
Abstract
Osmolytes are produced by various microorganisms as a defense mechanism to protect cells and macromolecules from damage caused by external stresses in harsh environments. Due to their useful stabilizing properties, these molecules are applied as active ingredients in a wide range of cosmetics and healthcare products. The metabolic pathways and biocatalytic syntheses of glycosidic osmolytes such as 2-O-α-d-glucosyl-d-glycerate often involve the action of a glycoside phosphorylase. Here, we report the discovery of a glucosylglycerate phosphorylase from carbohydrate-active enzyme family GH13 that is also active on sucrose, which contrasts the strict specificity of known glucosylglycerate phosphorylases that can only use α-d-glucose 1-phosphate as glycosyl donor in transglycosylation reactions. The novel enzyme can be distinguished from other phosphorylases from the same family by the presence of an atypical conserved sequence motif at specificity-determining positions in the active site. The promiscuity of the sucrose-active glucosylglycerate phosphorylase can be exploited for the high-yielding and rapid synthesis of 2-O-α-d-glucosyl-d-glycerate from sucrose and d-glycerate.
Key points
• A Xylanimonas protaetiae glycoside phosphorylase can use both d-glycerate and fructose as glucosyl acceptor with high catalytic efficiency
• Biocatalytic synthesis of the osmolyte 2-O-α-d-glucosyl-d-glycerate
• Positions in the active site of GH13 phosphorylases act as convenient specificity fingerprints
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The discovery of novel carbohydrate-active enzymes (CAZymes) is an area of broad interest due to the vital role that carbohydrates play in nature and in industry. The ongoing search for new CAZyme specificities continues to expand our understanding of natural metabolic pathways and frequently presents interesting opportunities for the synthesis and degradation of sugar-based compounds. However, the search is far from over. Large-scale sequencing efforts have led to an exponential growth of the number of protein sequences in the CAZy database, while only a fraction of these are ever experimentally characterized (Garron and Henrissat 2019; Drula et al. 2022). Considering the wide functional diversity that often exists even within one enzyme subfamily, it is not surprising that newly added sequences often receive an incorrect functional annotation. Rational approaches for enzyme discovery are very useful in that regard, as they can direct our attention to those CAZymes that are most likely to exhibit unique properties and, therefore, are worth studying in more detail.
A group of CAZymes that are particularly appealing for biotechnological applications are glycoside phosphorylases (GPs) (Li et al. 2022). These enzymes catalyze the breakdown of glycosidic bonds using inorganic phosphate to produce sugar 1-phosphates (i.e., phosphorolysis), but they can also catalyze the reverse reaction where such sugar 1-phosphates are used as glycosyl donor to form a new glycosidic bond (i.e., reverse phosphorolysis). Therefore, GPs are versatile synthetic tools that can be used to produce valuable carbohydrates, glycoconjugates, or glycosyl phosphates. The list of known GP specificities has grown considerably over the past few years, covering an increasingly large range of substrates and glycosidic linkages (Li et al. 2022). However, the spectrum of different phosphorylases remains narrow when compared to the enormous diversity of known glycoside hydrolases and glycosyltransferases.
Subfamily 18 of glycoside hydrolase family 13 (GH13_18) has proven to be an especially rich source of new GP specificities. The most famous member of this subfamily is sucrose phosphorylase (EC 2.4.1.7), of which the discovery dates back to the 1940s (Kagan and Latker 1942). This enzyme is quite promiscuous, allowing it to transfer the glucosyl moiety of sucrose not only to phosphate but also to a variety of different acceptor substrates with attractive yields. Hence, the transglycosylation activity of sucrose phosphorylase has been successfully exploited for the biocatalytic production of numerous glycosides and rare sugars (Franceus and Desmet 2020). In search for a more thermostable sucrose phosphorylase, we previously serendipitously identified a phosphorylase in GH13_18 that appeared to show a clear preference for sucrose 6F-phosphate over sucrose (EC 2.4.1.329) (Verhaeghe et al. 2014). This difference in specificity was found to be caused by few key sequence differences at positions in the active site of sucrose 6F-phosphate phosphorylase. The entire GH13_18 subfamily has since been thoroughly searched for other enzymes with diverging sequence motifs at those specificity-determining positions, leading to the discovery of phosphorylases that are not active on sucrose at all, but are instead dedicated to the (reverse) phosphorolysis of 2-O-α-glucosyl-d-glycerate (EC 2.4.1.352), 2-O-α-glucosylglycerol (EC 2.4.1.359), sucrose 6F-phosphate, or a substrate that is yet to be revealed (Franceus et al. 2017, 2018, 2019; Franceus and Desmet 2019; Tauzin et al. 2019). The availability of these enzymes has enabled the development of efficient new processes for the production of resveratrol glycosides, polyol glycosides, glucosylglycerol, and d-glycerate (Dirks-Hofmeister et al. 2015; Zhang et al. 2020, 2021, 2022).
Since the previous discovery of a novel specificity in GH13_18, the number of unique sequences in the subfamily has more than doubled. In this work, we report the characterization of a new member of GH13_18 with an unusual sequence motif at the specificity-determining positions in the active site and show that this enzyme can be applied for the high-yielding and rapid biocatalytic synthesis of 2-O-α-d-glucosyl-d-glycerate.
Materials and methods
Materials
All chemicals were obtained from Merck, unless noted otherwise. d-Glycerate was supplied as d-glyceric acid calcium salt dihydrate. The carbohydrates for the putative substrate screening were obtained from Biosynth. Primers were purchased from Integrated DNA Technologies.
Sequence analysis
The identifiers of proteins classified in GH13_18 were extracted from the CAZy database (http://www.cazy.org), and their amino acid sequences were obtained from GenBank. All duplicates were removed, leaving a total of ~ 1900 unique sequences. Clustal Omega was used to perform a multiple sequence alignment with default parameters (Sievers et al. 2011). A phylogenetic tree was constructed using RAxML v8.2.12 with the LG + I + G substitution model, and the tree was visualized using iTOL v5 (Stamatakis 2014; Letunic and Bork 2021).
Cloning, expression, and purification
The sequence of XpGP was codon-optimized for E. coli, synthesized and subcloned into a pET21a vector at the NheI and XhoI sites by Life Technologies (Belgium). The plasmid was used for transformation of E. coli BL21(DE3) electrocompetent cells. LB medium (500 mL in a 2-L flask; 10 g·L−1 tryptone, 5 g·L−1 NaCl, and 5 g·L−1 yeast extract) supplemented with 100 µg·mL−1 ampicillin was inoculated with an overnight culture (5 mL) containing these cells and incubated at 37 °C and 200 rpm until OD600 reached 0.6. The temperature was then lowered to 18 °C, and expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. After incubation for 16 h, cells were harvested by centrifugation, the supernatant was discarded, and the pellet was frozen at − 20 °C.
To extract and purify XpGP, the obtained pellet was thawed, resuspended in 8 mL lysis buffer (10 mM imidazole, 0.1 mM phenylmethylsulfonyl fluoride, 1 mg·mL−1 lysozyme, 50 mM phosphate-buffered saline; pH 7.4), and incubated on ice for 30 min. The lysate was sonicated three times for 2.5 min (Branson sonifier 450, level 3, 50% duty cycle), and the soluble fraction of the lysate was obtained by centrifugation (20,000 g for 30 min). The clarified lysate containing XpGP with a C-terminal His6-tag was purified by nickel-nitrilotriacetic acid chromatography following the instructions of the supplier (HisPur Ni–NTA; Thermo Fisher Scientific). The purified enzyme solution was concentrated, and the buffer was exchanged to 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) at pH 7.0 using an Amicon Ultra-15 centrifugal filter unit with a 30-kDa cutoff (Merck). The protein concentration was measured using a NanoDrop ND-1000 at 280 nm (Thermo Fisher Scientific) with the extinction coefficient (63,370 M−1 cm−1) and molecular weight (55.3 kDa) as determined by the ProtParam tool (https://web.expasy.org/cgi-bin/protparam/protparam).
Detection of reaction products
The release of inorganic phosphate from α-glucose 1-phosphate was monitored using the colorimetric phosphomolybdate assay (Gawronski and Benson 2004). The release of fructose from sucrose was monitored using the colorimetric bicinchoninic acid reducing sugars assay (Aerts et al. 2011). When using these assays to determine specific activities, one unit of activity was defined as the amount of enzyme that releases one µmol of the measured product (i.e., inorganic phosphate or fructose) per minute under the specified reaction conditions. All measurements of specific activities were performed in triplicate. Transglycosylation reactions starting from sucrose were monitored by high-performance anion exchange chromatography (HPAEC; Dionex ICS-600; Thermo Fisher Scientific) with a CarboPac PA20 pH-stable column and pulsed amperometric detection. The column was kept at 30 °C with a flow rate of 0.5 mL·min−1. The eluent composition was 30 mM NaOH for 8.5 min after which the NaOH concentration increased to 100 mM for 1.5 min. Next, the NaOAc concentration was gradually increased from 0 to 300 mM over 4 min. The eluent composition was kept constant for 0.5 min, gradually changed back to 30 mM NaOH and 0 mM NaOAc over 0.5 min, and kept constant for another 5 min. Before analysis, all reactions were inactivated by the low pH of the assay solution (phosphomolybdate assay) or by heating the sample at 95 °C for 5 min (HPAEC).
Characterization of XpGP
The activity of XpGP on various candidate glycosyl acceptors was evaluated by incubating 0.35 µM purified enzyme, 25 mM α-glucose 1-phosphate, and 50 mM putative substrate in 50 mM MOPS buffer at pH 7.0 and 30 °C for 30 min. A 50 µL-sample was analyzed using the phosphomolybdate assay, and a sample from a reaction without glycosyl acceptor was used as negative control.
The influence of pH on the activity of XpGP was determined by measuring the specific activity in reactions with 30 mM α-glucose 1-phosphate and 50 mM fructose at 30 °C in 50 mM acetate (pH 4.5–5.5), 2-morpholinoethanesulfonic acid (pH 5.5–6.5), MOPS (pH 7.0–7.5), or Tris (pH 8.0). The optimal temperature was determined using the same substrate concentrations in 50 mM MOPS at pH 7.0. Reaction samples were analyzed using the phosphomolybdate assay.
The thermodynamic stability of XpGP was evaluated by differential scanning fluorimetry in a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) using the FRET channel. Purified protein (22.5 µL of a 0.5 mg·mL−1 solution) was mixed with SYPRO Orange Protein Stain (2.5 µL of a 50X Stain solution). The temperature was increased from 20 to 99 °C at a rate of 0.5 °C·min−1, and fluorescence emissions were plotted as a function of temperature.
The kinetic stability of XpGP was evaluated by monitoring its residual specific activity at various time points during the incubation of 1 µM purified enzyme at 30 °C and at 45 °C in 50 mM MOPS, pH 7.0. Specific activities were determined with 30 mM α-glucose 1-phosphate and 50 mM fructose at 30 °C and pH 7.0, and reaction samples were analyzed using the phosphomolybdate assay.
Kinetic parameters were determined at the optimal pH (7.0) and temperature (30 °C) in 50 mM MOPS buffer. When varying the acceptor substrate concentration (fructose, d-glycerate, and l-glycerate), 30 mM α-glucose 1-phosphate was added as the cosubstrate, and reaction samples were analyzed using the phosphomolybdate assay. When varying the donor substrate concentration (sucrose), 100 mM phosphate was added as the cosubstrate, and reaction samples were analyzed using the bicinchoninic acid reducing sugars assay. The kinetic parameters were calculated by nonlinear regression of the Michaelis–Menten equation using SigmaPlot 14.5. For variant Q345A, the Michaelis–Menten equation was modified to incorporate substrate inhibition as follows: v0 = vmax⋅[S]/(KM + [S]⋅(1 + [S]/Ki)).
Mutagenesis
The Q5 mutagenesis kit (New England Biolabs) was used to generate the enzyme variants following the manufacturer’s instructions. The required primers were designed using the NEBaseChanger tool (Table S1).
Optimization of reaction conditions
Reactions for the synthesis of glucosylglycerate from sucrose and d-glycerate or l-glycerate were performed in Eppendorf tubes with a total reaction volume of 500 µM, without agitation, in 100 mM MOPS buffer at pH 7.0 and 30 °C. When monitoring the reaction over time, 5 µM purified XpGP was incubated with 300 mM sucrose and 300 mM d-glycerate. Samples were taken at various time points for 4 h and subsequently analyzed by HPAEC. When optimizing the substrate concentrations, the purified enzyme concentration was 3.7 µM, reactions were stopped after 4 h of incubation, and yields were determined by HPAEC. The conversion of sucrose to glucosylglycerate was monitored and quantified by determining the difference between the overall conversion of sucrose (i.e., release of fructose) and the contaminating hydrolytic conversion of sucrose (i.e., release of glucose).
NMR analysis
XpGP (5 µM) was incubated with 0.3 M sucrose and 0.3 M d-glycerate for 4 h at 30 °C. The reaction mixture was inactivated (5 min, 95 °C), centrifuged, and freeze-dried. 1H NMR and 13C NMR spectra were recorded at 400 and 100.6 MHz, respectively, on a Bruker Avance III HD Nanobay III instrument. Deuterated water was used as a solvent, and acetone was used as an internal chemical shift standard (δH 2.23 ppm and δC 31.5 ppm). All spectra were processed using TOPSPIN 3.6.3 software and compared to spectral data previously reported for 2-O-α-d-glucosyl-d-glycerate (Sawangwan et al. 2009).
Sequence information
The codon-optimized nucleotide sequence and the amino acid sequence of XpGP, including the C-terminal His6-tag, are shown in Table S2. The original nucleotide and amino acid sequences can also be found in GenBank under the accession numbers CP035493 and QAY71641.1, respectively. In UniProt, the amino acid sequence can be found with the identifier A0A4V0YGL6. Furthermore, the plasmid for expression of XpGP is publicly available at the BCCM/GeneCorner plasmid collection with accession number LMBP 13891.
Results
Sequence analysis and expression of an atypical glycoside phosphorylase
Protein sequences in subfamily GH13_18 contain a short stretch of specificity-determining residues that can be used to distinguish the different GP functions known to date (Franceus and Desmet 2020). This specificity fingerprint is found at positions 342–345 in the sucrose phosphorylase from Bifidobacterium adolescentis (BaSP). Structural analysis has shown that those positions are part of a dynamic loop that undergoes structural rearrangements during the catalytic cycle, while mutational studies have demonstrated that they are critically involved in substrate recognition in the + 1 subsite (Mirza et al. 2006; Verhaeghe et al. 2013). In BaSP, residues 342–345 establish important interactions with the fructosyl moiety of sucrose (Asp342), phosphate (Leu323 and Tyr344), or both (Gln345). By searching a multiple sequence alignment of subfamily GH13_18 for putative phosphorylases with a previously unreported sequence motif at the corresponding positions, we discovered a group of putative phosphorylases with an unusual LPHQ motif. Phylogenetic analysis showed that these proteins are most closely related to the BaSP-like sucrose phosphorylases, but do not share their characteristic D(L/I)YQ motif (Fig. 1). One representative from the group of atypical putative phosphorylases, originating from Xylanimonas protaetiae, was selected for expression and characterization. The putative glycoside phosphorylase from X. protaetiae (XpGP) with a C-terminal His6-tag was recombinantly expressed in Escherichia coli and purified to apparent homogeneity by affinity chromatography. Approximately 40 mg of purified protein could be obtained from a 500-mL culture.

Specificity-determining positions in CAZy subfamily GH13_18. a Simplified representation of the phylogeny of GH13_18. A sequence logo is shown of specificity-determining positions in subsite + 1 for each clade with characterized representatives (positions 342–345 in B. adolescentis sucrose phosphorylase and X. protaetiae glycoside phosphorylase). The clade containing the X. protaetiae GP is indicated by a blue box. b Homology model of the X. protaetiae GP with a glucose molecule in subsite -1, the catalytic residues (gold), and the specificity-determining residues in subsite +1 (blue)
Characterization of XpGP
To determine which substrate a novel CAZyme might be active on, earlier studies have drawn inspiration from the genomic context of the gene of interest. For example, genes encoding a glucosylglycerol phosphorylase are often situated next to other genes that are involved in glucosylglycerol metabolism, while those encoding a sucrose 6F-phosphate phosphorylase often reside in an operon with genes encoding a phosphofructokinase or sugar phosphotransferase system (Verhaeghe et al. 2014; Franceus et al. 2018). Unfortunately, scanning the genomic context of XpGP and its homologs did not reveal such clear patterns. Therefore, XpGP was screened on a diverse panel of candidate glucosyl acceptors in reactions with α-d-glucose 1-phosphate as the glucosyl donor (Table S3). Very high activity was observed in reactions with d-glycerate, indicating that XpGP is likely to be a glucosylglycerate phosphorylase. Several glucosylglycerate phosphorylases have already been discovered in GH13_18, although those are located in a different phylogenetic clade, they share low homology with XpGP (~ 30% sequence identity) and their sequences contain an entirely different characteristic sequence motif (Fig. 1). However, in contrast to all known glucosylglycerate phosphorylases from the same subfamily, which are known to have a very strict substrate specificity, XpGP also showed high activity in the presence of fructose as glycosyl acceptor.
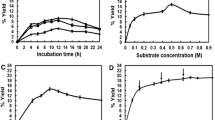
The optimal pH of XpGP was 7.0, which is comparable to that of other phosphorylases from the family (6.0–7.0) (Fig. S1A). Its optimal temperature for activity was 30 °C, matching the optimal growth temperature of X. protaetiae (28–30 °C) (Fig. S1B) (Heo et al. 2020). The apparent folding transition temperature (Tm) of XpGP was found to be 41 ± 1 °C by differential scanning fluorimetry. In addition, its kinetic stability was evaluated at 30 °C and 45 °C. At 30 °C, the enzyme retained 55% of its initial activity after 24 h of incubation. However, at 45 °C, only 40% of its activity was retained after 1 h of incubation, and it was fully inactivated after 4 h.
The kinetic parameters were determined in the synthesis direction of the reversible phosphorolysis reaction at the optimal pH and temperature, and the enzyme was found to exhibit Michaelis–Menten kinetics on the tested substrates (Table 1). The catalytic efficiency for d-glycerate (kcat/KM = 2.4 mM−1 s−1) was in line with that of several other GPs for their native acceptor substrate, further confirming the results from the substrate screening. XpGP showed a clear preference for d-glycerate over its enantiomer l-glycerate and over fructose. Further, the enzyme was confirmed to regioselectively synthesize 2-O-α-d-glucosyl-d-glycerate from sucrose and d-glycerate by NMR spectroscopy (Fig. S2). We also determined the kinetic parameters for sucrose in the phosphorolysis direction of the reaction and found that XpGP showed low affinity for this substrate (KM = 67 mM) in comparison to the values reported for known sucrose phosphorylases (KM = 1–15 mM). However, all previously reported glucosylglycerate phosphorylases are not active on sucrose at all (Franceus et al. 2017). In summary, the kinetic characterization of XpGP indicates that the enzyme can be regarded as a promiscuous sucrose-active glucosylglycerate phosphorylase (Fig. 2).

Reactions catalyzed by the Xylanimonas protaetiae glycoside phosphorylase from subfamily GH13_18. a Reversible phosphorolysis of 2-O-α-d-glucosyl-d-glycerate, b reversible phosphorolysis of sucrose, and c transglycosylation with sucrose and d-glycerate to form 2-O-α-d-glucosyl-d-glycerate
Mutational analysis
XpGP could successfully be distinguished from previously characterized phosphorylases by the presence of the atypical LPHQ sequence motif. The contribution of the residues in this sequence motif to substrate binding and catalysis was further examined by means of alanine scanning. All four amino acids (Leu342, Pro343, His344, and Gln345) were individually substituted with alanine to remove their side chain without disrupting the main chain conformation. Subsequently, the kinetic parameters for d-glycerate were determined for all variants (Table 2), revealing that all mutations resulted in significantly reduced turnover numbers. The L342A mutation showed the least detrimental effect, which is not surprising given the similar physicochemical properties of Leu and Ala. To more properly evaluate the importance of the Leu342 sidechain, we thus also replaced it by Asp, which is the residue found at the corresponding positions in sucrose phosphorylases. The L342D mutation did lower the affinity of XpGP for d-glycerate. The Q345A variant experienced a similar effect on affinity and also introduced clear substrate inhibition (Ki = 25 mM). Pro343 and His344 do not appear to be critically involved in the binding of d-glycerate. It has been described that the residues at the equivalent positions in sucrose phosphorylases primarily participate in phosphate binding, so it is likely that Pro343 and His344 play a similar role in XpGP (Verhaeghe et al. 2013). Curiously, the H334A mutation even improved the affinity of XpGP for d-glycerate at the cost of a severely reduced turnover number, resulting in the lowest catalytic efficiency overall.
One-step synthesis of glucosylglycerate from sucrose and glycerate
Sucrose phosphorylases have long been used for their ability to transfer the glycosyl moiety of sucrose to a diverse range of other molecules to form the corresponding glucosides in a single step. However, those alternative molecules are typically converted rather slowly, and d-glycerate is no exception (Aerts et al. 2011). Transglycosylation reactions with d-glycerate proceed at rates that are comparable to the contaminating hydrolytic reaction, where the enzyme transfers the glucosyl group of sucrose to water instead. Nevertheless, the Nidetzky group reported that an attractive yield (90% of d-glycerate converted) can be obtained with the sucrose phosphorylase from Leuconostoc mesenteroides (LmSP) by adding a 2.5-fold molar excess of sucrose, albeit after long incubation times (Sawangwan et al. 2009). Given the high activity of XpGP on both the donor sucrose and the acceptor d-glycerate, we hypothesized that it may be a more suitable catalyst for this process (Fig. 2c).
Incubating XpGP with equimolar amounts of sucrose and d-glycerate (300 mM) at 30 °C and pH 7.0 showed that the substrates were readily converted with a yield of 92% (Fig. 3). In contrast, LmSP can convert no more than 60% of d-glycerate at those concentrations because sucrose is gradually depleted due to hydrolysis (Sawangwan et al. 2009). Glucose was released by XpGP only in small amounts (< 1%), indicating that the undesired hydrolytic side reaction could effectively be suppressed by the presence of d-glycerate. Indeed, in the absence of any glycosyl acceptor, we found that XpGP hydrolyzed sucrose at a slightly higher rate (0.57 ± 0.04 U/mg) than LmSP did (0.30 ± 0.13 U/mg). Furthermore, transglycosylation reactions with XpGP proceeded considerably faster. Reactions with 5 µM XpGP and equimolar substrate concentrations reached maximal conversion after approximately 2 h, whereas the equivalent reaction with LmSP only converted 3% of sucrose at that time.

Conversion of 0.3 M sucrose and 0.3 M d-glycerate by 5 µM XpGP at 30 °C and pH 7.0 (▽, sucrose; ●, 2-O-α-d-glucosyl-d-glycerate; ○, glucose)
Next, the concentrations of sucrose and d-glycerate were varied and the yield was determined after 4 h of incubation (Table 3). The findings reaffirmed that significant hydrolysis of sucrose takes place only at low concentrations of d-glycerate. Hydrolytic activity was largely eliminated at a concentration of 300 mM, resulting in a yield of 92%. Providing a 1.33-fold molar excess of sucrose allowed reaching a yield of up to 98%. In comparison, the optimal reaction conditions with LmSP required a 2.5-fold molar excess of sucrose (Sawangwan et al. 2009). Further, l-glycerate was utilized as glycosyl acceptor far less efficiently than the preferred enantiomer, which may be a result of the lower affinity of XpGP for this substrate (Table 1).
Discussion
Microorganisms have evolved numerous strategies to survive or even thrive in extreme environments. One such defense mechanism involves the biosynthesis of organic molecules named compatible solutes or osmolytes that are accumulated to high intracellular levels in response to various stressful conditions (Empadinhas and da Costa 2011). 2-O-α-d-Glucosyl-d-glycerate is a widespread compatible solute that is known to improve osmoregulation and desiccation tolerance, especially during nitrogen starvation, and several enzymes involved in its metabolic pathways have been discovered over the last two decades (Nunes-Costa et al. 2017). Initially, it was believed that sucrose phosphorylases were also involved in glucosylglycerate metabolism. Genes encoding putative sucrose phosphorylases are frequently located near genes that encode the enzymes responsible for glucosylglycerate biosynthesis, and they were even found to be upregulated under nitrogen-limiting conditions (Ferreira et al. 2016). However, it was eventually discovered that those phosphorylases are not active on sucrose at all, but are instead strictly specific for glucosylglycerate (Franceus et al. 2017). In contrast, true sucrose phosphorylases are highly promiscuous enzymes that do show low transglycosylation activity with numerous alternative molecules, of which glycerate is just one example. The promiscuous sucrose-active glucosylglycerate phosphorylase reported in this study thus appears to combine the properties of the promiscuous sucrose phosphorylases and the strict glucosylglycerate phosphorylases.
Due to their unique and useful biological properties, osmolytes have attracted considerable interest for commercial applications (Becker and Wittmann 2020). The most popular example is ectoine, a non-proteinogenic amino acid that is widely used as cell protectant in skin or hair care products, or as a stabilizer of biomolecules in life sciences (Lentzen and Schwarz 2006). Glucosylglycerol, which is a structural analog of glucosylglycerate, is currently produced on industrial scale as well. The glycoside is synthesized by exploiting the transglycosylation activity of LmSP with sucrose as glycosyl donor and glycerol as acceptor, and it is applied as anti-aging ingredient and moisturizer in cosmetics (Goedl et al. 2008; Schagen et al. 2017). Although glucosylglycerate can be synthesized by a similar transglycosylation reaction with the same enzyme, this compound is not commercially available at this time, despite studies demonstrating that it outperforms other compatible solutes as a stabilizer of proteins at elevated temperatures, during storage, or during freeze-drying operations (Faria et al. 2008; Sawangwan et al. 2009, 2010).
The transglycosylation process from sucrose to d-glycerate catalyzed by XpGP offers a few clear advantages when compared to the corresponding processes for producing glycosidic osmolytes via LmSP. A high yield can be obtained with a vastly higher reaction rate, without requiring a large excess of substrate. However, the poor commercial availability of d-glycerate is an obvious hurdle that might hinder the large-scale implementation of the proposed process. The discovery of XpGP may stimulate the further development of practical biotechnological routes to generate d-glycerate from the cheap bulk feedstock glycerol, of which a few have already been explored before. Habe et al. investigated the ability of 162 acetic acid bacterial strains to accumulate d-glycerate when grown in media containing glycerol and found that Acetobacter tropicalis NBRC16470 can reach titers of 101.8 g/L with a 99% enantiomeric excess (Habe et al. 2009). Another promising strategy is the biocatalytic oxidation of glycerol by alditol oxidases. Although these enzymes prefer longer sugar alcohols such as sorbitol, the oxidase from Streptomyces coelicolor is capable of using glycerol as substrate, albeit with limited catalytic efficiency (Van Hellemond et al. 2009). Efforts to improve this promiscuous activity by directed evolution and semi-rational engineering have already yielded significantly improved variants, one of which has been integrated into an E. coli strain for the bioconversion of glycerol to optically pure d-glycerate with a titer of 30.1 g/L after 70 h (Gerstenbruch et al. 2012; Zhang et al. 2021). With further advances in strain engineering, protein engineering, or process optimization, the cost-effective production of 2-O-α-d-glucosyl-d-glycerate from sucrose and glycerol may finally become feasible, allowing this powerful osmolyte to be utilized for various interesting applications.
Data availability
All data that support the findings of this study are included within this paper. Raw data will be made available on reasonable request. The codon-optimized nucleotide sequence and the amino acid sequence of XpGP are shown in Table S2. The plasmid for expression of XpGP is publicly available at the BCCM/GeneCorner plasmid collection with accession number LMBP 13891.
References
Aerts D, Verhaeghe TF, Roman BI, Stevens CV, Desmet T, Soetaert W (2011) Transglucosylation potential of six sucrose phosphorylases toward different classes of acceptors. Carbohydr Res 346:1860–1867. https://doi.org/10.1016/j.carres.2011.06.024
Becker J, Wittmann C (2020) Microbial production of extremolytes — high-value active ingredients for nutrition, health care, and well-being. Curr Opin Biotechnol 65:118–128. https://doi.org/10.1016/j.copbio.2020.02.010
Dirks-Hofmeister ME, Verhaeghe T, De Winter K, Desmet T (2015) Creating space for large acceptors: rational biocatalyst design for resveratrol glycosylation in an aqueous system. Angew Chem 127:9421–9424. https://doi.org/10.1002/ange.201503605
Drula E, Garron M, Dogan S, Lombard V, Henrissat B, Terrapon N (2022) The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50:D571–D577. https://doi.org/10.1093/nar/gkab1045
Empadinhas N, da Costa MS (2011) Diversity, biological roles and biosynthetic pathways for sugar-glycerate containing compatible solutes in bacteria and archaea. Environ Microbiol 13:2056–2077. https://doi.org/10.1111/j.1462-2920.2010.02390.x
Faria TQ, Mingote A, Siopa F, Ventura R, Maycock C, Santos H (2008) Design of new enzyme stabilizers inspired by glycosides of hyperthermophilic microorganisms. Carbohydr Res 343:3025–3033. https://doi.org/10.1016/j.carres.2008.08.030
Ferreira C, Soares AR, Lamosa P, Santos MA, da Costa MS (2016) Comparison of the compatible solute pool of two slightly halophilic planctomycetes species, Gimesia maris and Rubinisphaera brasiliensis. Extremophiles 20:811–820. https://doi.org/10.1007/s00792-016-0868-0
Franceus J, Capra N, Desmet T, Thunnissen A-MWH (2019) Structural comparison of a promiscuous and a highly specific sucrose 6F-phosphate phosphorylase. Int J Mol Sci 20:3906. https://doi.org/10.3390/ijms20163906
Franceus J, Decuyper L, D’hooghe M, Desmet T, (2018) Exploring the sequence diversity in glycoside hydrolase family 13_18 reveals a novel glucosylglycerol phosphorylase. Appl Microbiol Biotechnol 102:3183–3191. https://doi.org/10.1007/s00253-018-8856-1
Franceus J, Desmet T (2020) Sucrose phosphorylase and related enzymes in glycoside hydrolase family 13: discovery, application and engineering. Int J Mol Sci 21:2526. https://doi.org/10.3390/ijms21072526
Franceus J, Desmet T (2019) A GH13 glycoside phosphorylase with unknown substrate specificity from Corallococcus coralloides. Amylase 32–40
Franceus J, Pinel D, Desmet T (2017) Glucosylglycerate phosphorylase, an enzyme with novel specificity involved in compatible solute metabolism. Appl Environ Microbiol 83:e01434-e1517. https://doi.org/10.1128/AEM.01434-17
Garron M-L, Henrissat B (2019) The continuing expansion of CAZymes and their families. Curr Opin Chem Biol 53:82–87. https://doi.org/10.1016/j.cbpa.2019.08.004
Gawronski JD, Benson DR (2004) Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal Biochem 327:114–118. https://doi.org/10.1016/j.ab.2003.12.024
Gerstenbruch S, Wulf H, Mußmann N, O’Connell T, Maurer KH, Bornscheuer UT (2012) Asymmetric synthesis of D-glyceric acid by an alditol oxidase and directed evolution for enhanced oxidative activity towards glycerol. Appl Microbiol Biotechnol 96:1243–1252. https://doi.org/10.1007/s00253-012-3885-7
Goedl C, Sawangwan T, Mueller M, Schwarz A, Nidetzky B (2008) A high-yielding biocatalytic process for the production of 2-O-(α-D-glucopyranosyl)-sn-glycerol, a natural osmolyte and useful moisturizing ingredient. Angew Chem Int Ed Engl 47:10086–10089. https://doi.org/10.1002/anie.200803562
Habe H, Shimada Y, Yakushi T, Hattori H, Ano Y, Fukuoka T, Kitamoto D, Itagaki M, Watanabe K, Yanagishita H, Matsushita K, Sakaki K (2009) Microbial production of glyceric acid, an organic acid that can be mass produced from glycerol. Appl Environ Microbiol 75:7760–7766. https://doi.org/10.1128/AEM.01535-09
Heo J, Kim S-J, Kim MA, Tamura T, Saitou S, Hamada M, Kim J-S, Hong S-B, Kwon S-W (2020) Lactococcus protaetiae sp. nov. and Xylanimonas protaetiae sp. nov., isolated from gut of larvae of Protaetia brevitarsis seulensis. Antonie Van Leeuwenhoek 113:1009–1021. https://doi.org/10.1007/s10482-020-01413-6
Kagan BO, Latker SN (1942) Phosphorolysis of saccharose by cultures of Leuconostoc mesenteroides. Biokhimiya 7:93–108
Lentzen G, Schwarz T (2006) Extremolytes: natural compounds from extremophiles for versatile applications. Appl Microbiol Biotechnol 72:623–634. https://doi.org/10.1007/s00253-006-0553-9
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296
Li A, Benkoulouche M, Ladeveze S, Durand J, Cioci G, Laville E, Potocki-Veronese G (2022) Discovery and biotechnological exploitation of glycoside-phosphorylases. Int J Mol Sci 23. https://doi.org/10.3390/ijms23063043
Mirza O, Skov LK, Sprogøe D, van den Broek L, a M, Beldman G, Kastrup JS, Gajhede M, (2006) Structural rearrangements of sucrose phosphorylase from Bifidobacterium adolescentis during sucrose conversion. J Biol Chem 281:35576–35584. https://doi.org/10.1074/jbc.M605611200
Nunes-Costa D, Maranha A, Costa M, Alarico S, Empadinhas N (2017) Glucosylglycerate metabolism, bioversatility and mycobacterial survival. Glycobiology 27:213–227. https://doi.org/10.1093/glycob/cww132
Sawangwan T, Goedl C, Nidetzky B (2009) Single-step enzymatic synthesis of (R)-2-O-α-D-glucopyranosyl glycerate, a compatible solute from micro-organisms that functions as a protein stabiliser. Org Biomol Chem 7:4267–4270. https://doi.org/10.1039/b912621j
Sawangwan T, Goedl C, Nidetzky B (2010) Glucosylglycerol and glucosylglycerate as enzyme stabilizers. Biotechnol J 5:187–191. https://doi.org/10.1002/biot.200900197
Schagen S, Overhagen S, Bilstein A (2017) New data confirm skin revitalizing and stress protection by Glycoin ® natural. Euro Cosmet 24–27
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Tauzin AS, Bruel L, Laville E, Nicoletti C, Navarro D, Henrissat B, Perrier J, Potocki-Veronese G, Giardina T, Lafond M (2019) Sucrose 6F-phosphate phosphorylase: a novel insight in the human gut microbiome. Microb Genomics 5:1–14. https://doi.org/10.1099/mgen.0.000253
Van Hellemond EW, Vermote L, Koolen W, Sonke T, Zandvoort E, Heuts DPHM, Janssen DB, Fraaije MW (2009) Exploring the biocatalytic scope of alditol oxidase from Streptomyces coelicolor. Adv Synth Catal 351:1523–1530. https://doi.org/10.1002/adsc.200900176
Verhaeghe T, Aerts D, Diricks M, Soetaert W, Desmet T (2014) The quest for a thermostable sucrose phosphorylase reveals sucrose 6’-phosphate phosphorylase as a novel specificity. Appl Microbiol Biotechnol 98:7027–7037. https://doi.org/10.1007/s00253-014-5621-y
Verhaeghe T, Diricks M, Aerts D, Soetaert W, Desmet T (2013) Mapping the acceptor site of sucrose phosphorylase from Bifidobacterium adolescentis by alanine scanning. J Mol Catal B Enzym 96:81–88. https://doi.org/10.1016/j.molcatb.2013.06.014
Zhang C, Chen Q, Fan F, Tang J, Zhan T, Wang H, Zhang X (2021) Directed evolution of alditol oxidase for the production of optically pure D-glycerate from glycerol in the engineered Escherichia coli. J Ind Microbiol Biotechnol 48:1–12. https://doi.org/10.1093/jimb/kuab041
Zhang T, Liu P, Wei H, Sun X, Zeng Y, Zhang X, Cai Y, Cui M, Ma H, Liu W, Sun Y, Yang J (2022) Protein engineering of glucosylglycerol phosphorylase facilitating efficient and highly regio- and stereoselective glycosylation of polyols in a synthetic system. ACS Catal 12:15715–15727. https://doi.org/10.1021/acscatal.2c05232
Zhang T, Yang J, Tian C, Ren C, Chen P, Men Y, Sun Y (2020) High-yield biosynthesis of glucosylglycerol through coupling phosphorolysis and transglycosylation reactions. J Agric Food Chem 68:15249–15256. https://doi.org/10.1021/acs.jafc.0c04851
Acknowledgements
This work was supported by Research Foundation – Flanders (FWO-Vlaanderen; postdoctoral fellowship J.F. with grant number 12ZD821N) and the Special Research Fund of Ghent University (grant number BOF/24J/2021/062). We would like to thank the Ghent University Core Facility “HTS for Synthetic Biology” for training, support and access to the instrument park.
Author information
Authors and Affiliations
Contributions
JF and TD conceived and designed the research. JF performed the sequence analysis and selected the enzyme. JF, MS, YA, and KB conducted experiments and analyzed data. JF wrote the manuscript with contributions of all authors. MD and TD supervised the work. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Franceus, J., Steynen, M., Allaert, Y. et al. High-yield synthesis of 2-O-α-d-glucosyl-d-glycerate by a bifunctional glycoside phosphorylase. Appl Microbiol Biotechnol 108, 55 (2024). https://doi.org/10.1007/s00253-023-12970-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-023-12970-x