
Abstract
A number of the inhibitors against programmed death protein 1 (PD-1) have been approved to treat recurrent or metastatic squamous cell carcinoma of head and neck (HNSCC). The interaction between PD-1 and its ligand (PD-L1) serves as an immune checkpoint that governs cytotoxic immune effectors against tumors. Numerous clinical trials of PD-1/PD-L1 inhibitors have so far been discordant about having sufficient PD-L1 expression in the tumor as a prerequisite for a successful anti-PD-1 treatment. On the other hand, vascular endothelial cells modulate immune activities through PD-L1 expression, and thus it is possible that the expressions of circulating endothelial cells (CECs) and circulating endothelial progenitor cells (CPCs) could affect antitumor immunity as well as neoangiogenesis. Here we investigated the potential involvement of PD-L1+ CECs and PD-L1+ CPCs in PD-1 blockade treatments for HNSCC patients. We measured CD8+ T cells, CECs, and CPCs in the peripheral blood of the HNSCC patients treated by anti-PD-1 therapies. We found that their PD-L1+ CPC expression before anti-PD1 therapies was strongly correlated with treatment responses and overall survival. Moreover, if the first infusion of PD-1 inhibitors reduced ≥ 50% PD-L1+ CPCs, a significantly better outcome could be predicted. In these patients as well as in an animal model of oral cancer, Pd-l1+ CPC expression was associated with limited CD8+ T-cell infiltration into the tumors, and anti-PD-1 treatments also targeted Pd-l1+ CPCs and increased CD8+ T-cell infiltration. Our results highlight PD-L1+ CPC as a potential regulator in the anti-PD-1 treatments for HNSCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized treatments of various cancers [1]. ICIs utilize antagonizing monoclonal antibodies (mAb) that specifically target cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein-1 (PD-1), or its ligand PD-L1 to restore the activity of immune effector cells, particularly CD8+ T cells (cytotoxic T lymphocytes, or CTL), in cancer patients [2]. In head and neck squamous cell carcinoma (HNSCC), PD-1 inhibitors benefit survival of metastatic patients who had unsuccessful responses with platinum [3, 4], albeit the overall response rates using PD-1 inhibitors are still around 13 ~ 18% [3, 5]. Because of the low response rates, high treatment cost, and immune-related adverse effects, it is urgent to identify biomarkers that could predict treatment responses to ICIs, which may be found in PD-L1 protein expression, tumor mutation burden, immune gene expression, and T-cell inflamed tumors [6,7,8]. Currently, there are no validated biomarkers that could predict or monitor responses of ICI therapies for HNSCC [3, 9]. Major obstacles to such biomarker development include dynamic variations associated with intratumor heterogeneity.
PD-1/PD-L1 blockade-induced antitumor immunity in the tumor microenvironment (TME), could result in different degrees of immune responses. There are factors in the TME that could attenuate trafficking of tumor-targeted T cells and suppress effector cell function [10]. These factors include stromal fibroblasts, endothelial cells (ECs), immune suppressor cells, and extracellular matrix that functions as a physical barrier. Among them, ECs express several immune checkpoint molecules that suppress effector T lymphocyte responses, such as PD-L1, T cell immunoglobulin and mucin-domain containing-3 (TIM3), B7-H3 and B7-H4 [11]. Specifically, PD-L1 and PD-L2 that inhibit effector T cell activation are selectively up-regulated in the ECs [12, 13]. Moreover, PD-L1-expressing ECs could recruit FoxP3 + T regulatory (Treg) cells to the TME to blunt anti-tumor activities and affect the prognosis of cancer patients [14]. On the other hand, bone marrow-derived circulating endothelial progenitor cells (CPCs) recruited to the TME could transdifferentiate into endothelial lineage and maintain the vascular integrity of neoangiogenesis [15, 16]. The CPCs endowed with anti-inflammatory properties may suppress cancer-targeting effectors [17] and form immunosuppressive vessels through the TNFα/TNFR2 axis [18]. These neovascular complexes could create an immune tolerance barrier that limits anti-cancer immune responses. Additionally, neovascularization in tumors is associated with tumor progression to higher grades of malignancy [19]. We therefore hypothesized that PD-L1-expressing circulating endothelial cells (CECs) and CPCs might dynamically modulate the effectiveness of PD-1/PD-L1 blockade treatments.
Here we demonstrated PD-1/PD-L1 blockade to be a promising therapy for patients with recurrent and metastatic HNSCC. Anti-PD-1 treatments activated CD8+ T cells in both responders and non-responders. Importantly, we found that the levels of PD-L1 in CPCs over the course of anti-PD-1 treatments could serve as a crucial biomarker for monitoring early treatment responses and predicting overall survival (OS).
Materials and methods
Patient subjects and clinical evaluation
This study was approved by the Mackay Memorial Hospital Institutional Review Board (21MMHIS287e), and was carried out in accordance with the principles of the Declaration of Helsinki. All patient subjects signed the informed consent to release their clinical data for this study. All participants with confirmed squamous cell carcinoma of the oropharynx, oral cavity, hypopharynx, or larynx, had been treated with local radiotherapy and cisplatin-based chemotherapy. Their PD-L1 expression in formalin-fixed tumor samples was assessed using the antibodies including PD-L1 IHC 22C3 (Dako, pharmDx), 28 − 8 (Dako, pharmDx), and Sp263* (Ventana, Roche Diagnostics). Human papillomavirus (HPV) was characterized by p16 (E6H4, Ventana) immunostain. The expression of PD-L1 was assessed by the tumor proportion score (TPS); only a TPS greater than 50% (or in more than 10% tumor cells [TC ≥ 10%]) meets the reimbursement criteria for ICI treatments set by the Taiwan National Health Insurance (TW-NHI), which were based on the recommendation of the European Medicines Agency (EMEA). As the ICI treatments are costly, all the HNSCC patient subjects who met the treatment reimbursement criteria were treated following the standard ICI guideline and protocols set by TW-NHI (eng.nhi.gov.tw/en/mp-2.html).
Forty-six HNSCC patients at the age of 18 years or older with recurrent or metastatic disease were eligible for anti-PD-1 immunotherapies. For measurement of vascular endothelial PD-L1 expression, 20 small vessels of endothelial cells in a tumor section were assessed. Responders or non-responders were determined according to the Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST V.1.1) [20]. Patients approved for anti-PD-1 therapies were treated with nivolumab 3 mg/Kg every 2 weeks or pembrolizumab 200 mg every 3 weeks intravenously until their conditions worsened. No patients withdrew due to drug intolerance or toxicity. Their peripheral blood (PB) samples were collected upon diagnosis and before every anti-PD-1 infusion treatment. The treatment responses evaluated by computed tomography (CT) every 8 weeks are categorized into complete response (CR), partial response (PR), and stable disease (SD) which lasts minimally 90 days during the treatment course. Patient information, including age, gender, clinical status, smoking, associated HPV status, prior treatments, metastatic stage, and types of the ICI treatments, were listed in Supplementary Table 1.
Flow cytometry
To quantify CD8+ T cell populations, 5 × 105 PB mononuclear cells were stained with the following fluorophore-conjugated mAbs: anti-CD3-FITC, anti-CD4-APC, anti-CD8-PE, and anti-CD107a-eFlour660. Anti-GrB-APC mAb was used to stain intracellular granzyme B (GrB). All data were compared with isotype controls and analyzed using Cell Quest Pro (BD Biosciences).
CECs were identified as CD31brightCD34+CD45−CD133− and CPCs were defined as CD31+CD34brightCD45dimCD133+ by multicolor flow cytometry (FACSCalibur, BD Biosciences), as described [21, 22]. Briefly, whole blood (3 mL) was directly stained with pre-conjugated anti-human mAbs—anti-CD31-FITC, anti-CD34-PE, anti-CD45-PercP, and anti-CD-133-PE (BD Biosciences) in the dark for 30 min, and then fixed with 4% paraformaldehyde. Selection and gating of CECs and CPCs were shown in Supplementary Fig. 1. Age-matched healthy adults were used as controls in each experiment (data not shown).
Cell culture
MILE SVEN 1 (MS1) mouse vascular endothelial cells (ATCC CRL2279) were engineered to stably express mouse Pd-l1 tagged with green fluorescence protein (GFP) (MR203953L4V, ORIGENE) or to stably express GFP alone (MS1GFP). The MOC-L1 oral squamous carcinoma cell is a C57BL/6 syngeneic cell line kindly provided by Dr. Chung-Ji Liu (MacKay Memorial Hospital) [23].
Immunohistochemistry and imaging for CD8+ T-cell distribution in tissues
CD8+ T cells were stained with mouse anti-human CD8 mAb (clone C8/114B, Dako) in paraffin-embedded tissue sections of HNSCC patients. Mouse tumor samples were cryosectioned and stained with PE-conjugated anti-mouse CD8 mAb (Miltenyi Biotec). Images of the stained sections were captured by TissueFAXS and analyzed by HistoQuest (TissueGnostics, Vienna, Austria). Cancer samples were categorized into “immune-desert”, “immune-excluded”, and “inflamed” phenotypes, based on CD8+ T-cell distribution [24, 25]. CD8+ T-cell density was measured in the center of the tumor (CT) and in the stroma-tumor borderline known as the invasive margin (IM), which ranges from 300 μm into the stroma to 50 μm into the tumor [26]. Besides TissueFAXS, fluorescently-labeled images were taken using TCS SP8 (Leica) coupled with the LAS X Software.
Establishing an animal model of the oral cancer
The animal study was approved by the Institutional Animal Care and Use Committee (IACUC of Taiwan MacKay Memorial Hospital). A total of 5 × 106 MOC-L1 cells mixed in 200 µl of Matrigel (BD Biosciences) were injected into the flank of each female C57BL/6 mouse (BioLASCO, Taiwan). From day 8 after the tumor graft, 1 × 105 CPCs expressing PD-L1 or lacking PD-L1 were injected intravenously into the tumor-bearing mouse per week for a total of 5 weeks. For immunotherapies, tumor-bearing mice were each intraperitoneally injected with 10 mg/Kg of anti-PD1 (clone RMP 1–14, BioXcell) weekly for 5 weeks. At the same time, control animals were injected with 10 mg/kg of IgG2a isotype (BioXcell). On the twelfth week after tumor cell inoculation, mice showed signs of poor health and weakness and were sacrificed. Their tumor volume was calculated using the formula: 0.52 x width2 x length.
Statistical analysis
To compare the means between two independent groups that were not normally distributed, the nonparametric Mann-Whitney U test was used. If two groups were normally distributed, Student’s t-tests were used for comparison. To compare three or more independent groups, one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons were used. The cutoff points for the most promising variables were optimized by time-dependent receiver operating characteristics (ROC) curves and the Youden index. Multivariate analysis was performed using logistic regression models to assess the influences of a number of variables. The Cox proportional hazards model was used for univariate and multivariate analyses of prognostic factors of OS and estimated hazard ratios (HRs). We used a stratified log-rank test to assess between-group differences in the OS. GraphPad Prism 8 (GraphPad Software) and SPSS 26 (IBM SPSS) were used for all analyses.
Results
Patient characteristics and responses
From June 2017 to December 2022, 46 heavily treated HNSCC patient subjects relapsed and were thus approved of anti-PD-1 therapies—nivolumab (n = 38) and pembrolizumab (n = 8), by the Taiwan National Health Insurance. All participants completed a 12-week therapy of anti-PD-1 infusion (including 4 cycles: C1, C2, C3 and C4). The patients who underwent anti-PD-1 therapies but still showed disease progression evaluated by CT were categorized as non-responders, and were switched to chemotherapies. Responders and non-responders showed similar demographics, disease characteristics, and PD-L1 levels (TPS ≥ 50 or TC ≥ 10%) (Supplementary Table 1). As of the final month of the study (December 2022), the median follow-up duration was 7.5 months. The median of overall survival of the nivolumab-treated responders was 36.1 months and that of the pembrolizumab-treated responders was 11.1 months. The overall response to either PD-1 inhibitors in our patients was 47.8% (SD 13.1%, PR 34.7%).
Differential CD8+ T-cell activities in patients treated with the PD-1 inhibitors
Upon release of granules like GrB and perforin [27], a lysosomal-membrane associated glycoprotein named CD107a is transferred to the plasma membrane, and CD107a on the cell surface could serve as a surrogate marker for granule exocytosis [28, 29]. To evaluate the activities of CD8+ T cells in the HNSCC patients, we measured CD3+CD8+ T-cell densities over the treatment course and found that they were not substantially different between responders and non-responders (Supplementary Fig. 2). In contrast, the surface CD107a and intracellular GrB levels in CD3+CD8+ T-cell subpopulations were remarkably different in each anti-PD-1 session before the infusion. Before the first anti-PD-1 infusion, the median values of CD107a+CD8+ T-cell counts (130 ± 65 /µL [responders] vs. 90 ± 63 /µL [non-responders], p = 0.018) and GrB+CD8+ T-cell counts (170 ± 103 /µL [responders] vs. 113 ± 90 /µL [non-responders], p = 0.041) were both significantly higher in responders than the non-responders (C1 in Fig. 1A and B). Interestingly, for the responders, their CD107a+CD8+ T cells increased and GrB+CD8+ T cells decreased throughout the anti-PD-1 treatment course. On the contrary, CD107a+CD8+ and GrB+CD8+ T-cell counts did not change significantly in the non-responders. Collectively, effective immune responses to PD-1 blockades were shown by the increase of surface CD107a levels on CD8 + T cells and by the depletion of intracellular GrB inside CD8+ T cells; these phenomena were evident in the responders and not in the non-responders.

CD107a+CD8+ and GrB+CD8+ T-cell subpopulations were associated with the outcome of ICI therapies in HNSCC patients. (A) CD107a+CD8+ T cell densities and (B) GrB+CD8+ T cell densities in responders and non-responders were determined by flow cytometric analyses at the indicated timepoints. The X-axis labels the point of each measurement right before an anti-PD1 infusion; an ICI treatment course constitutes of 4 anti-PD1 infusion sessions. The data from the timepoint “C1” provide the baseline values, or that before the first anti-PD1 infusion. Bars denote the median. Statistical significance was defined by *P < 0.05, **P < 0.01, ***P < 0.001. ns, non-significant. Data were analyzed using repeated measures 1-way ANOVA with Bonferroni’s multiple comparisons, or non-parametric Mann-Whitney U-test
CEC, CPC, PD-L1+CEC, and PD-L1+CPC were all reduced in responder patients
The dynamics of CECs and CPCs may serve as an indicator for cancer treatment responses and disease progression [15, 30, 31]. First, we analyzed whether CD107a+CD8+ and GrB+CD8+ T-cell expressions were correlated with the expressions of CECs and CPCs in each treatment (C1, C2, C3, C4). For the general matrix correlation analysis, these two T-cell subpopulations were not strongly correlated with CECs or CPCs (r < 0.7)(Supplementary Fig. 3). We therefore investigated whether anti-PD-1 treatments could change the dynamics of CEC and CPC numbers in the PB. As shown in Fig. 2A-B, the CEC counts before the anti-PD-1 treatments (C1 in Fig. 2) were comparable between responders and non-responders. Further, the numbers of CPCs were higher in responders (95 ± 50/µL, p = 0.029) than the non-responders (77 ± 41/µL) before the anti-PD-1 treatments. No significant differences in CEC and the CPC counts were found between responders and non-responders after each treatment session.

The circulating cell populations—CEC and CPC counts reflected differential responses to anti-PD1 treatments. By multicolor FACS analysis, (A) Viable CECs (CD31brCD34dimCD45−CD133−) and (B) CPCs (CD31+CD34brCD45dimCD133+) were identified in the PBMCs of the patient samples. The densities of (C) PD-L1+ CECs and (D) PD-L1+ CPCs in whole blood samples of the responders and the non-responders were further gated and measured. Bars denote the median. Statistical significance was defined by *P < 0.05, **P < 0.01, ***P < 0.001. Data were analyzed using one-way ANOVA with Bonferroni’s multiple comparisons, or non-parametric Mann-Whitney U-test
Given that ECs could selectively up-regulate PD-L1 and PD-L2 to inhibit activation of effector T cells in cancers [12, 13], we next explored whether anti-PD-1 treatments could affect PD-L1 levels in CECs and CPCs. We found that the numbers of PD-L1-expressing CECs and CPCs in patients’ PB were substantially higher in responders than the non-responders prior to anti-PD-1 therapies (PD-L1+ CECs: 53 ± 38/µL (responders) vs. 28 ± 26/µL (non-responders), p = 0.014); PD-L1+ CPCs: 54 ± 31/µL (responders) vs. 29 ± 28/µL (non-responders), p = 0.008).
The treatments with PD-1 inhibitors strikingly decreased PD-L1+ CECs and PD-L1+ CPCs more in responders than non-responders (Fig. 2C-D). Moreover, the differences of PD-L1+ CECs and PD-L1+ CPCs between responders and non-responders became overt after two anti-PD-1 infusions. We used ROC analyses to examine the impacts of these factors on anti-PD-1 treatment responses (Supplementary Table 2). Among these variables, the baseline counts of CD107a+CD8+ T cells, CPCs, PD-L1+ CECs, and PD-L1+ CPCs could impact the treatment responses significantly (Table 1). The joint effects of these significant variables were assessed using logistic regression analysis. As shown in Table 1, PD-L1+ CPC was the most effective factor in the anti-PD treatment responses (odds ratio 17.39, p = 0.015). As suggested by the Youden index, using 17/µL PD-L1+CPC as the cutoff could separate the patients who were most responsive to PD-1 inhibitors from the rest. In other words, anti-PD-1-treated HNSCC patients with higher baseline levels of PD-L1+ CPCs (≥ 17/µL) generally had higher rates of success in anti-PD-1 therapies.
Prediction of survival by anti-PD-1 responses, baseline PD-L1+CPC counts, and % reduction of PD-L1+CPCs after the first anti-PD-1 infusion
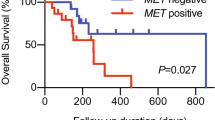
Among the 22 responders, the median time to show treatment responses was 4 months (ranging from one to six months). Based on the Cox model (Fig. 3A), 30% of the responders survived over 5 years. In contrast, all non-responsive patients succumbed in 2 years. The median OS was 35.0 months (95% CI: 1.5–66.5) for the responders and 3.8 months (95% CI 2.5–5.1) for the non-responders. We further applied the Cox regression model on baseline CD107a+CD8+ T cells, GrB+CD8+ T cells, CECs, CPCs, PD-L1+ CECs, and PD-L1+ CPCs, to find out which parameter could be the best predictor for the overall survival (Supplementary Table 3). We found that treated HNSCC patients with pretreated or baseline PD-L1+ CPCs ≥ 17/µL had a higher rate of survival (HR 0.28, 95% CI 0.11–0.87, p = 0.031). Indeed, the patients bearing baseline PD-L1+ CPCs ≥ 17/µL were predicted to survive for over 5 years with 20% probability. Conversely, if the patients bearing baseline PD-L1+CPCs < 17/µL, they were predicted not to survive for more than one year according to the log-rank test (Fig. 3B, p = 0.001).

Estimation of overall survival by the Cox model. Ticks in the graph label censoring, or the last time the patient was known to be alive. The responders were defined as having partial responses, i.e., to be stable after the first infusion of PD-1 inhibitors for minimal 90 days. Non-responders did not meet the criteria for the “responders” and showed cancer progression. The Cox model estimated survival according to stratification of (A) immunotherapy responses, (B) baseline PD-L1+CPC numbers, and (C) % reduction of PD-L1+ CPCs by the first anti-PD-1 infusion (Δ)
As shown in Fig. 3B, 97% of the participants died at the end of this study. The median survival for treated patients with PD-L1+CPCs ≥ 17/µL was 10.8 months (95% CI: 6.6–15.0), whereas that for patients with PD-L1+CPCs < 17/µL was 3.8 months (95% CI: 2.3–5.3). We also noted that the patients with ≥ 17/µL PD-L1+ CPCs and those with < 17/µL PD-L1+ CPCs showed different immune phenotypic distribution (Supplementary Fig. 4). Interestingly, the patients who were categorized in the unfavorable “immune-desert” group had significantly different outcomes depending on their baseline PD-L1+ CPCs (≥ 17/µL: 10.7 month survival; < 17/µL: 3.1 month survival by the log-rank test).
Furthermore, patients with ≥ 50% reduction of PD-L1+ CPCs by the first anti-PD-1 infusion (% ΔPD-L1+ CPCs in Fig. 3C) had a strikingly higher OS than the patients with < 50% reduction of PD-L1+ CPCs (p = 0.003, log-rank test); the median survival time was 23.6 months for the former (95% CI: 0–48.0), and 5.6 months for the latter (95% CI: 2.3–8.9). Taken together, these indicate that PD-L1+CPCs play a crucial role in influencing the outcomes of PD-1 blockade treatments in HNSCC patients. For adverse events (AEs) associated with PD-1 inhibitors, responders had higher incidence of AEs than non-responders (p = 0.045). In contrast, the frequencies of AEs in patients with baseline PD-L1+CPCs < 17/µL and ≥ 17/µL were not notably different (Supplementary Table 4).
Tissue residency of CD8+ T cells in tumors associated with PD-L1+ CPC expression
Since PD-L1+ CPC expression in ICI-treated HNSCC patients could affect treatment responses, we next explored whether the levels of intratumor vascular PD-L1+ ECs corresponded to the levels of PD-L1+ CPCs or PD-L1+ CECs in the PB. We measured % PD-L1+ ECs from 20 microvessels in each tumor sample. Figure 4A showed an image of low PD-L1+ EC expression from a patient with tongue cancer (left) and an image of high PD-L1+ EC expression from a patient with oropharynx cancer (right). By Spearman’s correlation analyses on the proportions of PD-L1+ CPCs among all CPCs and the proportions of PD-L1+ ECs among all ECs in the tumor (Fig. 4B), there was a remarkable correlation between the two (r = 0.6, p = 0.0015). For the patients with ≥ 17/µL PD-L1+CPCs, their %PD-L1+ ECs was significantly higher than that in the patients with < 17/µL PD-L1+ CPCs (Fig. 4C). Moreover, the number of PD-L1+ CPCs was well correlated with the number of CD8+ T cells attached to the small vessels (r = 0.62, p < 0.0001) (Fig. 4D). The densities of perivascular CTLs were also significantly higher in patients with ≥ 17/µL PD-L1+ CPCs (35.5 ± 4.7/mm2) than in patients with < 17/µL PD-L1+ CPCs (15.9 ± 3.6/mm2) (Fig. 4E). In contrast, there was no significant correlation between %PD-L1+ CECs and %PD-L1+ ECs (data not shown). The densities of CTLs in the central tumor (CT) region and in the invasive margin (IM) region were not substantially different between patients with ≥ 17/µL PD-L1+ CPCs and those with < 17/µL PD-L1+ CPCs (Supplementary Fig. 5).

The percentages of PD-L1+ CPCs were directly associated with the percentages of PD-L1+ ECs in intratumoral vessels and the expression of tissue-resident CD8 + T cells. (A) Two representative immunohistochemical images (×200 magnification; scale bar, 100 μm) of ECs stained with anti-PD-L1 antibody (dark brown) from patients expressing low PD-L1+ CPCs (left: < 17 /µL) and high PD-L1+ CPCs (right: ≥17 /µL). (B) The number of ECs was counted from 20 intratumoral microvessels per section for estimation of the percentage of PD-L1+ ECs in the same section. A direct correlation between the percentage of intratumoral PDL1 + ECs and the percentage of PDL1 + CPCs was found by Spearman’s correlation analyses. (C) The percentages of intratumoral PD-L1+ ECs were compared in the patients with low versus high levels of PD-L1+ CPC expression. (D) A direct correlation was observed between the expressions of PDL1+ CPCs and perivascular CD8+ T cells in the patients. (E) More perivascular CD8+ T cells were present in the patients with high levels of PD-L1+ CPC expression than with low levels of PD-L1+ CPC expression. Bars denote the median. Statistical significance was defined by *P < 0.05
Though the densities of CD8+ T cells in the CT and the IM areas were similar regardless of individual PD-L1+ CPC levels before anti-PD-1 therapies, their treatment responses were correlated with their individual baseline PD-L1+ CPC levels. We thus proposed that PD-1 blockade could alter CTL distribution, particularly in these patients with inherently high PD-L1+CPCs. We next used an animal model of oral cancer to investigate CTL localization in the tumor tissues before and after anti-PD-1 treatments.
Animal model of oral cancer
To explore how PD-L1+ CPCs could affect CD8+ T cells to defend cancer, we employed a mouse model of oral cancer. Four C57BL/6 female mice of 6 weeks old per group were assigned to be injected with mouse Pd-l1-transduced CPCs (GFP-Pd-l1 vector) or with GFP-transduced CPCs (GFP alone vector) intravenously. The other two groups were injected with the above-mentioned CPCs and subsequently treated with anti-mouse Pd-1 mAb twice a week for 3 weeks, before harvest of the tumor lumps for CD8+ T-cell quantitation. Figure 5A-B showed representative images of anti-CD8-stained tumor sections from mice injected with Pd-l1+ CPCs or Pd-l1− CPCs. The CPCs incorporated into the tumor vessels displayed green color. Figure 5C-D showed the effects of anti-Pd1 mAb on CD8+ T-cell infiltration into the oral tumor in the presence of Pd-l1+ CPCs or PD-L1− CPCs. As shown in Fig. 5E, in the absence of anti-PD-1 treatments, the CD8+ T-cell densities in tumors dissected from Pd-l1+CPCs-injected mice (12.0 ± 1.4/mm2) and from Pd-l1−CPCs-injected mice (9.4 ± 2.4/mm2) were comparable. Conversely, anti-Pd1 treatments significantly enhanced CD8+ T-cell infiltration more in the mice previously injected with PD-l1+ CPCs (17.4 ± 1.8/mm2) than with PD-l1− CPCs (12.2 ± 2.1/mm2). Moreover, the baseline perivascular densities of CD8+ T cells in the mice preinjected with Pd-l1+ CPCs (7.4 ± 1.1/mm2) were also significantly higher than those in the mice pre-injected with Pd-l1− CPCs (4.8 ± 1.3/mm2). After treatment with anti-Pd-1 antibody, the densities of perivascular CD8+ T cells increased significantly more in the mice pre-injected with Pd-l1+ CPCs (10.4 ± 1.2/mm2) than with Pd-l1− CPCs (7.4 ± 1.8/mm2) (Fig. 5F). The growth of oral cancer in these mice was observed, and anti-Pd-1 treatments indeed retarded their tumor growth (Fig. 5G). Concordant to patient data, murine anti-tumor responses triggered by anti-Pd-1 also was higher in the mice preinjected with Pd-l1+ CPCs (Fig. 5G). Taken together, anti-Pd-1 helped facilitate CD8+ T-cell infiltration into tumors especially in the mice with high Pd-l1+ CPC expression.

Effects of CPC-Pd-l1 influencing tissue-resident CD8+ T cells in mouse oral cancer. The murine oral cancer cell line MOC-L1 was grafted to mice subcutaneously, and mouse CPCs (1 × 105 cells) harboring Pd-l1-GFP expression or GFP alone were intravenously injected into the tumor-bearing mice once per week for 4 weeks. Representative immunofluorescence images (×400 magnification; scale bar, 50 μm) showed CD8+ T cells stained for CD8 (red), nuclei (blue), and vessels (green) were shown. (A-B) show tumor resections from tumor-bearing mice injected with (A) Pd-l1+ CPCs, or (B) Pd-l1− CPCs. (C-D) show tumor resections from tumor-bearing mice with subsequent injection with (C) Pd-l1+ CPCs or (D) Pd-l1− CPCs, and then treated with anti-Pd1 antibodies. Data of the control mice treated with rat IgG2a isotype were not shown. (E-F) show the densities of CD8+ T cells in (E) the central region of the tumor, or (F) the perivascular region of the tumor. (G) Tumor kinetics in MOC-L1-bearing mice that were preinjected with either Pd-l1+ CPCs or Pd-l1− CPCs weekly (green arrows) followed by anti-Pd-1 treatments (blue arrows) one week later. Results were presented in mean ± SEM. The data were summarized from two independent experiments. Bars denote the median. Statistical significance was defined by *P < 0.05, **P < 0.01, ***P < 0.001. Independent tumor growth curves were compared using one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons. Data were analyzed using paired or unpaired t test. ns, non-significant
Discussion
This study explored the potential roles of PD-L1+ CPCs during immunotherapies with PD-1/PD-L1 checkpoint inhibition in HNSCC patients. Here we found that the patients with more PD-L1+ CPCs before the anti-PD-1 therapies, as well as the patients whose PD-L1+ CPCs were eliminated more by the first anti-PD-1 infusion, showed longer survival. In the dissected tumor tissues from these patients, the number of CD8+ T cells located around the microvascular region of the tumor was directly proportional to the number of PD-L1+ ECs, as well as to the number of PD-L1+ CPCs before anti-PD-1 therapies. From our animal model of oral cancer, more CD8+ T cells were attracted to reside in the central tumor region after anti-PD-1 treatments, particularly in the mice preinjected with Pd-l1+ CPCs. In consistent with the mouse data, patients with more PD-L1+ CPCs (i.e., ≥ 17/µL) showed a better response to anti-PD-1 treatments. The patients whose PD-L1+ CPCs were eradicated most after the first infusion of PD-1 inhibitors also achieved a better outcome.
In line with previous reports of HNSCC immunotherapies using nivolumab [3] and pembrolizumab [32, 33], a durable response to PD-1 inhibitors were observed in 20 ~ 30% of the patients with higher PD-L1 expression in the tumors. The 5-year OS in our responders was approximately 30% (Fig. 3A). Interestingly, those patients who had high number of PD-L1+ CPCs before initiating the treatments were also more responsive to anti-PD-1 treatments (Table 1). Besides, baseline CD107a+ CD8+ T cells and GrB+ CD8+ T cells were also remarkably higher in the responders than the non-responders (Fig. 1). By multivariate logistic regression, expressions of PD-L1+ CPCs and cytotoxicity-associated CD107a+ T cells before the treatments were recognized as promising factors for prediction of the ICI responses (Table 1). Although dynamic alternations of PD-L1+ CECs were also significantly different between responders and non-responders (Fig. 2C), PD-L1+ CPC expression is the sole independent prognostic factor for survival identified by multivariate Cox regression (Fig. 3B; Supplementary Table 4). We thus focused on the role of PD-L1+ CPCs implicated in the ICI treatment responses.
Since expressions of PD-L1 and PD-L2 on the ECs could be enhanced by IFN-γ+ CD4 T cells [34,35,36], our further direction aims to examine whether PD-L1 expression on the CPCs could also be modulated by CD4 T cells through IFN-γ stimulation. Notably, bone marrow-derived CPCs recruited to form the abnormal tumor vessel architecture could hinder CD8+ T-cell infiltration [11, 37, 38] and immune effectors through up-regulation of PD-L1 and PD-L2 [12, 13].
The prognosis of HNSCC patients can be categorized into “cold, hot, and excluded” immunophenotypes [25]. Approximately 80% of the ICI-treated patients with < 17/µL PD-L1+ CPC were classified “immune-desert”, comparing to that about 51.4% of the ICI-treated patients with ≥ 17/µL PD-L1+CPCs were “immune-desert” (Supplementary Fig. 2). The different immune phenotypic distribution perhaps also contributed to their differences in survival. The hallmark of immune-desert tumors is mainly the deficiency of T cell infiltration. Low immunogenicity has the immune features like impaired antigen presentation [39, 40], downregulation of MHC class I [41, 42] and extracellular matrix production [43]. These may contribute to immune suppression. Extravasation of CTLs is another hurdle to eradicate the tumor cells.
Distribution of perivascular CD8+ T cells in tumor tissues was substantially different in patient samples with high versus low PD-L1+ CPC levels (Fig. 4D). Based on our animal model of oral cancer, tumor-bearing mice pre-injected with Pd-l1+CPCs presented conspicuously different microvascular co-localization of CTLs (Fig. 5B) as well as discernible CTL counts in the central tumor region (Fig. 5A) in contrast to the tumor-bearing mice injected with Pd-l1− CPCs. Conversely, CD8+ T-cell infiltration to both central and border areas of the tumor increased after anti-Pd-1 treatments. From the HNSCC patient data, those with larger reduction of PD-L1+ CPCs (i.e., > 50%) by the first anti-PD-1 infusion generally had a better outcome (Fig. 3C), which perhaps reflects a vascular normalization in the TME and subsequent restoration of the anti-tumor immunity. Currently, high-throughput molecular omics technologies including RNA-seq, proteomics, and metabolomics provide comprehensive understanding of the biological interplay among immune components in the TME [44, 45]. In our future direction, these systemic approaches could help elucidate the roles of BM-derived CECs and CPCs and how these could modulate PD-L1 expression and affect immune responses.
In conclusion, we found that both CD107a+ T cells and PD-L1+ CPCs influenced anti-PD-1 treatment responses in the HNSCC patients harboring high PD-L1 expression. As early efficacies of the PD-1 inhibitors were observed particularly in patients with over 50% reduction of PD-L1+ CPCs, the number of PD-L1+ CPCs could be deemed as an independent prognostic marker of survival. Our findings suggest that for anti-PD-1 immune therapies of HNSCC, individual PD-L1+ CPC count could be combined with his or her PD-L1 expression score in evaluating prognostic stratification. As this study summarized our 5-year data on the immune therapies of severe HNSCC patients, it is warranted to corroborate this finding in larger prospective HNSCC studies elsewhere. Whether PD-L1+ CPCs could also affect responses to immune checkpoint inhibitors in other types of cancers is also worth of future investigation.
Data availability
The data supporting the results reported in this study can be made available upon reasonable request to the corresponding author.
Abbreviations
- CEC:
-
circulating endothelial cell
- CPC:
-
circulating endothelial progenitor cell
- CTLA-4:
-
cytotoxic T-lymphocyte-associated protein-4
- EC:
-
endothelial cell
- ICI:
-
immune checkpoint inhibitor
- IFNγ:
-
interferonγ
- HNSCC:
-
head and neck squamous cell carcinoma
- MAb:
-
monoclonal antibody
- PB:
-
peripheral blood
- PBMC:
-
peripheral blood mononuclear cell
- PD-1:
-
programmed cell death protein-1
- PD-L1:
-
programmed cell death protein ligand-1
- RECIST:
-
response evaluation criteria in solid tumor
- TIM3:
-
T cell immunoglobulin and mucin-domain containing-3
- TME:
-
tumor microenvironment
- TNFα:
-
tumor necrosis factor-alpha
- TNFR2:
-
tumor necrosis factor receptor 2
- Treg:
-
regulatory T cell
References
de Miguel M, Calvo E (2020) Clinical challenges of Immune Checkpoint inhibitors. Cancer Cell 38(3):326–333. https://doi.org/10.1016/j.ccell.2020.07.004
Ribas A, Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359(6382):1350–1355. https://doi.org/10.1126/science.aar4060
Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L et al (2016) Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med 375(19):1856–1867. https://doi.org/10.1056/NEJMoa1602252
Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP et al (2016) Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 17(7):956–965. https://doi.org/10.1016/S1470-2045(16)30066-3
Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S et al (2017) FDA approval Summary: Pembrolizumab for the treatment of recurrent or metastatic Head and Neck Squamous Cell Carcinoma with Disease Progression on or after platinum-containing chemotherapy. Oncologist 22(7):873–878. https://doi.org/10.1634/theoncologist.2016-0496
Schalper KA, Kaftan E, Herbst RS (2016) Predictive biomarkers for PD-1 Axis therapies: the hidden treasure or a call for Research. Clin Cancer Res 22(9):2102–2104. https://doi.org/10.1158/1078-0432.CCR-16-0169
Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G (2017) The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 14(12):717–734. https://doi.org/10.1038/nrclinonc.2017.101
Oliva M, Spreafico A, Taberna M, Alemany L, Coburn B, Mesia R et al (2019) Immune biomarkers of response to immune-checkpoint inhibitors in head and neck squamous cell carcinoma. Ann Oncol 30(1):57–67. https://doi.org/10.1093/annonc/mdy507
Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N et al (2016) The Head and Neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 1(17):e89829. https://doi.org/10.1172/jci.insight.89829
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523(7559):231–235. https://doi.org/10.1038/nature14404
Lanitis E, Irving M, Coukos G (2015) Targeting the Tumor vasculature to enhance T cell activity. Curr Opin Immunol 33:55–63. https://doi.org/10.1016/j.coi.2015.01.011
Mazanet MM, Hughes CC (2002) B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol 169(7):3581–3588. https://doi.org/10.4049/jimmunol.169.7.3581
Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T et al (2003) Endothelial expression of PD-L1 and PD-L2 down-regulates CD8 + T cell activation and cytolysis. Eur J Immunol 33(11):3117–3126. https://doi.org/10.1002/eji.200324270
Liu S, Qin T, Liu Z, Wang J, Jia Y, Feng Y et al (2020) Anlotinib alters Tumor immune microenvironment by downregulating PD-L1 expression on vascular endothelial cells. Cell Death Dis 11(5):309. https://doi.org/10.1038/s41419-020-2511-3
Bertolini F, Shaked Y, Mancuso P, Kerbel RS (2006) The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer 6(11):835–845. https://doi.org/10.1038/nrc1971
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5(15):1597–1601. https://doi.org/10.4161/cc.5.15.3112
Lima Correa B, El Harane N, Gomez I, Rachid Hocine H, Vilar J, Desgres M et al (2021) Extracellular vesicles from human cardiovascular progenitors trigger a reparative immune response in infarcted hearts. Cardiovasc Res 117(1):292–307. https://doi.org/10.1093/cvr/cvaa028
Naserian S, Abdelgawad ME, Afshar Bakshloo M, Ha G, Arouche N, Cohen JL et al (2020) The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Commun Signal 18(1):94. https://doi.org/10.1186/s12964-020-00564-3
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8(8):579–591. https://doi.org/10.1038/nrc2403
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Ingram DA, Caplice NM, Yoder MC (2005) Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood 106(5):1525–1531. https://doi.org/10.1182/blood-2005-04-1509
Duda DG, Cohen KS, Scadden DT, Jain RK (2007) A protocol for phenotypic detection and enumeration of circulating endothelial cells and circulating progenitor cells in human blood. Nat Protoc 2(4):805–810. https://doi.org/10.1038/nprot.2007.111
Chen YF, Liu CJ, Lin LH, Chou CH, Yeh LY, Lin SC et al (2019) Establishing of mouse oral carcinoma cell lines derived from transgenic mice and their use as syngeneic tumorigenesis models. BMC Cancer 19(1):281. https://doi.org/10.1186/s12885-019-5486-7
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541(7637):321–330. https://doi.org/10.1038/nature21349
Ribbat-Idel J, Perner S, Kuppler P, Klapper L, Krupar R, Watermann C et al (2021) Immunologic cold squamous cell carcinomas of the Head and Neck are Associated with an unfavorable prognosis. Front Med (Lausanne) 8:622330. https://doi.org/10.3389/fmed.2021.622330
Burandt E, Blessin NC, Rolschewski AC, Lutz F, Mandelkow T, Yang C et al (2022) T-Cell density at the Invasive Margin and Immune Phenotypes Predict Outcome in Vulvar squamous cell Cancer. Cancers (Basel) 14(17). https://doi.org/10.3390/cancers14174246
Voskoboinik I, Whisstock JC, Trapani JA (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 15(6):388–400. https://doi.org/10.1038/nri3839
Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M et al (2003) Sensitive and viable identification of antigen-specific CD8 + T cells by a flow cytometric assay for degranulation. J Immunol Methods 281(1–2):65–78. https://doi.org/10.1016/s0022-1759(03)00265-5
Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M et al (2003) Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med 9(11):1377–1382. https://doi.org/10.1038/nm942
Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT et al (2005) Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol 23(31):8136–8139. https://doi.org/10.1200/jco.2005.02.5635
Ramcharan KS, Lip GY, Stonelake PS, Blann AD (2014) Effect of standard chemotherapy and antiangiogenic therapy on plasma markers and endothelial cells in Colorectal cancer. Br J Cancer 111(9):1742–1749. https://doi.org/10.1038/bjc.2014.491
Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr et al (2019) Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394(10212):1915–1928. https://doi.org/10.1016/s0140-6736(19)32591-7
Cohen EEW, Soulières D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ et al (2019) Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet 393(10167):156–167. https://doi.org/10.1016/s0140-6736(18)31999-8
Ma W, Pober JS (1998) Human endothelial cells effectively costimulate cytokine production by, but not differentiation of, naive CD4 + T cells. J Immunol 161(5):2158–2167
Kunitomi A, Hori T, Imura A, Uchiyama T (2000) Vascular endothelial cells provide T cells with costimulatory signals via the OX40/gp34 system. J Leukoc Biol 68(1):111–118
Lim WC, Olding M, Healy E, Millar TM (2018) Human endothelial cells modulate CD4(+) T Cell Populations and Enhance Regulatory T Cell Suppressive Capacity. Front Immunol 9:565. https://doi.org/10.3389/fimmu.2018.00565
De Bock K, Cauwenberghs S, Carmeliet P (2011) Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr Opin Genet Dev 21(1):73–79. https://doi.org/10.1016/j.gde.2010.10.008
Peske JD, Woods AB, Engelhard VH (2015) Control of CD8 T-Cell infiltration into tumors by vasculature and microenvironment. Adv Cancer Res 128:263–307. https://doi.org/10.1016/bs.acr.2015.05.001
Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V (2013) MHC class I antigen processing and presenting machinery: organization, function, and defects in Tumor cells. J Natl Cancer Inst 105(16):1172–1187. https://doi.org/10.1093/jnci/djt184
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS et al (2015) Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 33(11):1152–1158. https://doi.org/10.1038/nbt.3344
Ferris RL, Hunt JL, Ferrone S (2005) Human leukocyte antigen (HLA) class I defects in Head and Neck cancer: molecular mechanisms and clinical significance. Immunol Res 33(2):113–133. https://doi.org/10.1385/ir:33:2:113
Ferris RL, Whiteside TL, Ferrone S (2006) Immune Escape associated with functional defects in antigen-processing machinery in Head and Neck cancer. Clin Cancer Res 12(13):3890–3895. https://doi.org/10.1158/1078-0432.Ccr-05-2750
Desbois M, Udyavar AR, Ryner L, Kozlowski C, Guan Y, Dürrbaum M et al (2020) Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in Ovarian cancer. Nat Commun 11(1):5583. https://doi.org/10.1038/s41467-020-19408-2
Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ (2021) Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol 18(4):244–256. https://doi.org/10.1038/s41571-020-00449-x
Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14(1):11–31. https://doi.org/10.1038/nrclinonc.2016.60
Acknowledgements
The authors are grateful to all the patients who provided samples for this study. We thank Dr. Fang-Ju Sun for performing statistical analyses. This work was supported by MacKay Memorial Hospital Grant MMH-113-35.
Funding
This study was funded by the Department of Medical Research, MacKay Memorial Hospital.
Author information
Authors and Affiliations
Contributions
N.S. and C.C. collected clinical information and designed the figures and analyzed results. D.H. and K.C. performed IHC analysis. K.H. analyzed the data, discussed, and wrote the paper. K.C. performed immunofluorescence. C.K. performed flow cytometric analyses and cell experiments. N.S. and Y.C. contributed to clinical patient management. C.C. designed and supervised the study, wrote the paper and discussed together with K.H. All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Mackay Memorial Hospital (MMH) Institutional Review Board (21MMHIS287e), and was carried out in accordance with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Su, NW., Dai, SH., Hsu, K. et al. PD-L1-positive circulating endothelial progenitor cells associated with immune response to PD-1 blockade in patients with head and neck squamous cell carcinoma. Cancer Immunol Immunother 73, 3 (2024). https://doi.org/10.1007/s00262-023-03595-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-023-03595-0