symptoms from baseline at week 16 of treatment) (43.4 vs. 29.6%, p = 0.139), but showed significant benefit in reducing pruritus [15]. Nevertheless, whether RUX treatment is also safe and effective in pts with untreated PV is currently unknown.
We report here the results from the futility analysis of the PV cohort treated within the phase 2b clinical trial entitled “Ruxolitinib versus best available therapy in patients with high-risk polycythemia Vera or high-risk essential thrombocythemia” (Ruxo-BEAT; NCT02577926) to assess the efficacy and tolerability of ruxolitinib in untreated PV pts.
Methods
Ruxo-BEAT (NCT02577926) is a multicenter, open-label, two-arm phase 2b trial with a target population of 380 pts, divided into two strata of 190 untreated PV (a maximum of previous 6 weeks of HU, anagrelide, or interferon therapy was allowed) and 190 untreated or pretreated ET pts. All patients provided written informed consent, and the study was approved by the central ethics committee at RWTH Aachen University Hospital (EK 345/14) and at the local ethics committee of each center. The most relevant inclusion criteria were an age of 18 years of age or older, ECOG performance status of ≤ 2, a diagnosis of either PV or ET according to the WHO 2008 classification. Moreover, PV pts and ET pts had to be classified as high risk according to defined criteria as outlined below: for patients with high-risk PV or PV with indication for cytoreductive therapy due to progressive myeloproliferation, any one of the following had to be fulfilled ([16,17,18]): age > 60 years, previous documented thrombosis or thromboembolism, platelet count > 1500 × 109/L, poor tolerance of phlebotomy or frequent phlebotomy requirement, symptomatic or progressive splenomegaly, severe disease-related symptoms (according to the local investigator’s definition), or progressive leukocytosis with leukocyte counts > 20 × 109/L. The ET stratum of pts was not analyzed in the present futility analysis. Patients requiring prolonged use of oral corticosteroids with a dose of more than 20 mg per day (> 1 month) and patients with active splanchnic vein thrombosis within the last 3 months (included Budd-Chiari, portal vein, splenic and mesenteric thrombosis) were excluded. A detailed list of all in- and exclusion criteria can be found in the Supplementary Information.
Pts in each stratum were randomized in a 1:1 ratio to receive either RUX or best available therapy (BAT). Crossover from BAT to RUX was allowed in eligible patients after 6 months who did not achieve complete remission but still had sufficient hematologic reserves (platelet count ≥ 140 × 109/L, hemoglobin ≥ 10.0 g/dL, and ANC ≥ 1.5 × 109/L). Patients with PV in the RUX arm received a starting dose of 10 mg bid, which were allowed to be increased up to 20 mg bid due to insufficient efficacy. BAT included monotherapies of all commonly used medications (HU, anagrelide, interferon alpha, or others). BAT treatment was allowed to be changed during the course of the study. However, these patients were censored for primary endpoint analysis. The primary endpoint was the rate of complete clinico-hematologic response (CHR) at month 6 as defined by Barosi et al. Blood 2009 [19]. Secondary endpoints included the use of phlebotomy (recorded as the number of phlebotomies for the year before baseline and for 6 months during therapy, the latter of which was then calculated per year to be comparable with the pre-study phlebotomies), spleen size, patient-reported outcomes on specific MPN-associated symptoms using the MPN-SAF-TSS form (see appendix of the protocol), and overall survival. The MPN-SAF-TSS form is an abbreviated form of the former MPN-SAF questionnaire (9 out of 17 items) with the addition of one item from the so-called BFI questionnaire. The chosen items were the most clinically relevant items (concentration, early satiety, inactivity, night sweats, itching, bone pain, abdominal discomfort, weight loss, and fever) and in the MPN-SAF-TSS these items score on a 0 (absent/as good as it can be) to 10 (worst imaginable/as bad as it can be) linear analog self-assessment scale [20]. In the German version included in the present study, three additional symptoms regarding microvascular disturbances were added as well as a question on overall quality of life, while the question on fatigue and inactivity were combined.
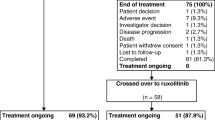
The current futility analysis of PV patients randomized to RUX, after 50 PV patients overall had been enrolled, was pre-specified in the trial protocol in order to exclude early failure or excessive toxicity. Of these 50 patients, 28 patients were randomized into the RUX arm. Closure of the PV subgroup was pre-specified if no favorable trend were observed for any of the following variables: (1) improvement (decrease) in the hematocrit level during 6 months of treatment or (2) improvement of one of the following three symptom variables assessed by physician’s judgement or via MPN-SAF during 6 months of treatment: pruritus, night sweats, or bone pain. Furthermore, the change in JAK2V617F allele burden during the first 6 months of treatment was analyzed as part of the scientific research program. Differences between screening (Hct) or baseline (all other variables) and end of month 6 (EOM6) (all variables) were calculated using McNemar’s test (for physician-assessed pruritus and night sweats) or the Wilcoxon signed rank test (all other variables). Probability values of < 0.05 were considered significant.
Results
Baseline variables
Twenty-eight pts with untreated PV were included in this analysis. Baseline data of these pts are listed in Table 1. Median time from the first diagnosis to initiation of treatment in the context of the RuxoBEAT trial was 7.6 months (IQR: 1.5–29.2 weeks). The most common indications for treatment were age over 60 years and previously documented thrombosis or thromboembolism (each in 13 patients).
Efficacy
At the time of data lock, 28 patients had received RUX for at least 6 months. Median time on drug during this first 6 months of treatment was 173 days (range 151–189). Efficacy variables at 6 months were assessed and compared to the screening/baseline variables, as pre-specified in the protocol for the futility analysis.
After 6 months, the median hematocrit level decreased from 46% (no missing values; range 34.6–61%) (IQR 42.98–47.08%) to 41.45% (no missing values; range 32–52%) (IQR 37.25–44.95%) (p = 0.000957) (Fig. 1). The median number of phlebotomies performed per year decreased from 4.0 (no missing values; IQR 1.00–6.00) to 0 (missing = 3; IQR 0–2.0) (p = 0.000493) (Fig. 1). Median JAK2V617F allele burden decreased from 44% (missing = 5; range 16–93%) to 34% (missing = 5; range 9–91%) (p = 0.000973). The percentage of patients with pruritus or night sweats, as assessed by the physician, decreased from 36 to 14% (no missing values; p = 0.07), and from 29 to 11% (no missing values; p = 0.063), respectively. Patient-reported outcome points on the MPN-SAF survey for pruritus decreased from median 2 (n = 26; missing = 2; range 0–10) (IQR 0–5) to 1 (range 0–5) (IQR 0–2) (p = 0.006), and there was a tendency for reduction of night sweat points (from median 1.5 (missing = 2; range 0–10) (IQR 0–5.25) to 0 (no missing values; range 0–7) (IQR 0–2.75); p = 0.101), while the points for bone pain remained unaltered (median 0 (missing = 2; range 0–10) (IQR 0–4) to 1 (no missing values; range 0–9) (IQR 0–2); p = 0.343) (Fig. 2). The median MPN symptom score (mean of all point values from the 12 symptom-oriented questions) decreased from a median value of 1.5 (missing = 2; range 0.09–7) (IQR 0.87–3.0) to a median value of 1.14 (no missing values; range 0–6) (IQR 0.48–2.55) after 6 months of therapy (p = 0.061).

Hematocrit levels and frequency of phlebotomies over time. (Left) Hematocrit (Hct) was assessed at screening and at 6 months of RUX treatment (EOM6) and decreased from 46% (no missing values; range 34.6–61%) (IQR 42.98–47.08%) to 41.45% (no missing values; range 32–52%) (IQR 37.25–44.95%) (p = 0.000957). (Right) The number of phlebotomies (PHL) was assessed during 12 months prior to baseline (BL) and during 6 months of treatment (EOM6). In order to be able to compare both time points, the number of PHL was doubled to calculate the number per 12 months. The median number of phlebotomies performed per year decreased from 4.0 (no missing values; IQR 1.00–6.00) to 0 (missing = 3; IQR 0–2.0) (p = 0.000493)

MPN-SAF symptom scores for pruritus, night sweats, and bone pain over time. Three symptoms (pruritus, night sweats, and bone pain) were assessed in patients at baseline (BL) and after 6 months of RUX treatment (EOM6), using the MPN-SAF questionnaire. The score for each individual patient is depicted. The median scores for pruritus decreased from median 2 (n = 26; missing = 2; range 0–10) (IQR 0–5) to 1 (range 0–5) (IQR 0–2) (p = 0.006), and a tendency for reduction was seen for the mean score of night sweats ((from median 1.5 (missing = 2; range 0–10) (IQR 0–5.25) to 0 (no missing values; range 0–7) (IQR 0–2.75); p = 0.101), while the mean score for bone pain remained unaltered (median 0 (missing = 2; range 0–10) (IQR 0–4) to 1 (no missing values; range 0–9) (IQR 0–2); p = 0.343)
Safety
During the first 6 months of therapy, 109 adverse events occurred in 24/28 patients. All of these AEs were grade 1 to 3, and no grade 4 or 5 events occurred. Eleven patients experienced at least one grade 3 AE, eight patients only grade 1 and 2 events, and five patients only experienced grade 1 events. Patients had a median of 3.0 events during the first 6 months of treatment (including the 4 without any such AEs). Of all 109 events, only 12 (11%) were grade 3, 31 (28%) were grade 2, and the majority (66/109 = 61%) were grade 1. Frequencies of AEs that occurred in more than 10% of the patients can be found in Table 2 (using system organ classes according to MedDRA level 14). The most frequent AE were general (fatigue), respiratory, dermatologic, renal and urinary, gastrointestinal, nervous system, cardiovascular disorders, infections, or musculoskeletal, connective tissue, or metabolic disorders (Table 2). Grade 3 AEs included two cases of liver enzyme increase, and one case each of compression fracture of spine with spinal canal stenosis, creatinine phosphokinase elevation, suspected coronary syndrome, urinary retention, herpes zoster, hot flashes, choroidal melanoma, macula edema, diabetic retinopathy, and hypertension.
Fifty of all 109 AEs were deemed not to be related to the study treatment according to the treating physician. Relationship to ruxolitinib in the remaining adverse events was determined to be related (n = 11), likely (n = 3), possible (n = 35), or unlikely (n = 10). AEs were more likely to occur early during the course of treatment, with 68.8% of all events occurring during the first 3 months of therapy.
Only two patients had to interrupt treatment because of AEs (one case of irritability, one case of back pain). Both of them were re-exposed and subsequently remained on drug treatment without any dose reduction. Four dose reductions were due to AEs (one case each of elevation of transaminases, thoracic pain, hypoglycemia, and headache). No patient permanently discontinued treatment during 6 months of therapy.
Eight severe adverse events (SAEs) occurred in five patients. However, only one SAE was interpreted as clearly related to ruxolitinib (herpes zoster), another case was considered possibly related (thoracic pain), while all other SAEs were considered to be unrelated according to the treating physician.
Disposition on treatment
All 28 patients began to study medication at the correct dose (10 mg BID). Within the first 6 months of study treatment, there were a total of seven dose reductions in six patients; two patients interrupted treatment because of AEs (see above). In addition, 11 dose escalations were reported in these patients.
None of the analyzed 28 randomized PV patients terminated the study within the first 6 months. There were no recorded protocol violations concerning informed consent, inclusion/exclusion criteria, or randomization.
Discussion
In this futility analysis, we analyzed interim data to decide whether enrollment of pts with untreated PV into the randomized Ruxo-BEAT trial is safe and effective. The futility analysis showed significant improvements for Hct, JAK2V617F allele burden, and pruritus. Moreover, there was a significant reduction in the number of phlebotomies. Consequently, the trial was recommended to be continued by the Data Monitoring Committee.
RUX treatment was well-accepted by patients, as indicated by the lack of dropouts in the first 6 months of treatment among the patients analyzed so far. Also, there were no new safety signals of adverse events of RUX treatment, when compared to those already described in the RESPONSE [21] and RESPONSE-2 [12] trials and the trials involving pts with myelofibrosis [22,23,24]. Importantly, no grade 4 or 5 AEs occurred. The treating physicians rated 54% of the AEs as unrelated or unlikely related to RUX. Of eight SAEs, only two were deemed definitely or possibly related to RUX (herpes zoster and thoracic pain). Thus, RUX treatment was well-tolerated in pts with previously untreated PV. Given that a short period (6 weeks) of prior treatment with HU, anagrelide, or interferon was allowed, this might have had an influence on the efficacy. However, only 25%, 0%, and 0% of pts, respectively, received such treatment. And if these patients were excluded from the analysis, there was still a significant improvement of Hct (from median 46 to 40.6%, p = 0.002; no missing values) and JAK2V617F allele burden (from median 74.5 to 61.5%, p = 0.000973; missing n = 5) in the remainder of the pts.
Sustained control of hematocrit levels is of major importance in PV pts, in order to decrease cardiovascular mortality (death from cardiovascular reasons or major thrombotic events) [8]. Our results show that RUX is effective in reducing Hct levels, thereby potentially decreasing the risk for thromboembolic events (the 6-month endpoint was too early for thromboembolic event assessment) and that this was possible without phlebotomy support. This might also have a positive influence on the quality of life of the patients, given that patients requiring frequent phlebotomies may suffer from phlebotomy-induced symptomatic iron deficiency.
The continuous measurement of JAK2 allele burden during therapy is an interesting means of non-invasive disease monitoring, albeit not yet clinical routine, as the consequences of the results and guidelines on how to act on changes are still lacking. In the RESPONSE trial, significant decreases in JAK2 allele burden due to RUX treatment were reported [25], and pts with a more pronounced JAK2 allele burden reduction tended to show stronger spleen volume reductions [25]. However, no correlations with other clinical outcome variables were described. In our study, it will be interesting to analyze the correlations of the JAK2 allele burden with relevant clinical outcome data (such as thromboembolic complications or transformation to post-PV MF), given that the PV pts in this trial were mostly untreated and had been enrolled at an earlier disease timepoint, possibly rendering them more susceptible to JAKi treatment than the pts enrolled in the RESPONSE trials.
RUX significantly reduced pruritus in our cohort. This is an important finding, since pruritus in PV may severely reduce the quality of life and is often not sufficiently treated by other drugs [26]. Moreover, pruritus was present in more than 20% of ropeginterferon-treated pts [27]. Furthermore, in our trial, RUX also showed a strong trend towards reduction of patient-reported night sweats, while patient-reported bone pain, an additional common symptom in PV pts [4], scores remained unaltered. In the RESPONSE trial publication, this symptom has not been reported separately [21], although the MPN-SAF including this item was used in this trial. In pts with myelofibrosis, RUX did lead to a significant reduction in bone pain compared to placebo in the COMFORT-I-trial [22].
This pre-specified analysis has certain limitations: due to the short time of RUX treatment and the low patient number, we have not yet studied whether there was any effect on the rate of thromboembolism. This will be important to analyze at later time points during the trial. Also, more abundant information on complete blood counts and symptoms will then need to be analyzed. Finally, since it is important to assess how RUX treatment compares to BAT in this cohort of untreated PV pts, a comparative analysis will be performed in the interim analysis and the final analysis of the trial.
First-line treatment of PV is rapidly evolving, particularly with the recent approval of ropeginterferon, and it will be interesting to evaluate whether RUX proves beneficial in the first-line setting. Also, the question of optimal second-line treatment after ropeginterferon is currently unclear but may involve RUX treatment. Finally, while cytoreductive treatment in PV is not currently recommended for low-risk PV pts, Barbui and colleagues [28] have raised the question if also low-risk PV pts might benefit from early intervention. The results from the interim analysis of the randomized “LOW-PV” clinical trial, which compares standard treatment (phlebotomies and low-dose ASA) with standard treatment plus ropegIFNa in pts with low-risk PV, showed a significant benefit regarding the primary endpoint (defined as maintenance of the median hct values of ≤ 45% without progressive disease during a 12-month period) for the ropegIFNa group. Due to the superiority of the ropegIFNa arm, recruitment had to be stopped prematurely, and the 2-year-follow-up data are eagerly being awaited. Possibly, if the trial remains positive, pts with low-risk PV will soon be recommended to receive early cytoreductive treatment, and this may invoke the question of whether to give RUX or HU in the second-line setting in low-risk PV pts or, alternatively, to expand our study to low-risk pts.
In conclusion, the futility analysis of the RuxoBEAT trial confirms that treatment with RUX in untreated PV pts is effective (regarding the above-mentioned endpoints), feasible, and well-tolerated. The trial is currently ongoing.
Data availability
Correspondence and requests for materials should be addressed to Steffen Koschmieder.
References
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434(7037):1144–1148
Hultcrantz M, Kristinsson SY, Andersson TM, Landgren O, Eloranta S, Derolf AR, Dickman PW, Bjorkholm M (2012) Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 30(24):2995–3001. https://doi.org/10.1200/JCO.2012.42.1925
Hultcrantz M, Wilkes SR, Kristinsson SY, Andersson TM, Derolf AR, Eloranta S, Samuelsson J, Landgren O, Dickman PW, Lambert PC, Bjorkholm M (2015) Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in Sweden between 1973 and 2005: a population-based study. J Clin Oncol 33(20):2288–2295. https://doi.org/10.1200/JCO.2014.57.6652
Scherber R, Dueck AC, Johansson P, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferarri ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Harrison CN, Radia D, Mesa RA (2011) The myeloproliferative neoplasm symptom assessment form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 118(2):401–408. https://doi.org/10.1182/blood-2011-01-328955
Grunwald MR, Burke JM, Kuter DJ, Gerds AT, Stein B, Walshauser MA, Parasuraman S, Colucci P, Paranagama D, Savona MR, Mesa R (2019) Symptom burden and blood counts in patients with polycythemia vera in the United States: an analysis from the REVEAL study. Clin Lymphoma Myeloma Leuk 19(9):579-584 e571. https://doi.org/10.1016/j.clml.2019.06.001
Barbui T, Tefferi A, Vannucchi AM, Passamonti F, Silver RT, Hoffman R, Verstovsek S, Mesa R, Kiladjian JJ, Hehlmann R, Reiter A, Cervantes F, Harrison C, Mc Mullin MF, Hasselbalch HC, Koschmieder S, Marchetti M, Bacigalupo A, Finazzi G, Kroeger N, Griesshammer M, Birgegard G, Barosi G (2018) Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia 32(5):1057–1069. https://doi.org/10.1038/s41375-018-0077-1
Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, Barbui T, European collaboration on low-dose aspirin in polycythemia vera I (2004) Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 350(2):114–124
Marchioli R, Finazzi G, Specchia G, Cacciola R, Cavazzina R, Cilloni D, De Stefano V, Elli E, Iurlo A, Latagliata R, Lunghi F, Lunghi M, Marfisi RM, Musto P, Masciulli A, Musolino C, Cascavilla N, Quarta G, Randi ML, Rapezzi D, Ruggeri M, Rumi E, Scortechini AR, Santini S, Scarano M, Siragusa S, Spadea A, Tieghi A, Angelucci E, Visani G, Vannucchi AM, Barbui T, Group C-PC (2013) Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 368(1):22–33. https://doi.org/10.1056/NEJMoa1208500
Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G, Barbui T (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23(10):2224–2232
Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, Rossiev V, Dulicek P, Illes A, Pylypenko H, Sivcheva L, Mayer J, Yablokova V, Krejcy K, Grohmann-Izay B, Hasselbalch HC, Kralovics R, Kiladjian JJ, Group P-PS (2020) Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol 7(3):e196–e208
Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Mesa R, He S, Jones MM, Garrett W, Li J, Pirron U, Habr D, Verstovsek S (2015) Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 372(5):426–435. https://doi.org/10.1056/NEJMoa1409002
Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, Callum J, Vannucchi AM, Sivgin S, Bensasson C, Khan M, Mounedji N, Saydam G (2017) Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 18(1):88–99. https://doi.org/10.1016/S1470-2045(16)30558-7
Kiladjian JJ, Zachee P, Hino M, Pane F, Masszi T, Harrison CN, Mesa R, Miller CB, Passamonti F, Durrant S, Griesshammer M, Kirito K, Besses C, Moiraghi B, Rumi E, Rosti V, Blau IW, Francillard N, Dong T, Wroclawska M, Vannucchi AM, Verstovsek S (2020) Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol 7(3):e226–e237. https://doi.org/10.1016/S2352-3026(19)30207-8
Alvarez-Larran A, Garrote M, Ferrer-Marin F, Perez-Encinas M, Mata-Vazquez MI, Bellosillo B, Arellano-Rodrigo E, Gomez M, Garcia R, Garcia-Gutierrez V, Gasior M, Cuevas B, Angona A, Gomez-Casares MT, Martinez CM, Magro E, Ayala R, Del Orbe-Barreto R, Perez-Lopez R, Fox ML, Raya JM, Guerrero L, Garcia-Hernandez C, Caballero G, Murillo I, Xicoy B, Ramirez MJ, Carreno-Tarragona G, Hernandez-Boluda JC, Pereira A, Group MPNS (2022) Real-world analysis of main clinical outcomes in patients with polycythemia vera treated with ruxolitinib or best available therapy after developing resistance/intolerance to hydroxyurea. Cancer 128(13):2441–2448. https://doi.org/10.1002/cncr.34195
Mesa R, Vannucchi AM, Yacoub A, Zachee P, Garg M, Lyons R, Koschmieder S, Rinaldi C, Byrne J, Hasan Y, Passamonti F, Verstovsek S, Hunter D, Jones MM, Zhen H, Habr D, Martino B (2017) The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Br J Haematol 176(1):76–85. https://doi.org/10.1111/bjh.14382
Eva Lengfelder GMB, Konstanze Döhner, Heinz Gisslinger, Martin Grießhammer, Steffen Koschmieder, Petro E. Petrides (2019) DGHO Onkopedia Guideline on polycythemia vera Accessed April 11th, 2021
Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, Hehlmann R, Hoffman R, Kiladjian JJ, Kroger N, Mesa R, McMullin MF, Pardanani A, Passamonti F, Vannucchi AM, Reiter A, Silver RT, Verstovsek S, Tefferi A, European L (2011) Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 29(6):761–770. https://doi.org/10.1200/JCO.2010.31.8436
Passamonti F (2009) New and old prognostic factors in polycythemia vera. Curr Hematol Malig Rep 4(1):19–24. https://doi.org/10.1007/s11899-009-0003-8
Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, Kiladjian JJ, Lengfelder E, McMullin MF, Passamonti F, Reilly JT, Vannucchi AM, Barbui T (2009) Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood 113(20):4829–4833. https://doi.org/10.1182/blood-2008-09-176818
Emanuel RM, Dueck AC, Geyer HL, Kiladjian JJ, Slot S, Zweegman S, te Boekhorst PA, Commandeur S, Schouten HC, Sackmann F, Kerguelen Fuentes A, Hernandez-Maraver D, Pahl HL, Griesshammer M, Stegelmann F, Doehner K, Lehmann T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Harrison CN, Radia D, Muxi P, Maldonado N, Besses C, Cervantes F, Johansson PL, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferrari ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Mesa RA (2012) Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 30(33):4098–4103. https://doi.org/10.1200/JCO.2012.42.3863
Vannucchi AM (2015) Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 372(17):1670–1671. https://doi.org/10.1056/NEJMc1502524
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM (2012) A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 366(9):799–807. https://doi.org/10.1056/NEJMoa1110557
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G (2012) JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 366(9):787–798. https://doi.org/10.1056/NEJMoa1110556
Al-Ali HK, Griesshammer M, Foltz L, Palumbo GA, Martino B, Palandri F, Liberati AM, le Coutre P, Garcia-Hernandez C, Zaritskey A, Tavares R, Gupta V, Raanani P, Giraldo P, Hanel M, Damiani D, Sacha T, Bouard C, Paley C, Tiwari R, Mannelli F, Vannucchi AM (2020) Primary analysis of JUMP, a phase 3b, expanded-access study evaluating the safety and efficacy of ruxolitinib in patients with myelofibrosis, including those with low platelet counts. Br J Haematol 189(5):888–903. https://doi.org/10.1111/bjh.16462
Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, Naim A, Paranagama D, Marker M, Gadbaw B, Kiladjian JJ (2017) Ruxolitinib reduces JAK2 p V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol 96(7):1113–1120. https://doi.org/10.1007/s00277-017-2994-x
Saini KS, Patnaik MM, Tefferi A (2010) Polycythemia vera-associated pruritus and its management. Eur J Clin Invest 40(9):828–834. https://doi.org/10.1111/j.1365-2362.2010.02334.x
Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R (2015) Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 126(15):1762–1769. https://doi.org/10.1182/blood-2015-04-637280
Barbui T, Vannucchi AM, De Stefano V, Masciulli A, Carobbio A, Ferrari A, Ghirardi A, Rossi E, Ciceri F, Bonifacio M, Iurlo A, Palandri F, Benevolo G, Pane F, Ricco A, Carli G, Caramella M, Rapezzi D, Musolino C, Siragusa S, Rumi E, Patriarca A, Cascavilla N, Mora B, Cacciola E, Mannarelli C, Loscocco GG, Guglielmelli P, Betti S, Lunghi F, Scaffidi L, Bucelli C, Vianelli N, Bellini M, Finazzi MC, Tognoni G, Rambaldi A (2021) Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol 8(3):e175–e184. https://doi.org/10.1016/S2352-3026(20)30373-2
Acknowledgements
We thank Kim Kricheldorf for support with the GSG-MPN bioregistry coordination, Carmen Fera, Michelle Haaße, Yvonne Schumacher, Amrei Pelzer, Christina Grohe, Irina Prell, Verena Deserno, and Rainer Schuckelt for support at the Center for Translational and Clinical Research Aachen (CTC-A), Kristina Pannen (née Feldberg) for logistic support with samples, and Britta Wagner for excellent secretarial assistance. We thank our colleagues and study staff at all RuxoBEAT study centers in Germany and our patients for participation in this trial. Additional centers participating in this study include Aschaffenburg (Martine Klausmann), Berlin (Philip LeCoutre), Chemnitz (Mathias Hänel), Düsseldorf University Hospital (Norbert Gattermann), Essen (Joachim R. Göthert), Mainz (Thomas Kindler), Mannheim (Andreas Reiter), Münster (Eva Eßeling), Nürnberg (Marinela Augustin), Duisburg (Sebastian Balleisen), Halle (Haifa Kathrin Al-Ali), Düsseldorf Marien-Hospital (Aristoteles Giagounidis), Siegburg (Stefan Fronhoffs), and Cologne (Christof Scheid). We thank Novartis for the financial support of the clinical trial and for providing the study medication. We thank the Deutsche Forschungsgemeinschaft (DFG) for supporting concomitant basic research associated with this entire trial within the Clinical Research Unit CRU344 (KO 2155/7-1, BR 1782/5-1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
SK designed the trial and the eCRF, led the trial as Principal Coordinating Investigator, contributed patient data, analyzed the data, wrote the manuscript, and edited the manuscript. SI designed the trial and the eCRF, managed the trial together with SK and THB, contributed patient data, analyzed the data, wrote the manuscript, and edited the manuscript. DWolf, FHH, AH, PS, MG, DWolleschak, UP, KD, PJJ, SP, MS, NVB, FS, MC, and DG contributed patient data, analyzed the data, and edited the manuscript. AM performed the JAK2 allele ratio assessments, analyzed the data, and edited the manuscript. MK acted as the CTC-A project manager for the trial, analyzed the data, and edited the manuscript. JFranklin, JFrank, and MH analyzed the data, performed the statistical analysis of the trial, and edited the manuscript. THB designed the trial, led the trial as Co-Principal Coordinating Investigator, contributed patient data, analyzed the data, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
This clinical trial received financial support from Novartis. Steffen Koschmieder received research grant/funding from Geron, Janssen, AOP Pharma, and Novartis; received consulting fees from Pfizer, Incyte, Ariad, Novartis, AOP Pharma, Bristol Myers Squibb, Celgene, Geron, Janssen, CTI BioPharma, Roche, Bayer, and PharmaEssentia; received payment or honoraria from Novartis, BMS/Celgene, Pfizer; received travel/accommodation support from Alexion, Novartis, Bristol Myers Squibb, Incyte, AOP Pharma, CTI BioPharma, Pfizer, Celgene, Janssen, Geron, Roche, AbbVie, Sierra Oncology, and Karthos; had a patent issued for a BET inhibitor at RWTH Aachen University; participated on advisory boards for Pfizer, Incyte, Ariad, Novartis, AOP Pharma, BMS, Celgene, Geron, Janssen, CTI BioPharma, Roche, Bayer, Sierra Oncology, and PharmaEssentia. Susanne Isfort reports consultancy for Ariad, Novartis, Pfizer, Incyte; honoraria from Ariad, BMS, Pfizer, Incyte, and Novartis; other payments (e.g., travel support) by Alexion, Amgen, Hexal, Mundipharma, Novartis, Pfizer, and Roche. Dominik Wolf reports research funding, consultancy, and honoraria for Novartis, Pfizer, AOP, BMS/Celgene, and Incyte. Florian H. Heidel reports research funding from Novartis, CTI, Celgene/BMS and consultancy for Novartis, CTI, BMS/Celgene, AOP Pharma, Pfizer, Incyte. Andreas Hochhaus reports research support by Novartis, BMS, Incyte, Pfizer. Philippe Schafhausen reports consulting/advisory role/honoraria/travel and accommodation support from Alexion, AOP Orphan, Blueprint Medicines, BMS/Celgene, Merck Serono, MSD, Novartis, Pfizer, Roche, and Sobi. Martin Griesshammer reports consultancy, honoraria, or Speakers Bureau for Novartis, AOP Orphan, BMS, AbbVie, Pfizer, Roche, Janssen, Gilead, AstraZeneca, and Sierra. Uwe Platzbecker reports consultancy for Abbvie, Amgen, BMS, Novartis, Curis, and Pfizer. Konstanze Döhner reports consulting/advisory role/honoraria from AbbVie, Celgene/BMS, Novartis, CTI BioPharma Corp, and Roche. Philipp Jost reports a consulting or advisory role, received honoraria, research funding, and/or travel/accommodation expenses from Ariad, Abbvie, Bayer, Boehringer, Novartis, Pfizer, Servier, Roche, BMS and Celgene, Pierre Fabre, Janssen / Johnson&Johnson, MSD. Frank Stegelmann reports honoraria from and consultancy for AOP Pharma, BMS/Celgene, Incyte, Novartis, and Pfizer. Martina Crysandt received honoraria from Astra Zeneca and travel accommodation support from Pfizer, Incyte, and Novartis. Deniz Gezer reports advisory board activity for Amgen, Celgene, GSK, and Takeda; travel grants for Amgen, Celgene, Janssen/Cilag, and Takeda; and was invited speaker for Amgen, Celgene, BMS, GSK, and Takeda. Martin Hellmich received funding from the sponsor of the study (RWTH Aachen University) for statistical planning and analysis. Tim H. Brümmendorf served as a consultant for Janssen, Merck, Novartis, and Pfizer, received research funding from Novartis and Pfizer, received honoraria from Novartis and Pfizer, and received other support (e.g., travel support) from Janssen, Merck, Novartis, and Pfizer.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koschmieder, S., Isfort, S., Wolf, D. et al. Efficacy and safety of ruxolitinib in patients with newly-diagnosed polycythemia vera: futility analysis of the RuxoBEAT clinical trial of the GSG-MPN study group. Ann Hematol 102, 349–358 (2023). https://doi.org/10.1007/s00277-022-05080-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-022-05080-7