
Abstract
Purpose
The adenosine A2A receptor has emerged as a therapeutic target for multiple diseases, and thus the non-invasive imaging of the expression or occupancy of the A2A receptor has potential to contribute to diagnosis and drug development. We aimed at the development of a metabolically stable A2A receptor radiotracer and report herein the preclinical evaluation of [18F]FLUDA, a deuterated isotopologue of [18F]FESCH.
Methods
[18F]FLUDA was synthesized by a two-step one-pot approach and evaluated in vitro by autoradiographic studies as well as in vivo by metabolism and dynamic PET/MRI studies in mice and piglets under baseline and blocking conditions. A single-dose toxicity study was performed in rats.
Results
[18F]FLUDA was obtained with a radiochemical yield of 19% and molar activities of 72–180 GBq/μmol. Autoradiography proved A2A receptor–specific accumulation of [18F]FLUDA in the striatum of a mouse and pig brain. In vivo evaluation in mice revealed improved stability of [18F]FLUDA compared to that of [18F]FESCH, resulting in the absence of brain-penetrant radiometabolites. Furthermore, the radiometabolites detected in piglets are expected to have a low tendency for brain penetration. PET/MRI studies confirmed high specific binding of [18F]FLUDA towards striatal A2A receptor with a maximum specific-to-non-specific binding ratio in mice of 8.3. The toxicity study revealed no adverse effects of FLUDA up to 30 μg/kg, ~ 4000-fold the dose applied in human PET studies using [18F]FLUDA.
Conclusions
The new radiotracer [18F]FLUDA is suitable to detect the availability of the A2A receptor in the brain with high target specificity. It is regarded ready for human application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The signaling molecule adenosine is an important modulator of neurotransmission, regulating physiological processes such as sleep, motor activity, or sensorimotor gating [1]. Consequently, modulating adenosine signaling is an emerging treatment option for neuropsychiatric and neurodegenerative disorders [1]. Adenosine regulates neurotransmission of glutamate, acetylcholine, γ-aminobutyric acid, and dopamine using four G-protein-coupled plasma membrane receptors - adenosine A1, A2A, A2B, and A3 receptors [1]. Whereas A1, A2B and A3 receptors are widely distributed throughout the brain, the A2A receptor is specifically expressed at high densities in the dorsal and ventral striatum, the main input structures of basal ganglia circuitry [2]. The binding of adenosine to the A2A receptor activates protein kinase A (PKA) via G protein–mediated stimulation of adenylyl cyclase and the corresponding increase in cyclic adenosine monophosphate (cAMP). In addition, PKA-independent pathways have also been reported [3, 4]. The A2A receptor forms dimers with itself and with further G-protein-coupled receptors, in particular D2, mGluR5, CB1, and A1 [3], which causes changes in A2A density in a wide range of neuropsychiatric and neurodegenerative diseases.
For instance, the measurement of mRNA levels indicated a reduced A2A receptor density in the caudate and putamen of patients with Parkinson’s disease (PD) [5], while increased A2A protein levels were found in PD patients with dyskinesias [6, 7] and in patients with Huntington’s disease (HD) [8]. Given that PD is characterized by a reduced mobility whereas PD patients with dyskinesias and HD patients show excessive movements, these findings collectively indicate that A2A is an important reporter - and modulator - of mobility in human basal ganglia. Accordingly, the A2A antagonist istradefylline has been approved for treatment of PD in the USA and in Japan [3, 9].
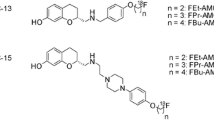
Molecular imaging of the A2A receptor by means of positron-emission tomography (PET) has the potential to quantitatively assess the receptor availability and changes thereof during the course of neuropsychiatric diseases’ pathological processes and determine optimal dosing regiments for drugs targeting A2A. Accordingly, research teams in both academia and industry have been working on the development of suitable PET radiotracers since more than 15 years. Initially, 11C-labeleld caffeine derivatives such as [11C]KF17837 [10], [11C]CSC [11], or [11C]KF21213 [12] were developed and investigated in animal models. With [11C]SCH442416, one of the first non-xanthine A2A receptor antagonists has been proven as suitable for in vivo imaging [13]. The first studies in humans assessing the distribution of A2A receptors in normal human brain with [11C]KF18446 (also known as [11C]TMSX, Fig. 1) [14] as well as the occupancy of the A2A receptor of the targeted drug candidate vipadenant with [11C]SCH442416 (Fig. 1) were published about 10 years ago [15]. At about the same time, the first 18F-labeled radiotracers were reported, e.g., analogs of SCH442416, such as [18F]MRS5425, (also known as [18F]FESCH, Fig. 1) [16, 17]. [18F]MNI-444 (Fig. 1) is another PET radiotracer that was used for imaging of the A2A receptor in healthy human subjects [18]. To assess the suitability of A2A receptor PET for the assessment of changes in the availability of A2A/D2 heterodimeric receptors in neurodegenerative diseases, our group performed dynamic PET studies in a rotenone-based mouse model of Parkinson’s disease with [18F]FESCH. However, the study yielded inconclusive data, at least in part due to the penetration of a non-negligible fraction of a single radiometabolite into the mouse’ brain [19]. Therefore, we intended to enhance the metabolic stability of [18F]FESCH by developing a deuterated isotopologue, FLUDA. To test the hypothesis that a selective substitution with deuterium improves the imaging properties of the novel A2A receptor–specific radiotracer [18F]FLUDA (Fig. 1), we performed a series of preclinical animal studies including in vivo metabolism and dynamic PET/MRI investigations in mouse and dynamic PET investigations in piglets to investigate target specificity and pharmacokinetics in different species. To further support the transfer of [18F]FLUDA to first-in-human studies, a preclinical acute toxicity study in rats has been commissioned as well.

Representative radiotracers for PET imaging of the A2A receptor and the herein reported [18F]FLUDA
Materials and methods
The full description of all procedures is provided in the Supplementary information (SI).
Chemical synthesis
FLUDA was synthesized by a microwave-assisted alkylation reaction of 4-[3-(5-amino-2-furan-2-yl-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-propyl]-phenol (desmethyl SCH442416) and 2-fluoroethyl-1,1,2,2-d4 4-methylbenzenesulfonate (3), that was previously prepared by tosylation of the commercially available ethane-d4-1,2-diol (1) and fluorination of ethane-1,2-diyl-d4 bis(4-methylbenzenesulfonate) (2, Scheme 1).

Synthesis of FLUDA, reagents and conditions. a p-TsCl, NEt3, CH2Cl2, 0 °C, 3 h, 68% yield. b TBAF, MeCN/THF, 90 °C, 15 min, 36% yield. c Desmethyl SCH442416, Cs2CO3, MeOH, microwave heating (1 h, 100 °C, 100 W), 37% yield
Radiosynthesis
[18F]FLUDA was prepared by a two-step one-pot manual radiosynthesis of the tosylate precursor 2 and the phenol precursor desmethyl SCH442416 with [18F]fluoride in anhydrous acetonitrile (MeCN) in the presence of potassium carbonate and Kryptofix 222 (K222) according to our optimized procedure for the radiosynthesis of [18F]FESCH (Fig. 2a) [19].

a Scheme of the two-step one-pot procedure for the radiosynthesis of [18F]FLUDA. b Representative chromatograms of isolation of [18F]FLUDA by semi-preparative HPLC. c Identification of [18F]FLUDA. HPLC chromatograms obtained by co-injection with the reference compound FLUDA
Physiochemical properties
The chemical stability of [18F]FLUDA was proven in saline, phosphate-buffered saline (PBS, pH 7.4) and n-octanol by incubation at 37 °C up to 60 min followed by radio-TLC and radio-HPLC analyses. The logD7.4 value was experimentally determined by the conventional shake-flask method using n-octanol and PBS as partition system (n = 4).
Animals
The animal experiments were performed with female CD-1 mice (10–12 weeks, 26–38 g) obtained from the Medizinisch-Experimentelles Zentrum (MEZ) at Universität Leipzig (Leipzig; Germany) and female piglets (6–12 weeks old, 14–18 kg) obtained from the Lehr-und Versuchsgut of the Faculty of Veterinary Medicine at Universität Leipzig (LVG; Oberholz; Germany).
In vitro binding assays
The test compounds (10 mM stock in DMSO) were incubated with crude cell membrane homogenates obtained from CHO cells stably transfected with human A2A receptor or human A1 receptor and with A2A receptor–specific [3H]ZM241385 or A1 receptor–specific [3H]DPCPX in an incubation buffer at room temperature (RT). The IC50 values were determined by non-linear regression analysis with GraphPad Prism 4.1 (GraphPad Inc.; La Jolla; CA), and Ki values estimated according to the Cheng-Prusoff equation with KD,ZM241385 = 0.8 nM and KD,DPCPX = 0.45 nM.
In vitro autoradiography
Cryosections from mouse and piglet brain were thawed, dried, and pre-incubated with buffer containing adenosine deaminase (ADA), and then with buffer containing ADA and [18F]FLUDA alone or together with 10 μM ZM241385 to assess non-specific binding of [18F]FLUDA or together with different concentrations of test compounds to estimate their binding affinity towards the A2A receptor. After washing and drying, the slides were exposed to a phosphor imager plate, and after scanning, the autoradiographic images were analyzed with the AIDA 2.31 software and inhibition curves created with GraphPad Prism 4.1 (GraphPad Inc., La Jolla, CA).
In vivo metabolism studies
In mice, [18F]FLUDA was administered i.v. as a bolus in the tail vein of awake animals (~ 36 MBq, n = 3). After 15 min, the animals were slightly anesthetized by isoflurane inhalation, and blood samples were taken by retro-orbital bleeding. The blood plasma was obtained by centrifugation. The brain was isolated immediately after cervical dislocation and homogenized in water. For analysis by RP-HPLC, brain homogenates and blood plasma were mixed with the 4-fold volumes of acetone/water (4/1; v/v), precipitated proteins removed by centrifugation, the supernatants were concentrated and analyzed by analytical RP-HPLC. For analysis by micellar-HPLC (MLC), brain homogenates and blood plasma were mixed with equal volumes of aqueous sodium dodecyl sulfate and directly injected into the MLC system.
In piglets, [18F]FLUDA was administered i.v. as a bolus in the auricular vein of the anesthetized animals (~ 203 MBq, n = 2). Arterial blood was sampled over 120 min and the blood plasma was obtained by centrifugation. For analysis by semi-preparative RP-HPLC, blood plasma was mixed with the twofold volumes of acetone/water (4/1; v/v), precipitated proteins were removed by centrifugation, and the supernatants were concentrated and analyzed by semi-preparative RP-HPLC.
Dynamic PET studies in mice
PET/MRI scans were performed using a preclinical PET/MRI system (PET/MRI 1Tesla; nanoScan®; MEDISO Medical Imaging Systems; Budapest; Hungary) in CD-1 mice under baseline (n = 4), control (vehicle, n = 8), and blocking (pre-administration of 2.5 mg/kg tozadenant (also known as SYN-115) or 1.0 mg/kg istradefylline (also known as KW-6002) i.v. 15 to 8 min before radiotracer n = 4, respectively) conditions. Dynamic whole-body animal PET scans were acquired during 60 min after i.v. administration of [18F]FLUDA (3.1–9.7 MBq, 0.7–4.5 nmol/kg). T1-weighted imaging was performed afterwards for anatomical orientation and attenuation correction. After reconstruction, PET images were analyzed in PMOD 3.9 (PMOD technologies LLC; Zurich; Switzerland), and volumes of interest were applied to the PET series to extract time-activity curves (TACs). TACs were expressed in standardized uptake value (SUV).
PET studies in piglets
PET scans were obtained on a clinical PET-System (ECAT Exact HR+; Siemens Healthcare GmbH; Erlangen; Germany) in piglets under control (vehicle, n = 1) and blocking conditions (2.5 mg tozadenant /kg i.v. 15 min before radiotracer followed by continuous infusion at 0.9 mg/kg/h for the duration of the study, n = 1). Dynamic PET scans were acquired during 90 min after i.v. administration of [18F]FLUDA (178–229 MBq; 0.08–0.16 fmol/kg, for control and blocking conditions respectively). After reconstruction, PET images were analyzed in PMOD 3.9 (PMOD technologies LLC; Zurich; Switzerland).
Toxicity studies in rats
The extended single-dose toxicity studies of FLUDA in male (n = 45) and female (n = 45) outbreed Wistar rats were performed in the Biological Testing Laboratory (BTL) in Russia (Study Number 678/19). The test item FLUDA was administered by a single bolus i.v. injection at doses of 6 and 30 μg/kg body weight (bw). Mortality, clinical pathology parameters (hematology and serum chemistry), organ weights, and microscopic tissue parameters were investigated 24 h and 2 weeks after treatment.
Results
Synthesis of the reference compound and affinity investigation
The deuterated derivative of FESCH was obtained by the use of a deuterated fluoroethyl building block according to the synthesis described in Scheme 1. Binding studies were performed by competitive radiotracer binding assays and revealed high-affinity binding of FLUDA towards the human A2A receptor with a Ki value of 0.74 ± 0.26 nM and negligible binding towards the human A1 receptor (Ki > 1 μM).
Radiosynthesis, lipophilicity, and chemical stability
[18F]FLUDA was obtained by a one-pot two-step radiofluorination as shown in Fig. 2 with a radiochemical yield of 19 ± 3% (end of bombardment = EOB), a radiochemical purity of ≥ 99%, and molar activities in the range of 72–180 GBq/μmol (end of synthesis = EOS) within a total synthesis time of 102 ± 4 min (n = 9). The shake-flask method was used to determine the logD7.4 value of [18F]FLUDA (2.01 ± 0.07). The incubation of [18F]FLUDA in saline, PBS (pH 7.4), and n-octanol has shown no degradation or defluorination.
In vivo metabolism
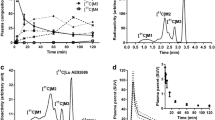
At 15 min after i.v. injection of [18F]FLUDA in mice and analysis, the parent fraction accounted for about 100% (RP-HPLC, recovery: 98%) and 95% (MLC) in brain (Fig. S2); and 71% (RP-HPLC, recovery: 86%, Fig. 3) and 56% (MLC, Fig. S1) in plasma. In piglets, the parent fraction accounted for about 47% in plasma at 16 min p.i (recovery: 89%, Fig. S1). The metabolic pattern in mice contained two fractions of radiometabolites, [18F]M1 and [18F]M2, not able to cross the blood-brain barrier (BBB). In contrast to the in vivo stability of [18F]FLUDA in mice, the plasma samples of piglets contained two additional radiometabolites, [18F]M3 and [18F]M4, supposed to have a similar structure to [18F]FLUDA based on their chromatographic behavior as shown in Fig. 3. Due to the PET studies in piglets (Fig. 7), these two radiometabolites are not expected to cross the BBB.

Representative RP-HPLC radiochromatograms of plasma samples after administration of [18F]FLUDA to a mouse and piglet
Quantitative in vitro autoradiography
The binding pattern of [18F]FLUDA in mouse brain is heterogeneous with the highest density of binding sites in the striatum, an A2A receptor–rich region. In A2A receptor–poor regions, such as cerebellum, midbrain, cortex, or thalamus, only negligible binding was detected (Fig. 4a, Fig. S3). About 90% of the binding of ~ 1 nM [18F]FLUDA in mouse striatum could be displaced by co-incubation with 10 μM ZM241385 (Fig. 4b). The binding sites in striatum were further characterized by saturation experiments which revealed a KD value of 4.30 ± 0.73 nM and a Bmax value of 556 ± 143 fmol/mg wet weight (Fig. 4d). Comparable results were obtained in a single experiment performed in cryosections of the pig brain to promote the preclinical evaluation of [18F]FLUDA in larger species (Fig. 5). The nearly exclusive binding of [18F]FLUDA in the striatum (Fig. 5a) was completely blocked by co-administration of ZM241385 (Fig. 5b). By homologous competition, the binding of FLUDA in the pig striatum was characterized with a KD value of 0.68 nM and a Bmax value of 218 fmol/mg wet weight (Fig. 5c).

Representative in vitro autoradiographic images of the binding pattern of [18F]FLUDA (0.93 nM) in horizontal mouse brain slices. The highest accumulation of activity in the striatum (a, red). The binding is completely blocked by co-administration of 10 μM of the A2A receptor antagonist ZM241385 (b). For annotation of the brain regions, the slices were Nissl-stained after autoradiography (c). St striatum, Cb cerebellum, and Cx cortex, Th thalamus. Representative competition curve (d). KD and Bmax calculated from the homologous completion of [18F]FLUDA with FLUDA (Cheng-Prusoff equation KD = IC50− [[18F]FLUDA] and Bmax = top – bottom ∙ (KD + [[18F]FLUDA])/[[18F]FLUDA] [20].

In vitro autoradiographic images of the binding pattern of [18F]FLUDA (0.64 nM) in sagittal pig brain slices. The highest accumulation of activity is in the striatum (a, red, St striatum, Cb cerebellum, Cx cortex, and Th thalamus). The binding is completely blocked by co-administration of 10 μM of the A2A receptor antagonist ZM241385 (b). Homologous competition curve (c)
Dynamic PET studies in mice
Representative PET images of [18F]FLUDA in a mouse brain are shown in Fig. 6a. Corresponding to the in vitro autoradiography, the highest uptake was detected in the A2A receptor–rich striatum. Also, the negligible accumulation of activity in A2A receptor–poor regions, such as cerebellum, is in agreement with the in vitro data. These general findings are confirmed by the analysis of the PET-derived regional time-activity curves (TACs) presented in Fig. 6b. In fact, a high specific-to-non-specific binding ratio was reached, with a maximum SUVrSt/Cb of 8.3 reflecting a high target selectivity (Fig. 6c). Furthermore, the continuous washout of activity from all brain regions confirms the absence of brain-penetrant radiometabolites. To validate the target specificity of [18F]FLUDA in vivo, the A2A receptor antagonist istradefylline (1 mg/kg) or tozadenant (2.5 mg/kg) were administered i.v. at 10 min before radiotracer injection. Maximum blocking was obtained by pre-treatment of istradefylline as reflected by the striatal SUV being similar to the cerebellar SUV under blocking conditions during the entire period of investigation (Fig. 6d–e). By tozadenant pre-treatment, the accumulation of activity in the striatum was reduced by 23.5% in comparison to the control (according to the area under the curve (AUC); AUCvehicle: 24.9 ± 8.7 SUV ∙ min; AUCtozadenant: 19.0 ± 4.3 SUV ∙ min from 0 to 60 min; Fig. S8). The activity accumulation in the cerebellum was not significantly affected by the blocking compound (Fig. 6e; AUCvehicle: 5.9 ± 3.0 SUV ∙ min; AUCtozadenant: 5.4 ± 1.3 SUV ∙ min from 0 to 60 min; Fig. S8), confirming the target specificity of [18F]FLUDA and indicating the suitability of the cerebellum as a reference region for A2A receptor imaging.

a Representative horizontal PET images (0–60 min) of [18F]FLUDA in the brain of CD-1 mice (striatum: red, cerebellum: yellow). b TACs at baseline for CD-1 mice (n = 4) in different brain regions after injection of [18F]FLUDA; TACs of c SUVrSt/Cb, d SUV striatum, and e SUV cerebellum: after pre-treatment with vehicle (red square, n = 8), tozadenant (2.5 mg/kg bw, blue circle, n = 4) and istradefylline (1.0 mg/kg bw, green triangle, n = 4)
Further analysis of the whole-body PET data obtained in control (vehicle-treated) animals revealed a high initial uptake in the small intestine and the liver, followed by a pronounced accumulation of activity in the former, along with a stable uptake in the kidney over the whole scanning time, indicating that [18F]FLUDA and/or its metabolites mainly undergo hepatobiliary excretion (Table S2; Fig. S9).
Dynamic PET studies in piglets
To further assist the transfer of [18F]FLUDA to clinical studies, we performed a pilot study to evaluate the pharmacokinetic profile of [18F]FLUDA by dynamic PET in piglets. The activity distribution in the brain of a control subject resembles the findings obtained in mouse—strong and nearly exclusive accumulation in the striatum and low signal in all other brain regions (Fig. 7a). The pre-administration of tozadenant (2.5 mg/kg) at 15 min before radiotracer followed by continuous infusion inhibited the accumulation of activity in the striatum by 50% (AUCvehicle: 42.1 SUV ∙ min; AUCtozadenant: 21.0 SUV ∙ min from 0 to 60 min), and in A2A receptor–poor regions, such as cerebellum by 17% (AUCvehicle: 24.0 SUV ∙ min; AUCtozadenant: 19.8 SUV ∙ min from 0 to 60 min; Fig. 7b).

a Representative horizontal PET images (0–60 min) of [18F]FLUDA in the brain of piglets (striatum: red, cerebellum: yellow). b TACs of SUVrSt/Cb of [18F]FLUDA in the pig brain after administration of vehicle (red square, n = 1) or blocking with tozadenant (blue circle, n = 1)
Single-dose toxicity study
In an extended single-dose toxicity study in rats, administration of FLUDA up to 30 μg/kg body weight, which is about 4000 times the estimated human dose and thus in accordance with the ICH guideline M3(R2), which recommends a minimum of a 50-fold the clinical exposure as the maximum dose for general toxicity studies in any species, did not cause any conspicuous features related to the hematology, clinical chemistry, necropsy, and histopathology analyses.
Discussion
The selective deuteration of the known A2A receptor radiotracer [18F]FESCH significantly improved the metabolic stability of the A2A receptor PET ligand [18F]FLUDA. Furthermore, the preclinical evaluation of [18F]FLUDA in both small and larger animals demonstrates high specific binding towards the adenosine A2A receptor in the brain, indicating the suitability of [18F]FLUDA for first-in-human studies.
In comparison to [18F]FESCH, [18F]FLUDA is much less susceptible to biotransformation. This is reflected by the much higher amount of the parent fraction in plasma at 15 min after i.v. injection in mice (41% [19] vs. 71%). The activity accumulating in the brain after i.v. injection of [18F]FLUDA corresponds almost completely to the parent compound. This finding is supported by the profile of the region-specific TACs obtained in dynamic PET studies with [18F]FLUDA in both mice (Fig. 6) and piglets (Fig. 7), which do not indicate any confounding accumulation of radiometabolites in the brain. In summary, deuteration of the fluoroethoxy group of [18F]FLUDA eliminates the main obstacle observed in the investigation of disease-related changes of the A2A receptor availability in preclinical studies using [18F]FESCH: significant amounts of brain-penetrant radiometabolites.
With respect to further more general properties such as affinity, selectivity, blood-brain barrier permeability, and clearance, [18F]FLUDA retains the positive characteristics of [18F]FESCH [16, 17, 19, 21]. The affinity of [18/19F]FLUDA towards human and mouse A2A receptors (Ki ~ 1 nM vs. KD ~ 4 nM) corresponds well to the results obtained for [18/19F]FESCH (Ki ~ 1 nM vs. KD ~ 5 nM; [19]) including a slight discrepancy between the two species that was reported previously (Table S1, Figs. S3, S4, S5, S6, S7) [22]. Regarding specificity, the accumulation of [18F]FLUDA resembles the expression pattern of the A2A receptor protein in both mouse brain and pig brain and both in vitro and in vivo (Figs. 4, 5, 6, and 7). Moreover, the accumulation of [18F]FLUDA can be blocked completely by structurally different selective A2A receptor antagonists (Figs. 6 and 7). The presence of an A2A receptor antagonist inhibited the accumulation of activity in the striatum almost completely but did not affect any other brain region in mice and affected the cerebellum by 17% in piglet. However, the high specificity of [18F]FLUDA, as well as the absence of effect of blocking compound on cerebellum activity accumulation in mice studies, suggests that this effect on the cerebellum of piglet is most likely due to inter-individual variability, and would need a larger-scale study to investigate its significance. Collectively, these findings indicate highly specific in vivo binding of [18F]FLUDA in the striata in both species.
The differences in the efficacy of the two A2A receptor ligands istradefylline and tozadenant to inhibit the binding of [18F]FLUDA in vitro and in vivo can be explained by known differences in the properties of these antagonists, in particular the remarkably low affinity of tozadenant towards mouse A2A receptors, which we determined in-house by [18F]FLUDA autoradiography (istradefylline: Ki (mouse A2A receptor) ~ 60 nM, Ki (piglet A2A receptor) ~ 15 nM; tozadenant: Ki (mouse A2A receptor) ~ 250 nM, Ki (piglet A2A receptor) ~ 10 nM; Table S1). Most of the preclinical studies that evaluated the specificity of A2A receptor–targeting PET radiotracers use istradefylline [17, 21, 23], whereas tozadenant has only been applied, to the best of our knowledge, in a single study in rhesus monkeys [24]. A head-to-head comparison as presented here for mice has not been reported so far. The higher efficacy of tozadenant in vivo in piglet in comparison to mouse (~ 50% displacement vs. ~ 25% displacement at 2.5 mg/kg followed by 0.9 mg/kg/h infusion and 2.5 mg/kg, respectively) can be explained by the dose-response results reported for the occupancy of [18F]MNI-444-labeled binding sites by tozadenant (47% vs. 95% at 1.5 mg/kg and 10.5 mg/kg, respectively; [24]). The observed difference in the maximum striatum-to-cerebellum ratio between mice (8.3) and piglet (2.1) is regarded to be caused by the different A2A receptor densities in the striatum, which were autoradiographically determined to be twofold higher in mice than in pigs [25].
By taking into consideration the density of the A2A receptor in the human striatum under physiologic conditions (e.g., 260–444 fmol/mg protein [26, 27]), we assume that the binding potential of [18F]FLUDA is suitable to quantify the receptor availability in humans. The highly specific binding of [18F]FLUDA towards the A2A receptor was confirmed by blocking studies conducted in vitro (Figs. 4 and 5) and in vivo (Figs. 6 and 7). The presence of A2A receptor antagonists inhibited the accumulation of activity in striatum almost completely but did not affect any other brain region. Accordingly, from the herein presented exploratory study, we assume that similar to the already clinically applied non-deuterated [18F]FESCH or [11C]preladenant, the quantification and kinetic modeling of PET data obtained with [18F]FLUDA might be facilitated by the use of the cerebellum as a reference region [21, 28]. However, full kinetic modeling with arterial input functions, ideally performed in a large animal and under conditions comparable to human studies, is required to validate the use of such region for the quantification of the availability of the A2A receptor by [18F]FLUDA PET.
Conclusion
[18F]FLUDA is a new A2A receptor–targeting PET radiotracer with promising preclinical results for clinical translation. The radiotracer can be prepared with high molar activities and in reasonable radiochemical yield (manuscript reporting automated synthesis under preparation). Highly affine and specific binding of [18F]FLUDA in vivo was demonstrated by PET studies performed in different species. The selective deuteration resulted in a high metabolic stability, and the identification of cerebellum as a reference region is assumed to facilitate the quantification of the availability of the A2A receptor in the brain and disease-related changes thereof in clinical routine. Furthermore, the results of a toxicity and a dosimetry study [29] indicate that the use of [18F]FLUDA in first-in-human imaging studies is safe.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Cunha RA. How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem. 2016;139:1019–55. https://doi.org/10.1111/jnc.13724.
Borroto-Escuela DO, Fuxe K. Adenosine heteroreceptor complexes in the basal ganglia are implicated in Parkinson’s disease and its treatment. J Neural Transm (Vienna). 2019;126:455–71. https://doi.org/10.1007/s00702-019-01969-2.
Domenici MR, Ferrante A, Martire A, Chiodi V, Pepponi R, Tebano MT, et al. Adenosine A2A receptor as potential therapeutic target in neuropsychiatric disorders. Pharmacol Res. 2019;147:104338. https://doi.org/10.1016/j.phrs.2019.104338.
Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. https://doi.org/10.1146/annurev.pharmtox.45.120403.095731.
Hurley MJ, Mash DC, Jenner P. Adenosine A2A receptor mRNA expression in Parkinson’s disease. Neurosci Lett. 2000;291:54–8. https://doi.org/10.1016/s0304-3940(00)01371-9.
Ramlackhansingh AF, Bose SK, Ahmed I, Turkheimer FE, Pavese N, Brooks DJ. Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology. 2011;76:1811–6. https://doi.org/10.1212/WNL.0b013e31821ccce4.
Villar-Menéndez I, Porta S, Buira SP, Pereira-Veiga T, Díaz-Sánchez S, Albasanz JL, et al. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol Dis. 2014;69:206–14. https://doi.org/10.1016/j.nbd.2014.05.030.
Varani K, Bachoud-Lévi AC, Mariotti C, Tarditi A, Abbracchio MP, Gasperi V, et al. Biological abnormalities of peripheral A2A receptors in a large representation of polyglutamine disorders and Huntington’s disease stages. Neurobiol Dis. 2007;27:36–43. https://doi.org/10.1016/j.nbd.2007.03.011.
Xu K, Bastia E, Schwarzschild M. Therapeutic potential of adenosine A2A receptor antagonists in Parkinson’s disease. Pharmacol Ther. 2005;105:267–310. https://doi.org/10.1016/j.pharmthera.2004.10.007.
Ishiwata K, Noguchi J, Toyama H, Sakiyama Y, Koike N, Ishii S, et al. Synthesis and preliminary evaluation of [11C]KF17837, a selective adenosine A2A antagonist. Appl Radiat Isot. 1996;47:507–11. https://doi.org/10.1016/0969-8043(95)00295-2.
Márián T, Boros I, Lengyel Z, Balkay L, Horváth G, Emri M, et al. Preparation and primary evaluation of [11C]CSC as a possible tracer for mapping adenosine A2A receptors by PET. Appl Radiat Isot. 1999;50:887–93. https://doi.org/10.1016/s0969-8043(98)00162-6.
Wang WF, Ishiwata K, Nonaka H, Ishii S, Kiyosawa M, Shimada J, et al. Carbon-11-labeled KF21213: a highly selective ligand for mapping CNS adenosine A2A receptors with positron emission tomography. Nucl Med Biol. 2000;27:541–6. https://doi.org/10.1016/s0969-8051(00)00126-8.
Todde S, Moresco RM, Simonelli P, Baraldi PG, Cacciari B, Spalluto G, et al. Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A2A receptor system using positron emission tomography. J Med Chem. 2000;43:4359–62. https://doi.org/10.1021/jm0009843.
Mishina M, Ishiwata K, Kimura Y, Naganawa M, Oda K, Kobayashi S, et al. Evaluation of distribution of adenosine A2A receptors in normal human brain measured with [11C]TMSX PET. Synapse. 2007;61:778–84. https://doi.org/10.1002/syn.20423.
Brooks DJ, Papapetropoulos S, Vandenhende F, Tomic D, He P, Coppell A, et al. An open-label, positron emission tomography study to assess adenosine A2A brain receptor occupancy of vipadenant (BIIB014) at steady-state levels in healthy male volunteers. Clin Neuropharmacol. 2010;33:55–60. https://doi.org/10.1097/WNF.0b013e3181d137d2.
Bhattacharjee AK, Lang L, Jacobson O, Shinkre B, Ma Y, Niu G, et al. Striatal adenosine A2A receptor-mediated positron emission tomographic imaging in 6-hydroxydopamine-lesioned rats using [18F]-MRS5425. Nucl Med Biol. 2011;38:897–906. https://doi.org/10.1016/j.nucmedbio.2011.01.009.
Khanapur S, Paul S, Shah A, Vatakuti S, Koole MJ, Zijlma R, et al. Development of [18F]-labeled pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH442416) analogs for the imaging of cerebral adenosine A2A receptors with positron emission tomography. J Med Chem. 2014;57:6765–80. https://doi.org/10.1021/jm500700y.
Barret O, Hannestad J, Vala C, Alagille D, Tavares A, Laruelle M, et al. Characterization in humans of 18F-MNI-444, a PET radiotracer for brain adenosine 2A receptors. J Nucl Med. 2015;56:586–91. https://doi.org/10.2967/jnumed.114.152546.
Schröder S, Lai TH, Toussaint M, Kranz M, Chovsepian A, Shang Q, et al. PET imaging of the adenosine A2A receptor in the rotenone-based mouse model of Parkinson’s disease with [18F]FESCH - synthesized by a simplified two-step one-pot radiolabeling strategy. Molecules. 2020;25. https://doi.org/10.3390/molecules25071633.
Harvey M. The GraphPad guide to analyzing radioligand binding data. Copyright© 1995-96 by GraphPad Software. http://www3.uah.es/farmamol/Public/GraphPad/radiolig.htm. Accessed 17 Aug 2020.
Khanapur S, Van Waarde A, Dierckx RAJO, Elsinga PH, Koole MJB. Preclinical evaluation and quantification of 18F-fluoroethyl and 18F-fluoropropyl analogs of SCH442416 as radioligands for PET imaging of the adenosine A2A receptor in rat brain. J Nucl Med. 2017;58:466–72. https://doi.org/10.2967/jnumed.116.178103.
Alnouri MW, Jepards S, Casari A, Schiedel AC, Hinz S, Müller CE. Selectivity is species-dependent: characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015;11:389–407. https://doi.org/10.1007/s11302-015-9460-9.
Zhou X, Boellaard R, Ishiwata K, Sakata M, Dierckx R, de Jong JR, et al. In vivo evaluation of 11C-Preladenant for PET imaging of adenosine A2A receptors in the conscious monkey. J Nucl Med. 2017;58:762–7. https://doi.org/10.2967/jnumed.116.182410.
Barret O, Hannestad J, Alagille D, Vala C, Tavares A, Papin C, et al. Adenosine 2A receptor occupancy by tozadenant and preladenant in rhesus monkeys. J Nucl Med. 2014;55:1712–8. https://doi.org/10.2967/jnumed.114.142067.
Sihver W, Schulze A, Wutz W, Stusgen S, Olsson RA, Bier D, et al. Autoradiographic comparison of in vitro binding characteristics of various tritiated adenosine A2A receptor ligands in rat, mouse and pig brain and first ex vivo results. Eur J Pharmacol. 2009;616:107–14. https://doi.org/10.1016/j.ejphar.2009.06.025.
Albasanz JL, Rodríguez A, Ferrer I, Martín M. Adenosine A2A receptors are up-regulated in Pick’s disease frontal cortex. Brain Pathol. 2006;16:249–55. https://doi.org/10.1111/j.1750-3639.2006.00026.x.
Wan W, Sutherland GR, Geiger JD. Binding of the adenosine A2 receptor ligand [3H]CGS 21680 to human and rat brain: evidence for multiple affinity sites. J Neurochem. 1990;55:1763–71. https://doi.org/10.1111/j.1471-4159.1990.tb04967.x.
Sakata M, Ishibashi K, Imai M, Wagatsuma K, Ishii K, Zhou X, et al. Initial evaluation of an adenosine A2A receptor ligand, 11C-preladenant, in healthy human subjects. J Nucl Med. 2017;58:1464–70. https://doi.org/10.2967/jnumed.116.188474.
Sattler B, Kranz M, Lai TH, Gündel D, Toussaint M, Schröder S, et al. Preclincal incorporation dosimetry of [18F]FLUDA - a novel 18F-labeled tracer for PET imaging of the expression of the adenosine A2A receptor (A2AR). J Nucl Med. 2020;61:1014.
Acknowledgments
We thank all colleagues of the Institute of Analytical Chemistry, Department of Chemistry and Mineralogy of Universität Leipzig (Leipzig; Germany), for NMR and HRMS measurements; Karsten Franke, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), for providing fluorine-18; Tina Spalholz, HZDR, for technical assistance; and Tatjana Sattler and all colleagues of the Department of Veterinary Medicine and Nuclear Medicine of University Hospital Leipzig (Leipzig; Germany) for the support of pig studies.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work (project no. 100226753) was funded by the European Regional Development Fund (ERDF) and Sächsische Aufbaubank (SAB).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Thu Hang Lai, Magali Toussaint, Rodrigo Teodoro, Sladjana Dukić-Stefanović, Friedrich-Alexander Ludwig, Barbara Wenzel, Daniel Gündel, Bernhard Sattler, Susann Schröder, and Winnie Deuther-Conrad. The first draft of the manuscript was written by Thu Hang Lai, Magali Toussaint, Winnie Deuther-Conrad, and Peter Brust, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All animal studies followed the international guidelines of animal care and were approved by Landesdirektion Leipzig (Reg.-Nr.: TVV 18/18; Reference number DD24.1-5131/446/19).
Consent to participate
Not applicable
Consent for publication
Not applicable
Code availability
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Preclinical Imaging
Supplementary Information
ESM 1
(PDF 1.95 mb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lai, T.H., Toussaint, M., Teodoro, R. et al. Improved in vivo PET imaging of the adenosine A2A receptor in the brain using [18F]FLUDA, a deuterated radiotracer with high metabolic stability. Eur J Nucl Med Mol Imaging 48, 2727–2736 (2021). https://doi.org/10.1007/s00259-020-05164-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-020-05164-4