
Abstract
The presence of IL-10, produced either by tumor cells or immunosuppressive cells, is frequently associated with a poor prognosis for cancer progression. It may also negatively impact anticancer treatments, such as immunotherapies, that otherwise would promote the activation of cytotoxic T cells capable of detecting and destroying malignant cells. In the present study, we evaluated a new adjuvant approach for anticancer immunotherapy using a plasmid vector encoding a soluble form of the IL-10 receptor (pIL-10R). pIL-10R was coadministered to mice with a DNA vaccine encoding the type 16 human papillomavirus (HPV-16) E7 oncoprotein genetically fused with glycoprotein D of herpes simplex virus (HSV) (pgDE7h). Immunization regimens based on the coadministration of pIL-10R and pgDE7h enhanced the antitumor immunity elicited in mice injected with TC-1 cells, which express HPV-16 oncoproteins. The administration of the DNA vaccines by in vivo electroporation further enhanced the anticancer effects of the vaccines, leading to the activation of tumor-infiltrating polyfunctional E7-specific cytotoxic CD8+ T cells and control of the expansion of immunosuppressive cells. In addition, the combination of immunotherapy and pIL-10R allowed the control of tumors in more advanced growth stages that otherwise would not be treatable by the pgDE7h vaccine. In conclusion, the proposed treatment involving the expression of IL-10R enhanced the antitumor protective immunity induced by pgDE7h administration and may contribute to the development of more efficient clinical interventions against HPV-induced tumors.
Similar content being viewed by others


Introduction
Cancer immunotherapies aim to boost the ability of the immune system to recognize and eliminate tumor cells. However, the therapeutic efficacy of such procedures may be compromised by tumor-induced immune escape mechanisms that inhibit the cytotoxic responses capable of eliminating cancerous cells [1]. Interleukin IL-10 (IL-10) has emerged as an important mediator of tumor-associated immune evasion mechanisms. IL-10 levels are elevated in the context of human papillomavirus (HPV) infection in both vaginal secretions from non-altered cervix and premalignant lesions of the cervix, and the IL-10 levels show a positive correlation with the grade of the lesion [2, 3]. IL-10 plays an important role in inhibiting antiviral responses, which favors the progression of premalignant lesions resulting in a predisposition for the development of uterine carcinoma [4, 5]. In addition, similar to other cancer types, HPV-positive cervical cancer, oral squamous cell carcinoma and head and neck cancer frequently show enhanced IL-10 expression, which is considered a prognostic indicator of poor clinical outcome [3, 6,7,8].
IL-10 contributes to the induction of immune tolerance, causing reduced expression of costimulatory and major histocompatibility complex class II (MHC-II) molecules in dendritic cells (DCs) and, therefore, inhibits DC differentiation and maturation; IL-10 also compromises DC migration to secondary lymphoid organs [9,10,11]. Increased levels of IL-10 in cancer patients correlate with the downregulation of MHC-II expression in cancer cells and the inhibition of T lymphocyte-mediated cytotoxicity [12]. Additionally, IL-10 promotes the expansion of systemic and tumor-infiltrating regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSCs), which creates a favorable environment for tumor progression and reduces local antigen-specific CD8+ T cell responses even in patients with detectable cellular immune responses [13, 14]. Therefore, the control of IL-10 levels represents a key step in tumor control, especially in advanced disease stages.
Previous evidence has demonstrated that silencing the gene encoding the IL-10 receptor (IL-10R) in DCs increases their antigen presentation and expression of costimulatory molecules, leading to enhanced tumor growth control in HPV-16 E6 and E7-expressing tumor cells (TC-1 cells) [15]. In addition, the combined silencing of IL-10 and TGF-β receptors in DCs significantly increases the number of activated E7-specific CD8+ T lymphocytes and enhances protective antitumor immunity in mice [16]. Another experimental approach developed to reduce the in vivo levels of IL-10 was based on the administration of a plasmid encoding the soluble extracellular region of the IL-10 receptor (pIL-10R) [17]. Such an approach led to increased survival and activation of proinflammatory responses in a murine melanoma model, but the strategy has not been tested as an adjuvant with other anticancer therapeutic interventions.
Our group has developed an immunotherapy against HPV-associated tumors based on immunization, either with a DNA vaccine or purified recombinant protein, with a chimeric protein derived from the fusion of the HPV-16 E7 oncoprotein with the HSV-1 glycoprotein D (gD) (pgDE7h) [18, 19]. The vaccines proved to be capable of activating E7-specific CD8+ T cell responses in mice and conferred therapeutic protection in mice grafted with tumor cells expressing HPV-16 oncoproteins [18]. However, this immunotherapeutic approach loses efficacy when it is applied to animals with tumors at more advanced stages. In light of this evidence, this study sought to evaluate the adjuvant effects of IL-10R expression on the immune responses mediated by pgDE7h. Our results demonstrate that the coadministration of pIL-10R and pgDE7h increased the activation and tumor infiltration of the E7-specific CD8+ T lymphocytes induced by immunotherapy and therefore generated more efficient control of tumors at more advanced stages.
Materials and methods
Plasmids
The pgDE7h vector contains a codon-optimized sequence encoding the gDE7 hybrid protein cloned into pUMVC3 (Aldevron, USA) [20]. pIL-10R encodes the soluble extracellular portion of the murine IL-10 receptor corresponding to amino acids 1–189 cloned into the NotI site of pcDNA3 (Invitrogen, USA) [17]. Plasmid purification was performed using a Giga Prep plasmid purification kit (QIAGEN, USA) according to the manufacturer’s instructions.
TC-1 cells
The TC-1 cell line is derived from primary lung epithelium cells of C57BL/6 mice that were transformed with c-Ha-ras- and HPV-16 E6- and E7-encoding genes [21]. TC-1 cells were cultured in RPMI (Gibco, USA) medium supplemented with 50 U/ml penicillin/streptomycin, 0.4 mg/ml G418, 10% fetal bovine serum (FBS) (Vitrocell, Campinas, São Paulo, Brazil) at 37 °C and 5% CO2. On the day of tumor grafting, the cells were treated with trypsin–EDTA (Vitrocell, São Paulo, Brazil) and suspended in serum-free RPMI medium at the proper concentrations for inoculation.
Tumor challenge
Six–eight-week-old C57BL/6 mice were transplanted with 7.5 × 104 TC-1 cells in 100 µl of RPMI medium by subcutaneous injection in the right dorsolateral region. Tumor growth was monitored 2–3 times a week with calipers, and survival was recorded for at least 60 days. The smaller (d) and larger (D) diameters of the tumors were measured, and the tumor volume was calculated according to the formula D × d2/2. Mice with tumors of approximately 1 mm3 were considered tumor-bearing mice, and mice with tumors greater than 700 mm3 were euthanized.
Immunization protocols
Mice were immunized by injection of one dose of pgDE7h (5 or 50 µg) by the intramuscular route (i.m.) in the tibialis anterior muscle or by electroporation (EP) 5 or 14 days after tumor grafting. For EP, a total of six 130 V 450 ms electrical pulses were applied using a NEPA21 Super Electroporator (NepaGeneCo., Ltd., Chiba, Japan). In the combined immunization regimen, mice received one or two doses of pIL-10R (50 µg) administered 5 days apart either by injection or EP. The first dose was coadministered with pgDE7h, and the second dose was delivered alone.
Intracellular cytokine staining (ICS) and MDSC detection
Peripheral blood mononuclear cell (PBMC) or spleen samples were collected 14 days after pgDE7h administration and incubated at a concentration of 106 cells per well for 12 h at 37 °C in a 5% CO2 atmosphere, in the presence of Brefeldin A (5 µg/ml) (GolgiPlug; BD Biosciences, USA) and IL-2 (5 ng/ml) and with or without MHC class I-specific E7-derived peptide (49RAHYNIVTF57) at a final concentration of 300 ng/well. In some experiments, 5 µg/ml anti-CD107a-FITC antibody (BD Biosciences, USA), 10 µg/ml monensin (GolgiStop; BD Biosciences, USA) and 2 µg/ml anti-CD28 antibody (BD Biosciences, USA) were added during the overnight stimulation. The cells were labeled with anti-CD8α-BV605 or -BB515-conjugated antibodies (BD Bioscience, USA). After fixation and permeabilization with Cytofix/Cytoperm (BD Biosciences, USA), the samples were intracellularly stained with anti-IFN-γ-PE or anti-IFN-γ-Alexa Fluor 700 and anti-TNF-α-PE-Cy7 antibodies (BD Bioscience, USA). MDSCs were tracked using anti-CD11b-FITC and anti-Gr1-PE antibodies (BD Bioscience). The samples were analyzed on an LSRFortessa® flow cytometer (BD Bioscience, San Jose, CA, USA), and the data were analyzed with FlowJo software. The analyses were performed after the initial exclusion of doublets using the FSC and SSC parameters. Subsequently, the percentages of CD8+ IFN-γ+, CD8+ TNF-α+ and CD8+ CD107a+ cells in the total CD8+ T cell population were determined. MDSCs were characterized as CD11bhigh GR-1int, CD11bhigh Gr1high or total CD11b+ Gr-1+.
Detection of tumor-infiltrating lymphocytes
Tumor samples were collected 28 days after tumor transplantation and digested with collagenase D (1 mg/mL) for 1 h at 37 °C and macerated to obtain a cell suspension. The samples were labeled with an APC-conjugated MHC class I dextramer carrying the E7-specific peptide (Immudex, USA) and surface stained with anti-CD8α-BB515 (BD Bioscience) and anti-CD45-PerCP Cy5.5 antibodies (BD Bioscience, USA). For some experiments, cells were fixed, permeabilized (Cytofix/Cytoperm; BD Biosciences, USA), and stained with anti-IFN-γ-PE monoclonal antibodies (mAb) (#554,412, BD Bioscience, USA) after stimulation overnight with IL-2 (5 ng/ml) in the presence of 49RAHYNIVTF57 peptide (300 ng/well). Samples were analyzed by flow cytometry using an LSRFortessa™ (BD Bioscience San Jose, CA, USA). Doublets were excluded using the FSC and SSC parameters, and CD45+ cells were subsequently analyzed to obtain the percentages of CD8+ IFN-γ+ or CD8+ dextramer E7-specific+ cells with the total CD8+ T cell population.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA). One- or two-way ANOVA was used with the Bonferroni multiple comparison test or Student’s t test was used for comparisons of unpaired samples. The log-rank test followed by the Mantel–Cox posttest was used for the survival analysis. p < 0.05 was considered statistically significant with a confidence interval of 95%.
Results
pIL-10R administration enhances the antitumor effects of pgDE7h
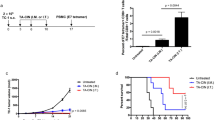
First, we determined whether the administration of pIL-10R would impact the therapeutic antitumor effects of pgDE7h. To address this question, mice were immunized with one dose of pgDE7h (50 µg) or underwent a combined immunization regimen consisting of two doses of pIL-10R (50 µg for each injection) injected 5 days apart by the intramuscular route (i.m.) starting five days after the TC-1 cell graft, and one dose of pgDE7h delivered at the same time as the first dose of pIL-10R (Fig. 1a). Mice immunized with pgDE7h showed delayed tumor growth compared with the sham-treated mice (PBS), and 40% of the vaccinated mice survived up to 2 months after the challenge, but all mice developed tumors (Fig. 1b–d). Mice immunized with two doses of pIL-10R showed reduced tumor size in comparison with the sham-treated mouse group (PBS), but by day 40 after challenge, all mice had to be euthanized due to the progression of the tumors (Fig. 1b, c). In contrast, mice immunized with pIL-10R combined with pgDE7h showed a significant decrease in tumor growth compared with the other mouse groups, and 60% of them survived until the end of the follow-up period (Fig. 1b, c). Tumor-free mice were only observed in the group that received the combined immunization regimen (pgDE7h plus pIL-10R) (Fig. 1d). These results indicate that the administration of pIL-10R enhanced the therapeutic antitumor effects of pgDE7h.

Administration of pIL-10R exhibits adjuvant effects on the tumor protection elicited by the pgDE7h vaccine. Mice were grafted with TC-1 tumor cells and i.m. immunized with one dose of pgDE7h 5 days later with or without two 50 µg doses of pIL-10R administered 5 days apart. Tumor growth was monitored 2–3 times per week for a period of 60 days. a Schematic immunization schedule. b Estimated tumor size over time (two-way ANOVA). c Percentage of surviving or d tumor-free mice over time (log-rank–Mantel–Cox). Data from 3 independent experiments (n = 10–15) (*) vs PBS, (#) vs pgDE7h. *p < 0.05, **p < 0.01, ***p < 0.001
EP administration enhances the antitumor effects of pgDE7h and pIL-10R and halts the expansion of immunosuppressive cells
In an attempt to increase the potency of the combined immunotherapeutic treatment, we delivered pgDE7h and pIL-10R to mice by in vivo EP. For this purpose, mice were injected with TC-1 cells and immunized with 5 µg of pgDE7h admixed with or without 50 µg of pIL-10R in a single dose regimen (Fig. 2a). As shown in Fig. 2b, mice immunized with pgDE7h showed partial tumor growth control and 80% survival up to 60 days after tumor challenge. However, mice immunized with pgDE7h and pIL-10R showed more pronounced tumor growth control and 100% survival (Fig. 2b, c). More importantly, complete tumor protection was observed in 90% of the mice vaccinated with pgDE7h/pIL-10R but only in 50% of the mice vaccinated with pgDE7h (Fig. 2c).

EP potentiates the effect of the combined immunotherapy with the pgDE7h vaccine and pIL-10R. Mice were injected with TC-1 tumor cells and immunized 5 days later using in vivo EP with one 5 µg dose of pgDE7h with or without one 50 µg dose of pIL-10R. Tumor growth was monitored 2–3 times per week for a period of 60 days. a Schematic immunization schedule. b Estimated tumor size over time (two-way ANOVA). c Percentage of surviving or d tumor-free mice over time (log-rank–Mantel–Cox). Data from 3 independent experiments (n = 10) (*) vs PBS, (#) vs pgDE7h. *p < 0.05, **p < 0.01, ***p < 0.001
In a second step, we aimed to evaluate the impact of the treatment in mice with more advanced tumors. The vaccine regimen was started 14 days after the tumor grafting, a point at which the tumors were palpable (1–2 mm2). Mice were immunized with a single dose of pgDE7h (5 µg) admixed with pIL-10R (50 µg) and followed by a second dose of pIL-10R administered 5 days later; both doses were delivered by EP (Fig. 3a). The combined therapy led to efficient tumor control, and 100% of the animals survived the challenge at the end of the observation period (60% tumor-free mice, Supplementary Fig. 1d) (Fig. 3b, c). Mice immunized only with pgDE7h showed delayed tumor development, and 80% of them were still alive at the end of the observation period (30% tumor-free, data not shown) (Fig. 3b, c). Immunization with pIL-10R alone was less efficient in tumor growth control, and mice treated with this approach showed slightly augmented survival compared to PBS-immunized mice (Fig. 3b, c). We also investigated whether a third dose of pIL-10R, administered 5 days after the second dose, would further improve the antitumor therapeutic protection. Mice that received 2 or 3 doses of pIL-10R showed no significant differences in their control of tumor growth or survival (Supplementary Fig. 1). By the end of the follow-up period, the tumors and spleens of mice given the combined immunization regimen were smaller than those from mice immunized with pgDE7h (Fig. 3d). The enlarged spleens in mice vaccinated with pgDE7h reflected the accumulation of MDSCs, as previously shown [22]. In accordance with the tumor protection results, mice immunized with pgDE7h and pIL-10R controlled the expansion of immunosuppressive cells. Administration of the combined immunotherapy led to a significant decrease in the frequency of the total CD11b+ GR-1+ and CD11b+ GR-1high cell populations in the spleens from vaccinated mice compared with those from mice immunized with pgDE7h (Fig. 3e). Taken together, these data indicate that immunization with pIL-10R delivered by EP enhances the therapeutic antitumor effects of pgDE7h, even in mice with pre-established tumors, and halts the expansion of MDSCs triggered by tumor progression.

Combination of pgDE7h and pIL-10R delivered by EP induces the regression of pre-established tumors and halts the accumulation of immunosuppressive cells. Mice were injected with TC-1 tumor cells and immunized 14 days later by in vivo EP with one 5 µg dose of pgDE7h with or without two 50 µg doses of pIL-10R administered 5 days apart. Tumor growth was monitored 2–3 times per week for a period of 60 days. a Schematic immunization schedule. b Estimated tumor size over time (two-way ANOVA). c Percentage of surviving mice over time (log-rank–Mantel–Cox). Data from 3 independent experiments (n = 10–15). d Representative pictures of tumors (top row) and spleens (lower row) harvested 28 days after the challenge from mice treated with the pgDE7h vaccine alone or in combination with pIL-10R. e Frequencies of total CD11b+ Gr-1+ or CD11b+ Gr-1high and CD11b+ Gr-1int subsets of MDSCs in the spleen were determined by flow cytometry (Student’s t test) (n = 6). (*) vs PBS, (#) vs pgDE7h. *p < 0.05, **p < 0.01, ***p < 0.001, ns not significant
Administration of pIL-10R promotes activation of polyfunctional tumor-infiltrating CD8+ T lymphocytes
Following the conditions described above, we next evaluated the induction of antigen-specific CD8+ T cell responses in mice given the pgDE7h/pIL-10R treatment by EP. As shown in Fig. 4a, b, a higher frequency of E7-specific IFN-γ-producing CD8+ T cells was detected in the blood of the mice given the combined therapy compared with that of mice given the sham treatment (PBS). Comparable results were observed in the spleens collected from the mice given the other immunization therapies. Notably, administration of pgDE7h/pIL-10R enhanced CD107a expression concomitantly with enhanced IFN-γ and TNF-α expression compared to the administration of pgDE7h (Fig. 4c). Furthermore, the combination of pgDE7h and pIL-10R enhanced the numbers of splenic IFN-γ/TNF-α- or IFN-γ/CD107a-producing CD8+ T cells compared with pgDE7h alone (Fig. 4c). When single cytokine production by CD8+ T lymphocytes was analyzed, we observed that the numbers of IFN-γ producing cells were increased in mice immunized with pgDE7h/pIL-10R compared with sham-treated mice (PBS), but the numbers of TNF-α producing cells were not increased and were instead similar in the two groups (Fig. 4c–e).

Coadministration of pIL-10R with pgDE7h enhances the activation of E7-specific CD8+ T cells. Mice were injected with TC-1 tumor cells and immunized 14 days later by in vivo EP with one 5 µg dose of pgDE7h with or without two 50 µg doses of pIL-10R administered 5 days apart by in vivo EP. a and b Blood samples and splenocytes c–e were harvested 28 days after immunization and stimulated overnight with the HPV-16 E7 Kb MHC class I-restricted immunodominant epitope peptide. The frequencies of IFN-γ-producing CD8+ T cells were determined by flow cytometry and are shown as representative dot plots a or percentages b. Unstimulated (-) IFN-γ-producing CD8+ T cell frequencies were below 0.2%. (n = 5–10) (ANOVA, posttest: Bonferroni). c The percentages of CD8+ T cells expressing IFN- γ, TNF-α and CD107a were determined by flow cytometry. d Frequencies of CD8+ T cells producing IFN-γ or e TNF-α (Student’s t test) (n = 6). *p < 0.05, **p < 0.01, ns not significant
Finally, we determined the presence of tumor-infiltrating E7-specific CD8+ T cells in mice given the combined immunization regimen. As indicated in Fig. 5a, compared with PBS-treated mice, mice immunized with pgDE7h and pIL-10R showed a significant increase in the influx of total and E7-specific CD8+ T cells to the tumor (Fig. 5a, b), and these cells were not detected in PBS- or pIL-10R-immunized mice (data not shown). In addition, approximately 15% of the total tumor-infiltrating CD8+ T cells were capable of producing IFN-γ upon ex vivo peptide stimulation (Fig. 5c, d). Collectively, these results indicate that the administration of pgDE7h/pIL-10R enhances the frequency and activation of systemic and tumor-infiltrating E7-specific CD8+ T cells, leading to more efficient control of tumor cell growth under experimental conditions.

Combination of pgDE7h and pIL-10R induces the infiltration of antigen-specific CD8+ T lymphocytes into the tumor site. Mice were injected with TC-1 tumor cells and immunized 14 days later by in vivo EP with one 5 µg dose of pgDE7h with or without two 50 µg doses of pIL-10R administered 5 days apart. The tumors were harvested 28 days after the challenge and analyzed by flow cytometry. a Number of total or b E7-specific CD8+ T cells that infiltrated the tumor site (ANOVA, Bonferroni). c Representative dot plots and d percentages of IFN-γ-producing CD8+ T cells after overnight stimulation with HPV-16 E7-derived peptide (Student’s t test). Unstimulated (-) IFN-γ-producing CD8+ T cell frequencies were below 0.3%. **p < 0.01, ***p < 0.001
Discussion
In clinical trials, immunotherapies have been shown to have therapeutic efficacy that supports the regression of precancerous HPV-associated lesions, but in advanced stages, the clinical efficacy still needs improvement. Targeting cancer-induced immunosuppressive mechanisms in parallel with the induction of immune responses to tumor antigens can provide a more appropriate environment for T cell priming and functioning. In light of this, in the present report, we described a novel experimental approach based on the coadministration of a DNA antitumor vaccine (pgDE7h) and a plasmid vector encoding the IL-10 receptor (pIL-10R) as a strategy to halt tumor-induced immunosuppressive mechanisms and, therefore, act as an adjuvant for antigen-specific immunotherapy. Our results demonstrated that the combination of pIL-10R with pgDE7h increased the activation of systemic and tumor-infiltrating E7-specific T lymphocytes and most importantly, the antitumoral therapeutic effects elicited by the DNA vaccine pgDE7h and thus may contribute to the development of more efficient anticancer immunotherapies.
Cytokines, such as IL-10, can be neutralized in vivo using monoclonal antibodies, interfering RNAs or plasmids expressing the corresponding cytokine receptor. In the present study, the decrease in circulating IL-10 levels caused by the injection of the plasmid encoding the soluble fraction of the IL-10 receptor in conjunction with pgDE7h immunization generated robust therapeutic antitumor effects in mice grafted with TC-1 tumor cells. In a melanoma mouse model, the administration of pIL-10R to tumor-bearing mice neutralized the IL-10 expression induced by the tumor and promoted more efficient activation of dendritic cells able to capture tumor antigens and trigger an antitumor inflammatory response [17]. Plasmid-based therapy offers some advantages, as DNA vaccines are considered safe, stable, relatively inexpensive and feasible for use in humans, particularly when administered by in vivo electroporation [23]. Furthermore, repeated administrations of anti-IL-10 neutralizing antibodies may have side effects, such as chronic gastrointestinal inflammation [24]. Other therapeutic strategies have been shown that blocking IL-10R can increase the potency of antitumoral effects. IL-10R gene silencing in dendritic cells has improved the cell functions related to the presentation of HPV-16 antigens, increased the expression of costimulatory molecules and controlled tumor growth in mice challenged with TC-1 cells [15]. Another study combining IL-10R and TGF-β-receptor silencing in dendritic cells demonstrated the presence of increased numbers of E7-specific CD8+ T lymphocytes and more pronounced antitumor effects [16]. Our results add another piece of information about the beneficial aspects of the reduction of IL-10 levels in the therapeutic control of tumors and describe a new feasible experimental approach that can induce similar IL-10-reducing effects and improve the potency of anticancer vaccines.
EP further improved the therapeutic protection elicited in mice immunized with pgDE7h/pIL-10R, as seen by the induction of a robust cellular antigen-specific response. These data suggest that EP can increase the expression of pIL-10R and the duration of the induced antitumor immunity. The use of EP for DNA vaccine delivery is known to increase the number of transfected cells and extend the antigen expression time, therefore enabling a stronger activation of protective immune responses [25]. Previous studies by our group have shown that under experimental conditions, EP promotes activation of antigen-specific CD8+ T cells and protection against HPV-associated tumors in mice [26]. Notably, the phase 2/3 clinical trial with the VGX-3100 DNA vaccine delivered by EP demonstrated that the immunization procedure is well tolerated by patients and permits the control of high-grade lesions in patients with cervical intraepithelial neoplasia [27]. This evidence supports the use of EP for the delivery of DNA vaccines and may contribute to the development of active immunotherapeutic strategies capable of controlling the growth of tumors in more advanced stages.
In the current study, coadministration of pIL-10R and pgDE7h followed by EP increased the magnitude of the activated CD8+ T cell responses and the number of cells simultaneously expressing IFN-γ, TNF-α and CD107a. The induction of polyfunctional CD8+ T cells is related to the control of HPV-associated cancer progression in murine models and in patients [28,29,30]. Accordingly, it has been suggested that the development of this type of cell correlates positively with the clinical efficacy of therapeutic vaccines against high-grade cervical lesions [31]. Other groups have shown that blocking IL-10/IL-10R signaling during antigen presentation favors the induction of immune responses with stronger CD8+ T cell responses and increased efficacy against tumors [15]. However, there is clinical evidence that IL-10 has a dual role in cancer therapy, as IL-10 can promote the enhancement of antigen-specific CD8+ cytotoxic T lymphocyte responses and expression of Th1 cytokines and can activate tumor-resident CD8+ T cells [32,33,34,35,36]. Our results provide experimental evidence that the neutralization of IL-10 by the expression of IL-10R improves the activation of polyfunctional CD8+ T lymphocytes in the presence of a vaccine vector capable of inducing a protective immune response. Thus, the conflicting results regarding the therapeutic effects of IL-10 may be, at least in part, explained by the specific features of the experimental model used in the present study. The coadministration of pIL-10R and pgDE7h clearly promoted an adjuvant effect, particularly in situations in which the tumors had reached larger sizes that downregulated the activation of the effector immune responses capable of recognizing and destroying the tumor cells. The main question to be answered in the future will be the fate of this new adjuvant treatment under clinical conditions.
In our experimental conditions, the expression of pIL-10R led to the enhanced migration of activated E7-specific CD8+ T cells into the tumor mass. Importantly, our findings suggest that the cells induced in this specific condition and present in the tumor microenvironment are not anergic and may contribute to tumor control. Administration of an anti-IL-10R mAb associated with immunization with E7-derived long peptides makes the tumor microenvironment more accessible for CD8+ T cell infiltration [37]. Nonetheless, other pathways that do not involve modulation by IL-10 can interfere with the activation and migratory behavior of T cells, leading to the blockade of cytotoxic activity at the tumor site. Checkpoint-blocking mAbs, such as those targeting PD-1/PD-L1 and CTLA4, are also alternatives to increase the potency of therapeutic vaccines [38, 39]. Our findings add a new and alternative approach that may impact both the activation of cytotoxic T cells and the immunosuppressive mechanisms triggered by tumor cells.
Together, the data presented in this study demonstrate that pIL-10R has an adjuvant effect when coadministered with the pgDE7h vaccine, improving both the protective antitumor immunity and therapeutic efficacy against HPV-associated tumors. The results represent a clinically translatable therapeutic approach capable of controlling immunological escape mechanisms induced by tumors and improving the mounting of specific cytotoxic responses to HPV-associated malignancies.
Abbreviations
- ATCC:
-
American Type Culture Collection
- CAPES:
-
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
- CEUA:
-
Ethics Committee on the Use of Animals in Experimentation
- c-Ha-Ras :
-
v-Ha-ras Harvey rat sarcoma viral oncogene homolog
- CNPq:
-
Conselho Nacional de Desenvolvimento Científico e Tecnológico
- CONCEA:
-
National Council for Control of Animal Experimentation
- E6:
-
Early protein 6
- E7:
-
Early protein 7
- EP:
-
Electroporation
- FAPESP:
-
Fundação de Amparo à Pesquisa do Estado de São Paulo
- FSC:
-
Forward scatter
- gD:
-
Glycoprotein D
- ICGEB:
-
International Centre for Genetic Engineering and Biotechnology
- mAb:
-
Monoclonal antibodies
- PBMC:
-
Peripheral blood mononuclear cell
- pgDE7h:
-
Vaccine plasmid composed of gD fused to HPV-16 E7
- pIL-10R:
-
Interleukin-10 receptor plasmid
- SSC:
-
Side scatter
References
Yaddanapudi K, Mitchell RA, Eaton JW (2013) Cancer vaccines: looking to the future. Oncoimmunology 2:1–7. https://doi.org/10.4161/onci.23403
Azar KK, Tani M, Yasuda H et al (2004) Increased secretion patterns of interleukin-10 and tumor necrosis factor-alpha in cervical squamous intraepithelial lesions. Hum Pathol 35:1376–1384. https://doi.org/10.1016/j.humpath.2004.08.012
Bermudez-Morales VH, Gutierrez LX, Alcocer-Gonzalez JM et al (2008) Correlation between IL-10 gene expression and HPV infection in cervical cancer: a mechanism for immune response escape. Cancer Invest 26:1037–1043. https://doi.org/10.1080/07357900802112693
Syrjänen S, Naud P, Sarian L et al (2009) Immunosuppressive cytokine Interleukin-10 (IL-10) is up-regulated in high-grade CIN but not associated with high-risk human papillomavirus (HPV) at baseline, outcomes of HR-HPV infections or incident CIN in the LAMS cohort. Virchows Arch 455:505–515. https://doi.org/10.1007/s00428-009-0850-7
Berti FCB, Pereira APL, Cebinelli GCM et al (2017) The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor Rev 34:1–13. https://doi.org/10.1016/j.cytogfr.2017.03.002
Alcocer-González JM, Berumen J, Taméz-Guerra R et al (2006) In Vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral Immunol 19:481–491. https://doi.org/10.1089/vim.2006.19.481
Chuang C-Y, Sung W-W, Wang L et al (2012) Differential Impact of IL-10 expression on survival and relapse between HPV16-positive and -negative oral squamous cell carcinomas. PLoS One 7:e47541. https://doi.org/10.1371/journal.pone.0047541
Bornstein S, Schmidt M, Choonoo G et al (2016) IL-10 and integrin signaling pathways are associated with head and neck cancer progression. BMC Genom 17:38. https://doi.org/10.1186/s12864-015-2359-6
Chen L, Qiu M, He W et al (2012) Functional study of immature dendritic cells co-transfected with IL-10 and TGF-beta 1 genes in vitro. Mol Biol Rep 39:6633–6639. https://doi.org/10.1007/s11033-012-1468-4
ten Broeke T, Wubbolts R, Stoorvogel W (2013) MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol 5:a016873. https://doi.org/10.1101/cshperspect.a016873
Maldonado RA, von Andrian UH (2010) How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 108:111–165. https://doi.org/10.1016/B978-0-12-380995-7.00004-5
García-Rocha R, Moreno-Lafont M, Mora-García ML et al (2015) Mesenchymal stromal cells derived from cervical cancer tumors induce TGF-β1 expression and IL-10 expression and secretion in the cervical cancer cells, resulting in protection from cytotoxic T cell activity. Cytokine 76:382–390. https://doi.org/10.1016/j.cyto.2015.09.001
Sevko A, Umansky V (2013) Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: Thick as thieves. J Cancer 4:3–11. https://doi.org/10.7150/jca.5047
Song X, Guo W, Cui J et al (2011) A tritherapy combination of a fusion protein vaccine with immune-modulating doses of sequential chemotherapies in an optimized regimen completely eradicates large tumors in mice. Int J Cancer 128:1129–1138. https://doi.org/10.1002/ijc.25451
Kim JH, Kang TH, Noh KH et al (2011) Blocking the immunosuppressive axis with small interfering RNA targeting interleukin (IL)-10 receptor enhances dendritic cell-based vaccine potency. Clin Exp Immunol 165:180–189. https://doi.org/10.1111/j.1365-2249.2011.04410.x
Ahn YH, Hong SO, Kim JH et al (2015) The siRNA cocktail targeting interleukin 10 receptor and transforming growth factor-β receptor on dendritic cells potentiates tumour antigen-specific CD8 + T cell immunity. Clin Exp Immunol 181:164–178. https://doi.org/10.1111/cei.12620
Marchi LHL, Paschoalin T, Travassos LR, Rodrigues EG (2011) Gene therapy with interleukin-10 receptor and interleukin-12 induces a protective interferon-γ-dependent response against B16F10-Nex2 melanoma. Cancer Gene Ther 18:110–122. https://doi.org/10.1038/cgt.2010.58
Diniz MO, Cariri FAMO, Aps LRMM, Ferreira LCS (2013) Enhanced therapeutic effects conferred by an experimental DNA vaccine targeting human papillomavirus-induced tumors. Hum Gene Ther 24:861–870. https://doi.org/10.1089/hum.2013.102
Porchia BFMM, Diniz MO, Cariri FAMO et al (2011) Purified herpes simplex type 1 glycoprotein D (gD) genetically fused with the type 16 human papillomavirus E7 oncoprotein enhances antigen-specific CD8 + T cell responses and confers protective antitumor immunity. Mol Pharm 8:2320–2330. https://doi.org/10.1021/mp200194s
Diniz MO, Lasaro MO, Ertl HC, Ferreira LCS (2010) Immune responses and therapeutic antitumor effects of an experimental DNA vaccine encoding human papillomavirus type 16 oncoproteins genetically fused to herpesvirus glycoprotein D. Clin Vaccine Immunol 17:1576–1583. https://doi.org/10.1128/CVI.00264-10
Lin KY, Guarnieri FG, Staveley-O’Carroll KF et al (1996) Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res 56:21–26
Diniz MO, Sales NS, Silva JR, Ferreira LCS (2016) Protection against HPV-16-associated tumors requires the activation of CD8 + effector memory T cells and the control of myeloid-derived suppressor cells. Mol Cancer Ther 15:1920–1930. https://doi.org/10.1158/1535-7163.MCT-15-0742
Lee S-H, Danishmalik SN, Sin J-I (2015) DNA vaccines, electroporation and their applications in cancer treatment. Hum Vaccin Immunother 11:1889–1900. https://doi.org/10.1080/21645515.2015.1035502
Ni G, Wang T, Walton S et al (2015) Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol 293:126–129. https://doi.org/10.1016/j.cellimm.2014.12.012
Gothelf A, Gehl J (2012) What you always needed to know about electroporation based DNA vaccines. Hum Vaccin Immunother 8:1694–1702. https://doi.org/10.4161/hv.22062
Sales NS, Silva JR, Aps LRMM et al (2017) In vivo electroporation enhances vaccine-mediated therapeutic control of human papilloma virus-associated tumors by the activation of multifunctional and effector memory CD8 + T cells. Vaccine. https://doi.org/10.1016/j.vaccine.2017.11.011
Trimble CL, Morrow MP, Kraynyak KA et al (2015) Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 386:2078–2088. https://doi.org/10.1016/S0140-6736(15)00239-1
De Rosa SC, Lu FX, Yu J et al (2004) Vaccination in humans generates broad T cell cytokine responses. J Immunol 173:5372–5380
Precopio ML, Betts MR, Parrino J et al (2007) Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med 204:1405–1416. https://doi.org/10.1084/jem.20062363
Rizzuto GA, Merghoub T, Hirschhorn-Cymerman D et al (2009) Self-antigen-specific CD8 + T cell precursor frequency determines the quality of the antitumor immune response. J Exp Med 206:849–866. https://doi.org/10.1084/jem.20081382
Kim TJ, Jin H-T, Hur S-Y et al (2014) Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat Commun 5:5317. https://doi.org/10.1038/ncomms6317
Li O (2013) The paradox of IL–10–mediated modulation in cervical cancer (Review). Biomed Reports. https://doi.org/10.3892/br.2013.69
Wilke CM, Wei S, Wang L et al (2011) Dual biological effects of the cytokines interleukin-10 and interferon-γ. Cancer Immunol Immunother 60:1529–1541. https://doi.org/10.1007/s00262-011-1104-5
Santin AD, Hermonat PL, Ravaggi A et al (2000) Interleukin-10 increases Th1 cytokine production and cytotoxic potential in human papillomavirus-specific CD8(+) cytotoxic T lymphocytes. J Virol 74:4729–4737
Wang Y, Liu X-H, Li Y-H, Li O (2013) The paradox of IL-10-mediated modulation in cervical cancer. Biomed Rep 1:347–351. https://doi.org/10.3892/br.2013.69
Li L, Ma Y, Liu S et al (2016) Interleukin 10 promotes immune response by increasing the survival of activated CD8 + T cells in human papillomavirus 16-infected cervical cancer. Tumor Biol 37:16093–16101. https://doi.org/10.1007/s13277-016-5466-3
Chen S, Ni G, Wu X et al (2016) Blocking IL-10 signalling at the time of immunization renders the tumour more accessible to T cell infiltration in mice. Cell Immunol 300:9–17. https://doi.org/10.1016/j.cellimm.2015.11.002
Ahrends T, Baba a N, Xiao Y et al (2016) CD27 agonism plus PD-1 blockade recapitulates CD4 + T-cell help in therapeutic anticancer vaccination. Cancer Res 76:2921–2931. https://doi.org/10.1158/0008-5472.CAN-15-3130
Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G (2013) Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 73:3591–3603. https://doi.org/10.1158/0008-5472.CAN-12-4100
Acknowledgements
We thankfully acknowledge the technical support of Eduardo Gimenes Martins and Carolina Bertelli.
Funding
This study was supported by funds from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and University of São Paulo (USP). Jamile R. Silva was a fellow supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Grant 2014/11073-1; Natiely S. Sales was a fellow supported by FAPESP, grant 2016/14344-1; Mariângela O. Silva was a fellow supported by FAPESP, Grant 2016/11397-7; Luana R. M. M. Aps was a fellow supported by FAPESP, Grant 2013/15360-2; Ana C. R. Moreno was a fellow supported by FAPESP, Grants 2015/16505-0 and 2016/00708-1 and supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Grant 560713; Mariana O. Diniz was a fellow supported by FAPESP, grant 2011/51218-0; Luís C. S. Ferreira was a fellow supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Grant 520931/1996-3. Elaine G. Rodrigues was a fellow supported by CNPq, Grant 308085/2015-9.
Author information
Authors and Affiliations
Contributions
JRS, MD and LCSF conceived the study and the experimental design. MOD supervised the experimental work. JRS, NSS, MOS, LRMMA, ACRM, and MOD carried out the experiments, processed the experimental data, and participated in the interpretation of the results. EGR discussed the results and contributed to the writing of the manuscript. JRS wrote the manuscript with support from MOD and LCSF.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All handling procedures were performed according to the protocol approved by the Ethics Committee on the Use of Animals (CEUA) of the Institute of Biomedical Sciences of the University of São Paulo (Brazil) on August 18, 2014 (project number 92/2014) and according to standard rules approved by the National Council for Control of Animal Experimentation (CONCEA), Brazil.
Animal source
Six–eight-week-old C57BL/6 mice were purchased from the Animal Breeding Center of the Institute of Biomedical Sciences of the University of São Paulo (Brazil) and housed in the Animal Facility at the same Institute.
Cell line authentication
The TC-1 cell line was originally created and kindly provided by Dr. T.C. Wu, Johns Hopkins University, Baltimore, MD, USA. The cells tested negative for mycoplasma by PCR and were cultured for no more than 2 weeks after thawing. The cells showed consistent morphologies and growth rates prior to use in in vivo experiments. The cells tested positive for the expression of the HPV-16 E6 and E7 proteins by Western blotting. The TC-1 cell line is not currently available from the ATCC bank.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Silva, J.R., Sales, N.S., Silva, M.O. et al. Expression of a soluble IL-10 receptor enhances the therapeutic effects of a papillomavirus-associated antitumor vaccine in a murine model. Cancer Immunol Immunother 68, 753–763 (2019). https://doi.org/10.1007/s00262-018-02297-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-02297-2