
Abstract
The protrusion (P) domain of the major structural protein VP1 of norovirus (NoV) is critical for the host’s immune response and receptor binding. Most heterologous P domains expressed in Escherichia coli or Komagataella pastoris (formally known as Pichia pastoris) form P particles consisting of 24 P monomers formed through intermolecular contact in the P regions and an end-linked cysteine tag. The small P particle is only found in P domains with terminal modifications. In this study, the NoV P domain of the most predominant NoV strain GII.4 isolated from Taiwan was expressed in K. pastoris. A high yield of NoV P was obtained using the high-cell density fermentation process in K. pastoris. A large amount of the small P particles and the trimer and dimer complexes formed by 12, 6, and 2 P monomers were observed in both the expression of the NoV P-His and P containing cysteine tag at the N-terminus. Dynamic light scattering and transmission electron microscopy analysis of the purified NoV P-His and P revealed that most of these small P particles are triangle-, square-, and ring-shaped with a diameter of 14–15 nm. The binding ability of purified NoV P-His and P to human histo-blood group antigen was confirmed by a saliva-binding assay. Without terminal modification, small P particles were formed in our study. The amino acid sequence analysis showed only four different amino acids (residue 84, 119, 136, and 313) between the P domain in this study and other investigated GII.4 strains suggesting that these amino acids might play an important role in the P particle formation. The small P particles formed by the Taiwan-native norovirus P domain overexpressed in K. pastoris may provide further information for morphogenesis studies and vaccine development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Norovirus (NoV) is one of the leading causes of acute nonbacterial gastroenteritis outbreaks worldwide (Li et al. 2012). To date, there is no effective way to control a NoV outbreak. Vaccination is the primary strategy to protect humans from infectious diseases. Due to the lack of a reliable and efficient cell culture system for producing inactivated or attenuated whole NoV vaccines, the development of NoV vaccines relies largely on virus-like particles (VLPs) formed by the major capsid protein VP1 or subviral particles formed by a specific domain of VP1 (Lucero et al. 2017).
The NoV virion is composed of 180 monomers (or 90 dimers) of VP1, which form an icosahedral structure. Previous studies have shown that the viral VP1, when expressed alone and purified from eukaryotic cells, was capable of self-assembly into VLPs that were morphologically and antigenically indistinguishable from the authentic virus (Lucero et al. 2017; Xia et al. 2007). The monomeric VP1 consists of two major domains: the interior shell (S) domain and the exterior protrusion (P) domain linked by a flexible hinge (Tan and Jiang 2012). In particular, the P domain is required for dimer interaction and can be further divided into two different subdomains, P1 and P2 (Bu et al. 2008; Chen et al. 2011). The P1 and P2 subdomains constitute the leg and the head, respectively, which are essential for the formation of the protruding P dimer formed by two P monomers. Notably, the commonly accepted noroviral receptors, although the binding affinity is much lower than that of VLPs. The HBGAs are complex carbohydrates present on red blood cells and the epithelia of digestive and respiratory cells; they are also found in free form in milk and saliva (Donaldson et al. 2008; Tan and Jiang 2005). Previously, Tan et al. showed that the P domain linked with short cysteine-containing peptides at either end significantly promoted the switch from P dimers to P particles, which are composed of 12 P dimers (Tan and Jiang 2005). Importantly, the binding affinity of the P particle to HBGA receptors was markedly enhanced compared to the P dimer and was approximately twofold higher than that of VLPs (Tan et al. 2011; Tan and Jiang 2005). These previous studies have suggested that P complexes may contain all elements required for viral receptor interaction and host immune response (Fang et al. 2013; Tan and Jiang 2012; Verma et al. 2016). The small P particles were only found by adding a flag tag (DYKDDDDK) at the C-terminus or by modifying the terminal RRR cluster to RPRPRP when expressed in E. coli (Tan et al. 2011).
Escherichia coli (Li et al. 2016; Tan et al. 2011; Tan and Jiang 2005; Xia et al. 2011) and Pichia pastoris which has been reclassified and renamed Komagataella pastoris (Tan et al. 2008) are two of the most commonly used expression hosts for the production of a variety of heterologous proteins including noroviral P protein. To our knowledge, the highest yields of E. coli under optimal conditions were 20 mg/L P protein in a soluble form (Tan and Jiang 2005; Tan and Jiang 2012) and 210 mg/L chimera P protein in the inclusion bodies (Fu et al. 2017). Unfortunately, a refolding scheme is usually required to correct P protein folding, and endotoxin risk has limited its potential applications. By contrast, K. pastoris retains advantages such as low costs and easy scale-up, as well as eukaryotic merit properties such as natural folding and post-translational modifications. K. pastoris is usually expressed at high yields under the methanol-induced AOX1 promoter; however, the highest yield of intracellular recombinant P protein reported to date was only 7.5 mg/L (Tan et al. 2008).
In this study, the gene of the NoV P domain isolated from the most predominant NoV strain GII.4 in Taiwan (Tang et al. 2013; Tsai et al. 2014) was cloned and expressed in K. pastoris. We demonstrated that high-level expression of the recombinant NoV P complexes was achieved in K. pastoris. The recombinant NoV P complexes were verified by LC-MS/MS and assayed by ELISA. In addition, the morphology of purified NoV P and P-His were analyzed by dynamic light scattering (DLS) and transmission electron microscopy, and its biological function was verified by a saliva-binding assay.
Materials and methods
Strains and media
Escherichia coli DH5α (GIBCO-BRL Life Technologies, Grand Island, NY, USA) was used for plasmid manipulation and grown in LB medium (Difco, Detroit, MI, USA). Komagataella pastoris KM71H (Arg+, Muts) and pPICZ B plasmids were purchased from Invitrogen (Invitrogen, Carlsbad, CA, USA). The buffered glycerol-complex medium (BMGY) for the K. pastoris culture was prepared according to the manufacturer’s instructions. The modified buffered methanol-complex medium (mBMMY) for induction was prepared as described in Chen et al. (2004).
Cloning of the P domain and plasmid construction
The cDNA clones of a human norovirus GII.4 strain, HuNV/GII.4/YJB1/2009/Chiayi, were used in this study. The NCBI GenBank accession number for the genome sequence of the viral strain is MG049692. The VP1 coding regions were amplified by PCR using the viral cDNA clones as templates. The P domain was amplified with or without a 6× histidine tag at the C-terminus deleting the shell domain and the hinge region from the VP1, which contains a short cysteine peptide (CNGRC) at the N-terminus. The PCR fragments were cloned into the EcoRI/XbaI site of pPICZ B (Invitrogen) to produce the expression plasmids pPICZ B-NoV P-His and pPICZ B-NoV P, respectively. The NoV P-His and NoV P expression plasmids were transformed into K. pastoris KM71H according to the manufacturer’s instructions. The primers used in this study are listed in Table 1.
Screening of transformants and flask culture conditions
The transformants harboring NoV P-His and NoV P were selected under zeocin (Invitrogen). Transformants were inoculated in a 5 mL BMGY medium at 30 °C and shaken at 250 rpm for 24 h. After centrifugation, the cells were resuspended in 1 mL mBMMY containing 1% methanol, grown at 30 °C, and induced by the addition of 1% methanol every 24 h. The resultant recombinant proteins were designated NoV P-His and P protein. After the analysis of total cell lysates by western blot, transformants with the highest yields were selected for further studies. The selected K. pastoris KM71H NoV P-His or P transformant was cultured in a 500-mL flask containing 100 mL BMGY supplemented with 1% (v/v) glycerol as a carbon source. Cells were grown at 30 °C and shaken at 250 rpm for 24 h until the OD600 reached approximately 25. After centrifugation, the cells were resuspended in 20 mL mBMMY containing 1% methanol, grown at 30 °C, and induced by the addition of 1% methanol every 24 h.
Fermentation
A three-phase fermentation process was performed including a glycerol batch phase, a glycerol-fed batch phase, and a methanol-induction phase. In the glycerol batch phase, the selected transformant was cultured in a 500-mL flask containing 100 mL BMGY at 30 °C until the OD600 reached 20. The 50 mL seed culture was added into a 5-L jar fermenter (Major Science, Saratoga, CA, USA) containing 2 L of BMGY supplemented with 4% (v/v) glycerol and 4.35 mL/L PTM 4 trace salts (2 g/L CuSO4 · 5H2O, 0.08 g/L NaI, 3 g/L MnSO4 · H2O, 0.2 g/L Na2MoO4 · 2H2O, 0.02 g/L H3BO3, 0.5 g/L CaSO4 · 2H2O, 0.5 g/L CoCl2, 7 g/L ZnCl2, 22 g/L FeSO4 · 7H2O, 0.2 g/L biotin, and 1 mL H2SO4). During fermentation, the temperature and pH were maintained at 30 °C and 6.0, respectively. For pH control, only ammonia solution [10% (v/v)] was used. The agitation rate was set to 800 rpm, and the air input rate was maintained at 4 vvm. The batch phase was maintained until the glycerol in the medium had been completely exhausted as indicated by a sudden increase in the level of dissolved oxygen. Two hundred fifty milliliters of 50% (v/v) glycerol containing 12 mL/L PTM 4 trace salts were fed during the glycerol batch-feeding phase. When the second spike of dissolved oxygen appeared, the culture was induced with 100% methanol supplemented with 12 mL/L PTM 4. Samples were collected for further analysis every 24 h. To calculate the CFU (colony-forming unit, a unit used to estimate the number of viable cells in the sample), tenfold dilutions are used in the plate count in the range of 30–300 CFU on a standard sized petri dish. After the cell colony formation, the number of yeast cell per milliliter of sample was calculated by dividing the number of colonies by the dilution factor.
Purification of NoV P-His protein
One gram of centrifuged yeast cells (wet weight) was resuspended in a 1.5 mL cell lysis buffer (50 mM sodium phosphate pH 7.4, 1 mM EDTA, 5% glycerol) and ground in liquid nitrogen with a mortar and pestle. The extracts were centrifuged at 12,000 g at 4 °C for 15 min to remove the debris, and the supernatants were applied to the HisTrap HP, Ni-NTA column (GE Healthcare Life Sciences, Pittsburgh, PA, USA). The NoV P-His protein was eluted with an elution buffer containing gradient imidazole concentrations.
Purification of NoV P protein
NoV P protein was purified according to Gaberc-Porekar and Menart (2001) in conjunction with metal-chelate affinity chromatography. After cell disruption, the extracts were centrifuged at 12,000 g at 4 °C for 15 min to remove the debris, and the supernatants were applied to the HisTrap HP, Ni-NTA column. The elution of HisTrap affinity at 100 mM imidazole was pooled and subjected to an anion exchanger, DEAE (GE Healthcare Life Sciences) in binding buffer (20 mM Tris-HCl, pH 8.5). The proteins were then eluted by an increasing salt gradient of elution buffer (1 M NaCl, 20 mM Tris-HCl, pH 8.5).
SDS-PAGE and western blot analysis
Protein extracts were subjected to a 12.5% SDS-PAGE and transferred using a wet transfer system (Bio-Rad) to the PVDF membrane (PerkinElmer, Waltham, MA, USA). Protein detection was performed using a homemade mouse anti-VP1 monoclonal antibody, anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase, GTX100118, GeneTex, Irvine, CA, USA), or anti-6xHis (HIT001R, Bioman, Jhonghe, Taipei, Taiwan) and followed by HRP-conjugated anti-mouse or anti-rabbit IgG antibody (PerkinElmer). The colorimetric detection was performed with the UVP AutoChemi Image System (UVP, Upland, CA, USA).
Indirect ELISA
Recombinant NoV P-His complex proteins were purified with a Ni column and used as the standard. Flat-bottomed 96-well PolySorp-Immunoplates (Nunc, Aarhus, Denmark) were coated with the P cell lysate (the supernatants of cell extracts) and NoV P-His antigen at 4 °C overnight. After gelatin-net blocking, 1:8000 diluted monoclonal anti-VP1 antibody was added to each well and incubated for 1 h at room temperature. HRP-conjugated goat anti-mouse IgG antibody (1:8000, PerkinElmer) was added to the plates as a secondary antibody and incubated for 1 h at room temperature. Signals were developed using TMB (BD OptEIA™, Franklin Lakes, NJ, USA), and the OD was measured at 450 nm. Total protein concentration was determined according to the Bradford method (Bio-Rad).
Gel filtration analysis
The Superose 6 Increase 10/300 GL filtration column (GE Healthcare), powered by an AKTA fast-performance liquid chromatography (FPLC) system (UPC 900/P-920, Amersham Pharmacia Biotech, Piscataway, NJ, USA), was used to analyze the molecular weight of the purified NoV P-His, NoV P, and P cell lysates. The gel filtration standard (Bio-Rad, catalog #151-1901) was used for the molecular weight (MW) determination. The running buffer contained 25 mM NaH2PO4, 0.15 M NaCl, pH 7.5.
Saliva-binding assay
Samples of human saliva phenotype B were boiled at 100 °C and centrifuged at 7000 g for 10 min. The supernatant was diluted 1:200 and 1:1000 with PBS. Flat-bottomed 96-well PolySorp-Immunoplates (Nunc, Aarhus, Denmark) were coated with the different dilutions of saliva at 25 °C overnight. After 5% nonfat milk blocking, NoV P or P-His proteins at 2.5–80 μg/mL in PBS were added to the wells and incubated for 1.5 h at 37 °C. The NoV proteins were detected using a 1:4000-diluted monoclonal anti-VP1 antibody for 1 h at 37 °C. HRP-conjugated goat anti-mouse IgG antibody (1:4000, PerkinElmer, Waltham, MA, USA) was added to the plates as a secondary antibody and incubated for 1 h at 37 °C. Signals were developed using TMB (BD OptEIA ™, Franklin Lakes, NJ, USA), and the OD was measured at 450 nm. Total protein concentration was determined by the Bradford method (Bio-Rad). Three independent experiments were conducted for each concentration point.
Identification of NoV P by LC-MS/MS
Spots excised from the stained gels were processed according to the standard mass spectrometry sample preparation protocol (Gharahdaghi et al. 1999; Shevchenko et al. 1996). In-gel digestion of proteins was carried out using MS-grade Trypsin Gold (Promega, Madison, WI, USA) overnight at 37 °C. Tryptic digests were extracted using 10 μL Milli-Q water initially followed by two extractions with a total of 20 μL 50% acetonitrile/0.1% trifluoroacetic acid. The combined extracts were dried in a vacuum concentrator at room temperature and then dissolved in 1 μL of 5% acetonitrile and 0.5% trifluoroacetic acid.
Protein analysis was performed using an LTQ-Orbitrap mass spectrometer (Thermo Scientific, Basingstoke, UK). The MS/MS signal was analyzed by the MASCOT searching engine (www.matrixscience.com). The search parameters were defined as follows: enzyme, trypsin; fixed modification, pyridylethylation; variable modifications, oxidation; peptide MS tolerance, ± 20 ppm; fragment MS tolerance, ± 1 Da, and allowance of one missed cleavage.
Particle size analysis and transmission electron microscopy (TEM)
Gel filtration-purified NoV P or P-His fraction eluted at 15 mL was prepared for particle size analysis and TEM. Particle size analysis was performed by dynamic light scattering (DLS) using a nanoparticle analyzer HORIBA SZ-100 (Horiba, Kyoto, Japan) at a fixed scattering angle of 90° at room temperature. For TEM, protein was negatively stained with 2% uranyl acetate and observed with TEM (Hitachi HT7700, Tokyo, Japan) at an accelerating voltage of 100 kV.
Results
Expression and purification of NoV P-His from flask cultures
The SDS-PAGE and western blotting analysis of total cell lysates of K. pastoris NoV P-His from flask cultures are illustrated in Fig. 1A. The NoV P-His protein production increased with the methanol induction time. The whole cell lysates of K. pastoris NoV P-His were purified using a Ni-NTA column. As shown in Fig. 1B, most of the host cell proteins were removed by the elution buffer containing 50 and 100 mM imidazole, and the NoV P-His complexes were eluted by 300 mM imidazole. The purity of the NoV P-His protein reached 95%. The purified NoV P-His protein was used as an indirect ELISA standard and an HBGA-binding assay test.

NoV P-His protein production and purification. A Time course of P-His protein expression from flask cultures. Each sample of 10 μg protein was separated on 12.5% SDS-PAGE and immunoblotted with anti-His and anti-GAPDH antibodies. The anti-GAPDH (MW ~ 36 kDa) was used as an internal control. The lower duplicate gel stained with Coomassie blue was used to confirm proper separation and equal loading of whole cell lysates. The “cultivation” represents the time of culture and the “methanol” represents the time of methanol induction. B Purification of NoV P-His fusion protein using the Ni-NTA column. The NoV P-His fusion protein was washed and eluted by increasing the concentration of imidazole. Lanes O and OC indicate the whole cell lysates and protein flow through the solution. Lanes 50 to 300 indicate the wash and elution solution containing 50, 100, and 300 mM imidazole, respectively
Characterization of NoV P-His complexes
The polymer formation of the purified NoV P-His protein was analyzed by gel filtration. Each eluted fraction from the Superose 6 Increase column was identified using Bio-Rad’s gel filtration standards. As illustrated in Fig. 2A, the majority of NoV P-His proteins were eluted between 670 kDa and 158 kDa indicating that NoV P-His proteins likely formed small P particles or trimers of the P dimer, composed of 12 or 6 copies of the P domain. The fraction of gel filtration-purified NoV P-His eluted at 15 mL was used for analysis of the particle size and morphology. As shown in Fig. 2B, the diameter of gel filtration-purified NoV P-His protein determined by DLS was approximately 15 nm, which is in agreement with TEM data. Furthermore, the purified NoV P-His protein exhibited triangle, square, and ring shapes as shown in Fig. 2C. These results indicated that the majority of NoV P-His proteins were likely to form triangular or square small P particles with a diameter of 10–15 nm containing 12 copies of the P domain. These observations are consistent with findings in Tan et al. (2011).

Characterization of purified NoV P-His. A Size-exclusion chromatography of purified NoV P-His using a Superose 6 Increase column. The blue dashed line is a standard protein used to determine each elution fraction’s molecular weight. The black line is the profile pattern on size-exclusion chromatography of the purified NoV P-His. An SDS-PAGE analysis of the peak fractions is shown on the left side of the gel filtration curves. B Dynamic light scattering assay. The particle size distribution of gel filtration-purified NoV P-His eluted at 15 mL was measured by DLS. C TEM analysis. The morphology of gel filtration-purified NoV P-His eluted at 15 mL was observed by TEM. The scale bar is 20 nm
Saliva-binding assay of NoV P-His
The NoV GII.4 VLPs have been identified by their interaction with epitopes A/B or O-secretor types in saliva samples (Tan et al. 2008; Yang et al. 2010). To confirm the biological function of NoV P-His produced by K. pastoris, saliva-binding assays using purified NoV P-His particles were performed as shown in Fig. 3. The binding patterns showed that the binding activity was positively correlated with saliva concentrations at the fixed NoV P-His concentration and NoV P-His concentrations at the fixed saliva concentration indicating that the NoV P-His complexes contained domains essential for saliva binding.

The saliva-binding assay. B-type saliva (solid line 1:200, dashed line 1:1000) were coated on plates and incubated with different concentrations of NoV P-His particles proteins
NoV P protein expression in flasks and fermenter cultures
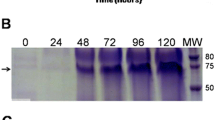
The SDS-PAGE and western blotting analysis of total cell lysates of K. pastoris NoV P from flask cultures are illustrated in Fig. 4. Similar to NoV P-His proteins, the NoV P protein production increased with the methanol induction time, and the highest NoV P protein concentration occurred at 96 h after induction. The representative results of the NoV P protein production in K. pastoris using a fermenter are shown in Fig. 5A. The fed-batch phase was initiated after the initial glycerol had been exhausted as evidenced by an abrupt increase in dissolved oxygen. After the fed-batch phase, the viable-cell concentration reached 8 × 109 cfu/mL and did not change significantly until the end of the experiment. To facilitate the de-repression of the AOX1 promoter, 0.5 mL/L methanol was added to the culture medium prior to the glycerol depletion. The methanol feed rate was set at 1 mL/h/L after the first hour, increased step-wise to 1.4, 1.8, 2.2, 2.6, and 3.0 mL/h/L every 30 min, and maintained at 3.0 mL/h/L until the end of induction. The SDS-PAGE and western blotting analysis of total cell lysates of K. pastoris NoV P from fermentation cultures are shown in Fig. 5B. A small amount of NoV P protein was first detected at the end of glycerol feeding, then increased with the induction time. After 120 h of induction, the NoV P protein levels reached approximately 220 mg/L based on an indirect ELISA assay using purified P-His as a standard. The base feeding paralleled both methanol feeding and P protein production, which is consistent with a previous study (Tsai and Huang 2008).

Time course of NoV P protein expression from flask cultures. Each sample of 10 μg protein was separated on 12.5% SDS-PAGE and immunoblotted with the anti-NoV and anti-GAPDH antibodies. The anti-GAPDH (MW ~ 36 kDa) was used as an internal control. The lower duplicate gel stained with Coomassie blue was used to confirm proper separation and equal loading of whole cell lysates. The “cultivation” represents the time of culture and the “methanol” represents the time of methanol induction

NoV P protein production from fermenter cultures. A Time course of the fermentation parameter, feeding, and cell amount on P transformants in large-scale expression. Fermentation parameter on DO (red line), pH (black line), viable cell concentration (CFU/mL) (yellow circle), feeding amount on methanol (blue line), and base (green line) are plotted against the cultivation time. B Time course of P protein on large-scale expression. Each sample of 4 μg protein was separated on 12.5% SDS-PAGE and immunoblotted with anti-NoV and anti-GAPDH antibodies. The anti-GAPDH (MW ~ 36 kDa) was used as an internal control. The lower duplicate gel stained with Coomassie blue was used to confirm proper separation and equal loading of whole cell lysates. The “cultivation” represents the time of culture and the “methanol” represents the time of methanol induction
Characterization of NoV P complexes and verification by LC-MS/MS
To verify the conformation of NoV P, the polymer formation of K. pastoris NoV P from fermentation culture whole cell lysates was analyzed by gel filtration as illustrated in Fig. 6A. The results of the western blotting and SDS-PAGE analysis of fractions eluted from the gel filtration are shown in Fig. 6B. Similar to NoV P-His proteins, the anti-NoV signals at 35 kDa were detected between the eluted fractions at 14 and 16 mL (Fig. 6B). This suggests that the majority of NoV P proteins were eluted between 670 kDa and 158 kDa indicating that NoV P proteins were also likely to form small P particles or trimers of the P dimer, composed of 12 or 6 copies of the P domain. No P particles with a molecular weight larger than 670 kDa or particles composed of 24 P domain copies were found. The 35 kDa protein of fraction at 16 mL was extracted from the gel, digested with trypsin, analyzed by LC-MS/MS, and identified by comparative analysis of the peptide masses using the Mascot search software with human norovirus protein based on sequences in NCBI Entrez (ATN44699.1). Figure 6C shows that 73% of the peptide fragments matched the P protein sequences. Based on the analysis of the western blot and LC-MS/MS from gel filtration, the majority of NoV P protein consisted of small P particles and trimers of the P dimer.

Characterization of NoV P. A Size-exclusion chromatography pattern. The blue dashed line is standard protein used to determine each elution fraction’s molecular weight. The black line is the profile pattern on size-exclusion chromatography of recombinant NoV P whole cell lysates. B SDS-PAGE and western blot analysis. Each elution fraction of 12 μl was separated on 12.5% SDS-PAGE and immunoblotted with anti-NoV antibody. The western blot analysis showed a major signal monomeric unit of 35 kDa, while minor fractions of 70 kDa (dimer) and over 180 kDa (multimer) were also present. The duplicate gel stained with Coomassie blue was used to examine chromatographic profiles of the whole cell lysate on the SDS-PAGE pattern. C LC-MS/MS identification. The elution fraction (from (B) SDS-PAGE) of NoV P whole cell lysates at the 35 kDa dense band (red arrow) was identified by LC-MS/MS. The result showed 73% coverage (red bold letters) with the NoV P peptide sequence.
Characterization of purified NoV P complexes
The polymer formation of the purified NoV P protein was analyzed by gel filtration as illustrated in Fig. 7A. Each eluted fraction from the Superose 6 Increase column was identified using Bio-Rad’s gel filtration standards. The results of the SDS-PAGE analysis of fractions eluted from the gel filtration are shown in Fig. 7B. Similar to NoV P-His proteins, the purified NoV P proteins were also likely to form small P particles composed of 12 copies of the P domain. The fraction of gel filtration-purified NoV P eluted at 15 mL was also used for analysis of the particle size and morphology. Fig. 7C shows that the diameter of purified NoV P protein was approximately 14 nm, which is in agreement with the observation of TEM exhibiting triangle, square, and ring shapes (Fig. 7D). These results indicated that the majority of NoV P proteins were also likely to form square or ring small P particles with a diameter of 10–15 nm containing 12 copies of the P domain.

Characterization of purified NoV P. A Size-exclusion chromatography of purified NoV P using a Superose 6 Increase column. The blue dashed line is a standard protein used to determine each elution fraction’s molecular weight. The black line is the profile pattern on size-exclusion chromatography of the purified NoV P. B SDS-PAGE and western blot analysis. Each elution fraction was separated on 12.5% SDS-PAGE and immunoblotted with anti-NoV antibody. C Dynamic light scattering assay. The particle size distribution of gel filtration-purified NoV P eluted at 15 mL was measured by DLS. D TEM analysis. The morphology of gel filtration-purified NoV P eluted at 15 mL was observed by TEM. The scale bar is 20 nm.
Saliva-binding assay of NoV P
The biological activity of purified NoV P particles is illustrated in Fig. 8. The binding patterns showed that the binding activity was positively correlated with saliva concentrations at the fixed NoV P concentration and NoV P concentrations at the fixed saliva concentration indicating that the NoV P particles contained domains essential for saliva binding.

The saliva-binding assay. B-type saliva (solid line 1:200, dashed line 1:1000) was coated on plates and incubated with different concentrations of NoV P particle proteins.
Sequence alignment of the different GII.4 strains
From the P domain sequence alignment with 16 GII.4 strains circulated during 1987 to 2008 (Yang et al. 2010), the NoV strain isolated from Taiwan was most similar to the GII.4 2006b cluster (Supplementary Fig. S1 and S2). These 16 NoV proteins formed P particles composed of 24 copies of the P domain. The small P particles and the trimer complexes composed of 12 and 6 copies of the P domain, were only found in the most investigated GII.4 strain VA387 (AY038600) with C-terminal modification (Tan et al. 2011). Figure 9 shows the alignment of P domain amino acid sequences of the GII.4 strain 06Y06bC2 (ADF50099.1), the most investigated GII.4 strain VA387 (AY038600), and the strain in this study (MG049692). Only four different amino acids (residue 84, 119, 136, and 313) were found between ADF50099.1 and MG049692. Intriguingly, small P particles were formed in our study without terminal modification. By comparing the P domain sequence AY038600 with MG049692, differences in 6 amino acids in the P1 domain and 25 in the P2 domain were found. Notably, those four different amino acid residues were also found in AY038600 suggesting that these four point mutations might play a crucial role in P particle size.

Multiple NoV P domains sequence alignment. Strains are as follows: this study (MG049692), 06Y06bC2 (ADF50099.1) (Yang et al. 2010), and VA387 (AY038600). Boundaries between P1 and P2 subdomains are indicated by black arrows. Site I, II, and III indicated the three major regions of HBGA-binding interfaces. Amino acids with the red bold letter and “★” at the bottom indicate dissimilarity.
Discussion
Recombinant NoV P particles are useful in vaccine development and in the study of virus-host interaction. Most reported P particles or chimera P particles were expressed in E. coli, and the yields were 20 mg/L of the P protein in a soluble form (Tan and Jiang 2012) and 210 mg/L of the chimera P protein in inclusion bodies (Fu et al. 2017). In this study, we elevated the NoV P protein expression level to 220 mg/L in a soluble form by optimizing K. pastoris fermentation conditions and provided a cornerstone for the development of tag-free P protein purification schemes.
Most of the P particles assembled from other GII.4 NoV proteins expressed by E. coli or K. pastoris tend to form P particles; however, our data revealed that the polymer formation, both for the NoV P and P-His, was smaller than reported in previously published data (Tan et al. 2011; Tan and Jiang 2012; Yang et al. 2010). Previous studies reported that P particle formation and stability were affected by two factors: intermolecular contact in the P regions and a short chain of cysteine residues at either end of the P domain (Tan et al. 2011; Tan and Jiang 2012). Until now, the mechanism for multiple intermolecular interactions in P particle formation was unclear. Tan et al. reported that the small P particle containing six P dimers is formed after modifications at the C-terminus of the P domain and revealed similar antigenic and HBGA-binding properties to those of the parental VLP and P particles. They found that the three repeat arginine R-clusters (RRR-clusters) in the P1 regions at the C-terminus are crucial for P particle formation and HBGA-binding (Tan et al. 2011). Furthermore, when an R-cluster was replaced by a histidine cluster, neither large nor small P particles were found; however, three P dimers (~ 200 kDa complexes) and P dimers were formed, and no HBGA-binding ability remained even with the cysteine tag (Tan et al. 2011). These results indicate that the R-cluster is critical for P particle formation. However, the role of other amino acids in P particle formation and the binding ability of HBGAs should not be excluded. In this study, small P particles with HBGA-binding ability were formed by the NoV P or P-His lacking terminal modifications expressed in K. pastoris. Although the intermolecular interactions during P particle formation were unclear, the role of residues 84, 119, 136, and 313 deserve further investigation to decipher their role in particle formation.
In conclusion, we overexpressed norovirus P proteins native to Taiwan in K. pastoris and found that NoV P tended to form smaller P particles than those found in other GII.4 strains. The analysis of amino acid sequences suggests that four amino acid residues might play an essential role in the formation of small P particles.
References
Bu W, Mamedova A, Tan M, Xia M, Jiang X, Hegde RS (2008) Structural basis for the receptor binding specificity of Norwalk virus. J Virol 82(11):5340–5347. https://doi.org/10.1128/JVI.00135-08
Chen Y, Tan M, Xia M, Hao N, Zhang XC, Huang P, Jiang X, Li X, Rao Z (2011) Crystallography of a Lewis-binding norovirus, elucidation of strain-specificity to the polymorphic human histo-blood group antigens. PLoS Pathog 7(7):e1002152. https://doi.org/10.1371/journal.ppat.1002152
Chen CC, Wu PH, Huang CT, Cheng KJ (2004) A Pichia pastoris fermentation strategy for enhancing the heterologous expression of an Escherichia coli phytase. Enzym Microb Technol 35(4):315–320. https://doi.org/10.1016/j.enzmictec.2004.05.007
Donaldson EF, Lindesmith LC, Lobue AD, Baric RS (2008) Norovirus pathogenesis: mechanisms of persistence and immune evasion in human populations. Immunol Rev 225:190–211. https://doi.org/10.1111/j.1600-065X.2008.00680.x
Fang H, Tan M, Xia M, Wang L, Jiang X (2013) Norovirus P particle efficiently elicits innate, humoral and cellular immunity. PLoS One 8(4):e63269. https://doi.org/10.1371/journal.pone.0063269
Fu L, Jin H, Yu Y, Yu B, Zhang H, Wu J, Yin Y, Yu X, Wu H, Kong W (2017) Characterization of NoV P particle-based chimeric protein vaccines developed from two different expression systems. Protein Expr Purif 130:28–34. https://doi.org/10.1016/j.pep.2016.09.015
Gaberc-Porekar V, Menart V (2001) Perspectives of immobilized-metal affinity chromatography. J Biochem Biophys Methods 49(1–3):335–360
Gharahdaghi F, Weinberg CR, Meagher DA, Imai BS, Mische SM (1999) Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis 20(3):601–605. https://doi.org/10.1002/(SICI)1522-2683(19990301)20:3<601::AID-ELPS601>3.0.CO;2-6
Li Y, Fu L, Hu Y, Jin H, Zheng Y, Yin Y, Wu H, Yu X, Kong W (2016) Establishment of a novel method without sequence modification for developing NoV P particle-based chimeric vaccines. Protein Expr Purif 121:73–80. https://doi.org/10.1016/j.pep.2016.01.003
Li J, Predmore A, Divers E, Lou F (2012) New interventions against human norovirus: progress, opportunities, and challenges. Annu Rev Food Sci Technol 3:331–352. https://doi.org/10.1146/annurev-food-022811-101234
Lucero Y, Vidal R, O'Ryan GM (2017) Norovirus vaccines under development. Vaccine 36:5435–5441. https://doi.org/10.1016/j.vaccine.2017.06.043
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68(5):850–858. https://doi.org/10.1021/ac950914h
Tan M, Fang P, Chachiyo T, Xia M, Huang P, Fang Z, Jiang W, Jiang X (2008) Noroviral P particle: structure, function and applications in virus-host interaction. Virology 382(1):115–123. https://doi.org/10.1016/j.virol.2008.08.047
Tan M, Fang PA, Xia M, Chachiyo T, Jiang W, Jiang X (2011) Terminal modifications of norovirus P domain resulted in a new type of subviral particles, the small P particles. Virology 410(2):345–352. https://doi.org/10.1016/j.virol.2010.11.017
Tan M, Huang P, Xia M, Fang PA, Zhong W, McNeal M, Wei C, Jiang W, Jiang X (2011) Norovirus P particle, a novel platform for vaccine development and antibody production. J Virol 85(2):753–764. https://doi.org/10.1128/JVI.01835-10
Tan M, Jiang X (2005) The P domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. J Virol 79(22):14017–14030. https://doi.org/10.1128/JVI.79.22.14017-14030.2005
Tan M, Jiang X (2012) Norovirus P particle: a subviral nanoparticle for vaccine development against norovirus, rotavirus and influenza virus. Nanomedicine (Lond) 7(6):889–897. https://doi.org/10.2217/nnm.12.62
Tang MB, Chen CH, Chen SC, Chou YC, Yu CP (2013) Epidemiological and molecular analysis of human norovirus infections in Taiwan during 2011 and 2012. BMC Infect Dis 13:338. https://doi.org/10.1186/1471-2334-13-338
Tsai CT, Huang C-T (2008) Overexpression of the Neocallimastix frontalis xylanase gene in the methylotrophic yeasts Pichia pastoris and Pichia methanolica. Enzym Microb Technol 42:459–465. https://doi.org/10.1016/j.enzmictec.2008.01.018
Tsai CN, Lin CY, Lin CW, Shih KC, Chiu CH, Chen SY (2014) Clinical relevance and genotypes of circulating noroviruses in northern Taiwan, 2006-2011. J Med Virol 86(2):335–346. https://doi.org/10.1002/jmv.23728
Verma V, Tan W, Puth S, Cho KO, Lee SE, Rhee JH (2016) Norovirus (NoV) specific protective immune responses induced by recombinant P dimer vaccine are enhanced by the mucosal adjuvant FlaB. J Transl Med 14(1):135. https://doi.org/10.1186/s12967-016-0899-4
Xia M, Farkas T, Jiang X (2007) Norovirus capsid protein expressed in yeast forms virus-like particles and stimulates systemic and mucosal immunity in mice following an oral administration of raw yeast extracts. J Med Virol 79(1):74–83. https://doi.org/10.1002/jmv.20762
Xia M, Tan M, Wei C, Zhong W, Wang L, McNeal M, Jiang X (2011) A candidate dual vaccine against influenza and noroviruses. Vaccine 29(44):7670–7677. https://doi.org/10.1016/j.vaccine.2011.07.139
Yang Y, Xia M, Tan M, Huang P, Zhong W, Pang XL, Lee BE, Meller J, Wang T, Jiang X (2010) Genetic and phenotypic characterization of GII-4 noroviruses that circulated during 1987 to 2008. J Virol 84(18):9595–9607. https://doi.org/10.1128/JVI.02614-09
Funding
This work was supported by the Ministry of Science and Technology, Taiwan, ROC (MOST-105-2313-B-002-043, MOST-106-2313-B-002-030).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 979 kb)
Rights and permissions
About this article

Cite this article
Chen, YL., Chang, PJ. & Huang, CT. Small P particles formed by the Taiwan-native norovirus P domain overexpressed in Komagataella pastoris. Appl Microbiol Biotechnol 102, 9707–9718 (2018). https://doi.org/10.1007/s00253-018-9331-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9331-8