
Abstract
In aquatic ecosystems, zooplankton-associated bacteria potentially have a great impact on the structure of ecosystems and trophic networks by providing various metabolic pathways and altering the ecological niche of host species. To understand the composition and drivers of zooplankton gut microbiota, we investigated the associated microbial communities of four zooplankton genera from different seasons in the Baltic Sea using the 16S rRNA gene. Among the 143 ASVs (amplified sequence variants) observed belonging to heterotrophic bacteria, 28 ASVs were shared across all zooplankton hosts over the season, and these shared core ASVs represented more than 25% and up to 60% of relative abundance in zooplankton hosts but were present at low relative abundance in the filtered water. Zooplankton host identity had stronger effects on bacterial composition than seasonal variation, with the composition of gut bacterial communities showing host-specific clustering patterns. Although bacterial compositions and dominating core bacteria were different between zooplankton hosts, higher gut bacteria diversity and more bacteria contributing to the temporal variation were found in Temora and Pseudocalanus, compared to Acartia and Synchaeta. Diet diatom and filamentous cyanobacteria negatively correlated with gut bacteria diversity, but the difference in diet composition did not explain the dissimilarity of gut bacteria composition, suggesting a general effect of diet on the inner conditions in the zooplankton gut. Synchaeta maintained high stability of gut bacterial communities with unexpectedly low bacteria-bacteria interactions as compared to the copepods, indicating host-specific regulation traits. Our results suggest that the patterns of gut bacteria dynamics are host-specific and the variability of gut bacteria is not only related to host taxonomy but also related to host behavior and life history traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The microbial community associated to their hosts has multiple functions for ecological processes in both aquatic and terrestrial ecosystems [1,2,3,4]. For individual organisms, symbiotic bacteria provide additional trophic pathways, interact with the immune system, assimilate ambient nutrients, and mitigate pathogens or parasites [5, 6]. For ecosystems, bacterial symbiosis not only links to host niche occupation but also contributes to substance circulation at a large scale, such as nitrogen cycling and methane emission [7,8,9]. In aquatic ecosystems, bacterioplankton and zooplankton are key nodes in trophic networks that perform critical ecological functions by transforming, concentrating, and channeling carbon and essential nutrients across trophic levels [10, 11]. The concentration of bacteria associated to zooplankton is magnitudes higher than that of free-living bacterioplankton, creating bacterial activity hotspots that enhance nutrient cycling within the trophic network [12, 13]. Considering the significant biomass of global aquatic zooplankton and bacteria, the association of these organisms may significantly contribute to aquatic substance cycling [14, 15]. Determining the gut bacteria composition and dynamics is an important first step to understanding the role of zooplankton-associated bacteria in aquatic ecosystems. While the drivers and mechanisms of bacterial symbionts for plants and terrestrial animals are often described with theoretical frameworks [16, 17], drivers and spatiotemporal dynamics of the bacterial community associated to zooplankton hosts are not well known.
Consistent with other organisms, the functions of zooplankton-bacterial symbiont interactions are diverse. The gut microbiome may benefit their zooplankton hosts in the acclimation to different environments, breaking down indigestible diet compounds, degrading toxic substances, and combating parasitism [18,19,20]. Bacteria may also interact with the host immune system and even change the morphology of host guts, as shown for Daphnia and juvenile squid Euprymna [21, 22]. In return, hosts provide relatively stable gut conditions as a refuge for the symbionts, keeping bacteria away from stressful and fluctuating environmental factors [18]. The stability of the gut environment is host-specifically regulated, and the dynamics of symbiotic gut bacteria are controlled by a complex network of interactions [13, 18, 23]. These interactions involve bacteria traits related to the adaptive capacity to the host conditions, host-related mechanisms, such as habitat filtration of the immune systems or diet, and microbiome-related mechanisms, such as resource competition and antagonism [16, 24].
Although various factors regulate symbiont composition, the dynamics of zooplankton symbiotic bacteria follow a general metacommunity framework, corresponding to the bacteria recruitment process [13, 16,17,18, 25]. Seed bank and community assembly theory emphasize the importance of inoculation of background bacteria that shape the structure of the symbiotic community in various organisms [22, 26]. The inoculated bacteria subsequently experience screening of the host’s inner environment, interaction with local bacterial communities, and regulation of host metabolism. Hence, in both vertebrates and invertebrates, symbionts show environmental-specific (i.e., geological and seasonal) and host-specific (i.e., subpopulation and genotype) patterns [2, 19, 27,28,29,30,31,32]. In widely studied organisms, such as humans, hosts with similar traits tend to have bacterial communities with a similar set of dominant species [31, 33, 34]. Clustering of zooplankton-associated bacterial communities was also observed in field samples of zooplankton [27], but the zooplankton gut environment is likely less stable than the mammalian gut systems and is assumed to lack consistency of dominating bacteria species [28]. In addition, higher diversities of symbiotic bacteria correlate with higher resistance to disturbance because more diverse bacteria have more complete usage of niches in the gut environment [17, 34]. While these theories are important for understanding drivers affecting the dynamics of bacterial communities, they remain to be tested for specific zooplankton taxa.
To better understand the dynamics of zooplankton gut symbionts under the scope of general symbiosis ecology, we investigated bacterial compositions and diversity within a range of zooplankton hosts across a temporal gradient. We hypothesized that (H1) zooplankton-associated bacterial communities form clusters of different dominant species across host genera and that the variability of bacterial communities is host-specific. Because diet is one main factor shifting symbiotic bacteria composition in many organisms [19, 23, 31, 33, 35] and zooplankton have diverse feeding behaviors and diet compositions [36, 37], we assumed that (H2) the zooplankton bacteria dynamics are driven by feeding selectivity on phytoplankton. To test our hypotheses, the succession of and effects from prey selectivity for zooplankton-associated gut bacteria were tested for copepod and rotifer genera using Illumina sequencing of the 16S rRNA gene in the Baltic Sea. For H1, we described the temporal variation of bacteria composition by illustrating host and seasonal clustering and alpha diversity for zooplankton-associated bacteria. Besides dominating bacteria, important bacteria during community fluctuations were determined by estimating the contribution of the different bacteria to the temporal variance of the symbiotic community in the different zooplankton hosts. We further examined bacteria correlation patterns to identify the stability of bacteria-bacteria interactions as an indication of distinct bacterial succession in the zooplankton host genera. For H2, we tested the effects of each diet component on the alpha diversity of bacterial communities and the correlation between the similarity of diet composition and the similarity of bacterial communities.
Materials and Methods
Sampling and Zooplankton Sorting
Samples were taken from the offshore monitoring station Landsort Deep (BY31, 58° 35′ N, 18° 14′ E) in the northern Baltic Sea proper in June and August 2017 and March 2018. Zooplankton were collected from vertical hauls at 0–30 m, 30–60 m, and 60–100 m with a 90-µm-WP2 net. Water samples were taken with 10-L Niskin bottles every 5 m from 0 to 30 m and every 10 m from 30 to 100 m depth, and equal volumes from each depth strata were pooled before further analysis. The samples were filtered with 25 mm filters placed in Swinnex holders (Merck/Millipore) with 0.2 and 2 µm polycarbonate and 20 µm nylon filters to separate planktonic communities. Filters were stored under − 80 °C until DNA extraction. Water samples filtered with different filtration sizes were combined in downstream analyses.
Four genera of zooplankton that were present throughout the sampling period were sorted by stereomicroscopy from the depth strata where they were most abundant, including adult stages of the copepods Temora longicornis and Pseaudocalanus spp. from 30 to 60 m depth, the copepod Acartia spp. and rotifer Synchaeta baltica from 0 to 30 m depth. Each individual zooplankton was rinsed with a bleach solution (~ 1%) and, if necessary, appendages were detached to remove potential ectosymbionts. Four to eight replicates with three to six individuals each were sorted per genus and month for DNA sequencing.
DNA Extraction and Metabarcoding
DNA of filtered water samples was extracted with the DNeasy Plant Mini Kit (Qiagen), whereas zooplankton samples were lysed by bead beating using 1 mm glass beads, followed by DNA extraction with the QIAamp DNA Micro Kit (Qiagen). Universal primers 341F (CCTACGGGNGGCWGCAG) and 805R (GACTACHVGGGTATCTAATCC) were used for PCR-amplification of the V3-V4 region of the 16S rRNA gene. The PCR amplicon libraries were sequenced with MiSeq (MSC 2.5.0.5/RTA 1.18.54) pair-end set-up (2 × 300 bp, v.3, Illumina, San Diego, California). A detailed description of sampling and metabarcoding analysis is provided by Zamora-Terol et al. (2020) and Novotny et al. (2021). DNA sequences and associated metadata have been deposited in the European Nucleotide Archive (ENA) under accession no. PRJEB39191.
Data Analysis
Demultiplexing the output data from Illumina sequencing was performed with BCL2FASTQ2 software (Illumina, ver. 2.20.0.422), and primers were trimmed by CUTADAPT ver. 1.18 [38]. Subsequently, raw amplicons were analyzed by the DADA2 pipeline into tables of amplicon sequence variants (ASVs) per sample. The detailed parameters used in each step are described in the paper of Novotny et al. [36]. The taxonomy annotation of ASVs was performed by Naïve Bayesian Classifier for rRNA taxonomic assignment within the DADA2 pipeline. A combination of the SlLVA database [39] and the PhytoREF database [40] was used as a reference for the taxonomic assignment of prokaryote and photoautotrophic organisms. ASVs that failed to be annotated were marked with “x” in the corresponding taxonomy level (i.e., Rickettsialesxxx represents an unidentified species under order Rickettsiales). To focus the analysis on heterotrophic bacterial phyla, autotrophic bacteria were excluded based on information from the literature [30, 41,42,43]. Samples with sequencing yield < 500 bp were discarded.
Downstream data analysis was done in R 4.2.1 [44] with the phyloseq 1.40.0 package for data filtration [45]. We filtered and included ASVs that exist in all replicates of the zooplankton and water samples and where each ASV contributed to at least 1% relative abundance in each individual sample. Rarefaction curves indicated that the species number in all retained samples approached a plateau. To identify host and seasonal effects, we performed the PerMANOVA analysis based on the Bray–Curtis distance (the Bray–Curtis dissimilarity among bacterial communities as the response variable and zooplankton hosts and sampling seasons as the explanatory variables). Additionally, the interaction between the zooplankton host and sampling season was tested with Bonferroni-corrected P-values using the vegan 2.6–4 package [46]. For testing the assumption of dispersion homogeneity, we used the PerMDISP test. To visualize host and seasonal clustering patterns of bacterial composition, we used nonparametric multidimensional scaling (NMDS) with the Bray–Curtis distance. Bacteria host and temporal variation of alpha and beta diversity were calculated as the Shannon index and Bray–Curtis distance based on the relative abundance of ASVs for each sample. To identify key bacteria contributing to temporal variance in each zooplankton, the contribution of each ASV to the Bray–Curtis dissimilarity between bacterial communities was calculated with simper function (available in the vegan package), including a permutation process for significance analysis. We selected core ASVs, defined as those occurring throughout the sampling period in each zooplankton host and water. To compare bacterial correlation patterns across zooplankton genera, correlation patterns between core ASVs shared by all zooplankton were calculated across months, using the Kendall correlation test in the R package stats with log-transformed relative ASV abundances.
We also used generalized linear models (GLM) with a quasi-Poisson error distribution (log link function) for the correlation of the Shannon index of bacterial communities and the diet components of each zooplankton genus across the entire sampling period. The diet composition was calculated as a selectivity index based on DNA metabarcoding of the major phytoplankton taxonomic groups, including chlorophytes, diatoms, filamentous cyanobacteria, picocyanobacteria, and other phytoplankton as suggested by Novotny et al. [37]. To investigate if the core microbiota of each zooplankton host was influenced by diet, we tested correlations between the Bray–Curtis dissimilarity matrixes of the diet and the gut bacteria using the Mantel test with Spearman’s rank correlation.
Results
Bacterial Community Clustering
A total of 7,281,735 reads with 7878 ASVs was obtained from Illumina sequencing for all water and zooplankton samples. After ASV filtration, we obtained 4,089,094 reads of 143 ASVs belonging to heterotrophic bacteria from both 34 water and 68 zooplankton samples (40,089 ± 26,599 reads/sample). Based on the ASV composition, NMDS ordination suggested that the bacterial communities clustered according to host and month (Fig. 1). The variances of bacterial communities across hosts and months were both significant, yet host differences explained more variance than month (PerMANOVA, Table 1). However, as the dispersion of the bacterial communities differed among zooplankton hosts (Fig. 1a, PerMDISP, F(4,97) = 3.09, P = 0.019), a non-homogenous variance may interfere with these results. While the cluster dispersion was similar between months (Fig. 1b, F(2,99) = 0.18, P = 0.84), the interaction between zooplankton host and sampling season was significant (PerMANOVA, F(8, 87) = 2.63, P = 0.001). Homogeneous variance of all 14 zooplankton-by-month clusters was found when excluding the Synchaeta March sample cluster (PerMDISP, F(13,83) = 1.76, P = 0.063)), suggesting host-specific temporal variance of the bacterial communities. Tukey’s HSD suggested that Synchaeta had the lowest dispersion, indicating less variable bacterial communities compared to the copepods. Despite the heterogeneity of the dispersion, host clustering patterns shown by NMDS ordination supported the importance of host identity for bacterial community composition, as was suggested by PerMANOVA.

NMDS plots of the bacterial communities across a water and zooplankton hosts and b months. Zooplankton genera and water are represented with colors, and months with symbols. Ellipses following the t-distribution of the NMDS scores are included (March with dashed, June with dotted, and August with solid lines)
Bacterial Diversity Across Water and Zooplankton Hosts
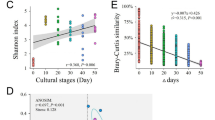
The analysis of bacteria diversity showed host-specific temporal dynamics of symbiotic bacteria. Estimates of alpha diversity divided samples into three categories: filtered water, Temora and Pseudocalanus, and Acartia and Synchaeta (Fig. 2a). Filtered water samples had the highest diversity (pairwise t-test with Bonferroni’s correction, P < 0.001), Temora and Pseudocalanus had similar (P = 1.00) and intermediate diversity, whereas Acartia and Synchaeta had the lowest diversity (P < 0.005). Notable is that the diversity of the latter two was higher in August (Acartia, P = 0.008) and June (Synchaeta, P = 0.01). High beta diversity (Bray–Curtis index > 0.5) between sampling months of all zooplankton hosts and the water samples, except for a low beta diversity in Synchaeta between March and August (P < 0.001), indicated shifts in bacterial community over the season (Fig. 2b). Estimates of beta diversity within months suggested variation of the bacterial community between zooplankton individuals, with the lowest variation for Synchaeta in March (P < 0.001), similar variability for all zooplankton and water samples in June, and less variation for Temora and Synchaeta in August (P < 0.001) (Fig. 2c).

Alpha and beta diversity patterns of bacterial communities. a Alpha diversity (Shannon) of water and zooplankton hosts across months. b Pairwise beta diversity (the Bray–Curtis dissimilarity) of zooplankton hosts between two sampling months. c Beta diversity of water and zooplankton hosts within month. The symbols represent the mean and the error bars standard deviations. Note that the error bar of water in March (c) is missing because of only two water samples
Bacteria Associated to Zooplankton
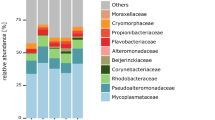
In total, we found 129 core ASVs present throughout the sampling period in all zooplankton hosts and water samples, which represented between 50 and 75% of total bacterial read abundance. However, only 28 core ASVs were shared across all zooplankton hosts, and the shared ASVs were present at low relative abundance in the filtered water (Fig. 3a). These shared core ASVs represented more than 25% of relative bacterial abundance in all zooplankton, and more than 60% in Synchaeta, dominated by the classes Gammaproteobacteria, Bacterioida, and Alphaproteobacteria (Fig. 3a). Among the core ASVs in each individual zooplankton host and filtered water samples, water had the highest number (109 ASVs), followed by Pseudocalanus (79), Temora (71), Synchaeta (54), and Acartia (44) which corresponds to the higher alpha diversity in Pseudocalanus and Temora compared with Synchaeta and Acartia (Fig. 2).

Relative abundance of shared and core ASVs compared to water (inner ring). a Relative abundance of shared ASVs present in all zooplankton hosts across months at class level (outer rings) compared to water. b Relative abundance of core ASVs across months (outer ring) for each zooplankton host and their relative abundance in water samples (inner ring)
Among the 98 core ASVs found in zooplankton hosts, the dominance of certain ASVs showed host-specific distribution patterns (Fig. 3b). Acartia was mainly associated to Burkholderiaceae RS62-marine group (ASV 2, 35% of total relative abundance ± 26 SD), Flavobacterium (ASV 3, 12% ± 19 SD), and an unknown species of Rhodobacteraceae (ASV 16, 12% ± 14 SD); Pseudocalanus to an unknown species of Rhodobacteraceae (ASV 14, 18% ± 21 SD) and Burkholderia-Caballeronia-Paraburkholderia (ASV 1, 19% ± 21 SD); and Temora to Flavobacterium ASV 3 (15% ± 18 SD), whereas Synchaeta was mainly associated to Burkholderia-Caballeronia-Paraburkholderia (ASV 1, 50% ± 27 SD) and Candidatus megaira (ASV 13, 12% ± 11SD) (Table 2). These dominating core ASVs significantly contributed to the variance of bacterial communities between zooplankton genera, in addition to a few less dominant ASVs in specific zooplankton (Simper analysis, Table S1), suggesting that the less dominating bacteria existed across zooplankton genera were also host-specific. About 75% of the temporal gut microbiota dissimilarity in each zooplankton host can be explained by less than five ASVs in Acartia and Synchaeta, while for Temora and Pseudocalanus over 10 ASVs are required to explain bacterial community dynamics (Fig. 4), suggesting that the bacteria have more dominating effects on the variance between season in Acartia and Synchaeta.

Accumulation curves (the Bray–Curtis dissimilarity) of ASVs for each zooplankton host, indicating the number of ASVs contributing to dissimilarity
Correlation Within Bacterial Microbiota
Correlation patterns between the shared 28 core ASVs within zooplankton hosts across the season showed different patterns between zooplankton hosts (Fig. 5). In Acartia and Pseudocalanus, the bacteria were divided into two groups that were negatively correlated, suggesting co-occurrence within groups and mutual exclusion between groups. Co-occurrence or positive correlations include ASVs belonging to Proteobacteria, Actinobacteria, Firmicutes, Verrucomicrobia, Acartia, and Bacteroidetes. In Temora, positive correlations across ASVs were similarly observed but with less distinctive groups, while few significant bacterial correlations were observed in Synchaeta (Fig. 5). The correlation patterns indicate that each zooplankton host had unique gut bacterial interactions and that the correlation pattern of Synchaeta was more similar to the water samples, suggesting weak host regulation.

Kendall’s rank correlation coefficients of 28 core bacteria in a water, b Acartia, c Temora, d Synchaeta, and e Pseudocalanus. Correlations are based on the log-transformed relative abundance of ASVs. Asterisks indicate a significant correlation at P < 0.05. Y-axis labels are the combination of Phylum abbreviates and Family names of the corresponding ASVs on the x-axis. The ASVs were ordered hierarchically based on the correlation coefficient
Correlation Between Bacterial Alpha Diversity and Diet Composition
Zooplankton diets consisting of diatoms and filamentous cyanobacteria were negatively correlated with the alpha bacteria diversity in all zooplankton hosts (Table 3). Yet, the Mantel test suggested that diet differences never explained the dissimilarity of gut bacteria composition between zooplankton hosts (r = − 0.02, P = 0.75). These results suggest that the zooplankton diet affects the diversity of the gut bacterial communities but not the specific succession outcome of bacterial communities’ composition.
Discussion
Invertebrates like zooplankton are expected to develop less stable gut conditions and more strongly fluctuating gut bacterial communities compared to mammals [22, 28]. Yet, in this study, host differences showed a stronger effect than seasonal differences on the gut bacterial community. Despite that distinct gut bacterial community compositions were found in different zooplankton hosts, compositional clustering patterns, temporal changes of diversity, and bacteria contributing to the temporal variance in the gut communities detected resemblances among the four zooplankton genera that were inconsistent with the taxonomic relations of the hosts. Distinct symbiotic bacteria distribution patterns in zooplankton indicate the importance of host-related traits in shaping gut bacteria composition.
Gut Bacteria Dynamics of Temora and Pseudocalanus With High Diversity
Clustering patterns suggest different bacterial compositions among the zooplankton, but also several similarities between particularly Temora and Pseudocalanus. These two copepod genera had a higher alpha diversity and more bacteria species contributing to the temporal variance than did Synchaeta and Acartia (Figs. 2B and 4 and Table 2). Higher diversity and temporal variance of Temora and Pseudocalanus are possibly related to their deeper distribution in the water column and their vertical migration behavior [36, 47], exposing the zooplankton to a wider range of bacterioplankton across the different water layers [48]. In addition to their vertical migration behavior, the resemblance of their bacteria diversity can potentially be explained by a similar feeding behavior, although the dominating core bacteria and community composition differed between Temora and Pseudocalanus. Both genera had a diet composed of Cyanobiaceae and Chlorophyta during March [36], which may establish similar gut environments of the zooplankton during early life stages, eventually leading to the observed bacterial communities in the adults. The importance of bacterial inoculation at the early life stages with long-term effects was previously documented in squid larvae and human infants [22, 34], and may be a common phenomenon.
Acartia and Synchaeta Had Less Diverse Community With Less Bacteria Contributing to the Temporal Variance
Acartia and Synchaeta show less vertical migration and are mainly present in the upper 30 m water column, above the thermocline depth [36]. This more constrained vertical distribution probably exposes Acartia and Synchaeta to stable bacteria seed banks for gut bacteria inoculation because earlier studies suggest that the distribution and diversity of bacterioplankton are significantly related to water-column stratification [49]. Acartia had in comparison with the other copepods an unexpectedly low diversity of gut bacteria and fewer bacteria contributing to the temporal variation of gut communities, although the bacteria diversity increased during August. Higher diversity in August may be related to the presence of different Acartia species, while A. tonsa mainly dominates during the early season [50]. Instead of other copepods, Acartia more resembled the rotifer, Synchaeta. The diet analysis also showed that Acartia and Synchaeta shared a diet with high proportions of filamentous cyanobacteria, which may have contributed to the low bacteria diversity and fewer bacteria contributing to the temporal variance in their gut communities [36, 51]. This effect of diet components on gut bacteria is supported by the negative correlation between diatom and filamentous cyanobacteria with gut microbiota diversity for the entire zooplankton community. Filamentous cyanobacteria potentially played a crucial role in shaping gut microbiome in zooplankton in Acartia and Synchaeta. Mantel’s tests however showed that the similar gut bacteria composition was unrelated to the diet similarity. These findings suggest a general influence from the diet on niche structure, as indicated by diversity and number of key bacteria, rather than effects on specific bacteria composition in zooplankton gut from field studies. This conclusion is in contrast to the expected direct influence of diet phytoplankton on specific gut bacteria composition as observed in laboratory studies [20, 23, 35] In nature, diverse bacteria with redundant functionality from ambient environments may fill niches in the zooplankton gut and interfere with the correlation between diet and specific bacteria composition [16, 52].
Correlation Patterns Reflected Host Genus-Related Regulations
Gut bacteria correlation patterns distinguished the rotifer from the copepods. Two groups of bacteria with within-group positive correlations were generally found in the copepod hosts with negative correlations between these bacteria groups. This correlation pattern suggests a combination of competition and antagonism along with mutualistic interactions in copepod guts, agreeing with research on mammalian gut bacteria [1, 17, 32]. Notably, our correlation results were based on the seasonal change of abundance, and it is debatable how important bacterial interactions and random processes are for the fluctuations. The suggested importance of bacteria interactions, nevertheless, indicates that further studies on the functional annotation of co-occurring bacteria are needed, and the correlation patterns suggest two alternative gut symbiont regimes in copepods during the sampling seasons.
The core bacteria ASVs were less grouped over time in Synchaeta whose correlation pattern was instead closer to the pattern in water. This similarity suggests a strong environmental influence on the bacterial community, but this conclusion is contradicted by the low clustering dispersion and low beta diversity in Synchaeta which instead suggest a higher stability of gut bacteria over the season. The dynamics of gut bacteria in Synchaeta can also be seen as contradictory with general ecology theory which suggests that stable communities require a diverse community with a high level of interactions [16, 17, 29, 34, 53]. The bacterial community pattern of Synchaeta suggests unique regulation of rotifer gut symbionts, probably from specific regulation related to the host species. The host-dependent gut microbiota has been found in zooplankton in the laboratory, and multiple underlying mechanisms were proposed, such as the response of the host immune system, or other physical or chemical host screen mechanisms favoring specific dominating bacteria [1, 5, 19, 54]. The dominating bacteria of Synchaeta, Burkholderia-Caballeronia-Paraburkholderia, widely exists in invertebrates, mammals, and plants [55, 56] and includes species with different pathogenic or symbiotic capacities. Burkholderia-Caballeronia-Paraburkholderia was also found in Pseudocalanus; in this case, in combination with diverse and correlated bacterial communities, but whether the same strains of Burkholderia-Caballeronia-Paraburkholderia are present in Synchaeta and Pseudocalanus remains to be verified.
In conclusion, we find that the gut microbiome of zooplankton showed host-specific clustering patterns. Each host genus had unique bacteria contributing to the temporal variability of gut communities, and the temporal succession of gut microbiota was also different between genera. Each of the copepods had two bacteria groups with positive within-group correlations, while the rotifer had less correlated bacterial communities that more resembled the ambient water. Temporal variance of bacterial composition and bacterial correlation showed different similarities of gut bacteria patterns of zooplankton, which may be related to the ecological niche and taxonomy of the hosts. To further investigate the distribution of gut bacteria associated with zooplankton, the next step would be to classify the bacteria into functional groups in order to examine distinct bacteria with redundant functions. For better elucidation of underlying mechanisms of bacteria dynamics and host taxonomy, transcriptomic and proteomic analysis can be further implemented on the entire holobiont, drawing a functional network of bacteria and host metabolism. Overall, our results from this field study suggest that gut bacteria dynamics are not just related to host taxonomy but are also affected by host behavior and life history traits, such as feeding patterns.
Data Availability
The original dataset is available in the European Nucleotide Archive (ENA) under accession no. PRJEB39191. The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Hooper LV, Midwedt T, Gordon JI (2002) How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 22:283–307
Engel P, Moran NA (2013) The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev 37:699–735
Rosenberg E, Koren O, Reshef L et al (2007) The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol 5:355–362
Xiao F, Zhu W, Yu Y et al (2021) Host development overwhelms environmental dispersal in governing the ecological succession of zebrafish gut microbiota. NPJ Biofilms Microbiomes 7:. https://doi.org/10.1038/s41522-020-00176-2
Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science (1979) 336:1268–1273. https://doi.org/10.1126/science.1223490
Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human–microbe mutualism and disease. Nature 449:811–818. https://doi.org/10.1038/nature06245
Borges RM (2017) Co-niche construction between hosts and symbionts: ideas and evidence. J Genet 96:483–489. https://doi.org/10.1007/s12041-017-0792-9
Oldroyd GED, Murray JD, Poole PS, Downie JA (2011) The rules of engagement in the legume-rhizobial symbiosis. Annu Rev Genet 45:119–144. https://doi.org/10.1146/annurev-genet-110410-132549
Ellis JL, Dijkstra J, Kebreab E et al (2008) Aspects of rumen microbiology central to mechanistic modelling of methane production in cattle. J Agric Sci 146:213–233. https://doi.org/10.1017/S0021859608007752
Zamora-Terol S, Novotny A, Winder M (2020) Reconstructing marine plankton food web interactions using DNA metabarcoding. Mol Ecol 29:3380–3395. https://doi.org/10.1111/mec.15555
Fenchel T (2008) The microbial loop – 25 years later. J Exp Mar Biol Ecol 366:99–103. https://doi.org/10.1016/j.jembe.2008.07.013
Shoemaker KM, Moisander PH (2017) Seasonal variation in the copepod gut microbiome in the subtropical North Atlantic Ocean. Environ Microbiol 19:3087–3097. https://doi.org/10.1111/1462-2920.13780
Grossart HP, Riemann L, Tang KW (2013) Molecular and functional ecology of aquatic microbial symbionts. Front Microbiol 4:59. https://doi.org/10.3389/fmicb.2013.00059
Sogin ML, Morrison HG, Huber JA et al (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci 103:12115–12120. https://doi.org/10.1073/pnas.0605127103
Ward BA, Dutkiewicz S, Follows MJ (2014) Modelling spatial and temporal patterns in size-structured marine plankton communities: top–down and bottom–up controls. J Plankton Res 36:31–47. https://doi.org/10.1093/plankt/fbt097
Christian N, Whitaker BK, Clay K (2015) Microbiomes: unifying animal and plant systems through the lens of community ecology theory. Front Microbiol 6:869. https://doi.org/10.3389/fmicb.2015.00869
Walter J, Maldonado-Gómez MX, Martínez I (2018) To engraft or not to engraft: an ecological framework for gut microbiome modulation with live microbes. Curr Opin Biotechnol 49:129–139
Tang KW, Turk V, Grossart HP (2010) Linkage between crustacean zooplankton and aquatic bacteria. Aquat Microb Ecol 61:261–277
MacKe E, Callens M, De Meester L, Decaestecker E (2017) Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nat Commun 8:. https://doi.org/10.1038/s41467-017-01714-x
Gorokhova E, El-Shehawy R, Lehtiniemi M, Garbaras A (2021) How copepods can eat toxins without getting sick: gut bacteria help zooplankton to feed in cyanobacteria blooms. Front Microbiol 11:. https://doi.org/10.3389/fmicb.2020.589816
Cooper RO, Cressler CE (2020) Characterization of key bacterial species in the Daphnia magna microbiota using shotgun metagenomics. Sci Rep 10:. https://doi.org/10.1038/s41598-019-57367-x
Nyholm SV, McFall-Ngai MJ (2004) The winnowing: establishing the squid - vibrios symbiosis. Nat Rev Microbiol 2:632–642
Tang K, Dziallas C, Hutalle-Schmelzer K, Grossart HP (2009) Effects of food on bacterial community composition associated with the copepod Acartia tonsa Dana. Biol Lett 5:549–553. https://doi.org/10.1098/rsbl.2009.0076
Prosser JI, Bohannan BJM, Curtis TP et al (2007) The role of ecological theory in microbial ecology. Nat Rev Microbiol 5:384–392. https://doi.org/10.1038/nrmicro1643
Miller ET, Svanbäck R, Bohannan BJM (2018) Microbiomes as metacommunities: understanding host-associated microbes through metacommunity ecology. Trends Ecol Evol 33:926–935. https://doi.org/10.1016/j.tree.2018.09.002
Hannes P, Ruben S (2008) An evaluation of methods to study the gut bacterial community composition of freshwater zooplankton. J Plankton Res 30:997–1006. https://doi.org/10.1093/plankt/fbn061
De Corte D, Srivastava A, Koski M et al (2018) Metagenomic insights into zooplankton-associated bacterial communities. Environ Microbiol 20:492–505. https://doi.org/10.1111/1462-2920.13944
Eckert EM, Anicic N, Fontaneto D (2021) Freshwater zooplankton microbiome composition is highly flexible and strongly influenced by the environment. Mol Ecol 30:1545–1558. https://doi.org/10.1111/mec.15815
Dillon RJ, Vennard CT, Buckling A, Charnley AK (2005) Diversity of locust gut bacteria protects against pathogen invasion. Ecol Lett 8:1291–1298. https://doi.org/10.1111/j.1461-0248.2005.00828.x
Romero J, Ringø E, Merrifield DL (2014) The gut microbiota of fish. Aquaculture nutrition: gut health, probiotics and prebiotics, 75–100. https://doi.org/10.1002/9781118897263.ch4
Arumugam M, Raes J, Pelletier E et al (2011) Enterotypes of the human gut microbiome. Nature 473:174–180. https://doi.org/10.1038/nature09944
Coyte KZ, Rakoff-Nahoum S (2019) Understanding competition and cooperation within the mammalian gut microbiome. Curr Biol 29:R538–R544. https://doi.org/10.1016/j.cub.2019.04.017
Wu GD, Chen J, Hoffmann C et al (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science (1979) 334:105–108. https://doi.org/10.1126/science.1208344
Koenig JE, Spor A, Scalfone N et al (2011) Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 108:4578–4585. https://doi.org/10.1073/pnas.1000081107
Li Y, Xu Z, Liu H (2021) Nutrient-imbalanced conditions shift the interplay between zooplankton and gut microbiota. BMC Genomics 22:. https://doi.org/10.1186/s12864-020-07333-z
Novotny A, Zamora-Terol S, Winder M (2021) DNA metabarcoding reveals trophic niche diversity of micro and mesozooplankton species. Proc R Soc B Biol Sci 288:20210908. https://doi.org/10.1098/rspb.2021.0908
Novotny A, Serandour B, Kortsch S, et al (2023) DNA metabarcoding highlights cyanobacteria as the main source of primary production in a pelagic food web model. Sci Adv 9:. https://doi.org/10.1126/sciadv.adg1096
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10. https://doi.org/10.14806/ej.17.1.200
Pruesse E, Quast C, Knittel K et al (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. https://doi.org/10.1093/nar/gkm864
Decelle J, Romac S, Stern RF et al (2015) PhytoREF: a reference database of the plastidial 16S rRNA gene of photosynthetic eukaryotes with curated taxonomy. Mol Ecol Resour 15:1435–1445. https://doi.org/10.1111/1755-0998.12401
Tomova I, Lazarkevich I, Tomova A et al (2013) Diversity and biosynthetic potential of culturable aerobic heterotrophic bacteria isolated from Magura Cave, Bulgaria. Int J Speleol 42:65–76. https://doi.org/10.5038/1827-806X.42.1.8
Hosokawa S, Kuroda K, Narihiro T et al (2021) Cometabolism of the superphylum Patescibacteria with anammox bacteria in a long-term freshwater anammox column reactor. Water (Basel) 13:208. https://doi.org/10.3390/w13020208
Rinninella E, Raoul P, Cintoni M et al (2019) What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. https://doi.org/10.3390/microorganisms7010014
R Core Team (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/
McMurdie PJ, Holmes S (2013) phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. https://doi.org/10.1371/journal.pone.0061217
Oksanen J, Simpson G, Blanchet F, Kindt R, Legendre P, Minchin P, O'Hara R, Solymos P, Stevens M, Szoecs E, Wagner H, Barbour M, Bedward M, Bolker B, Borcard D, Carvalho G, Chirico M, De Caceres M, Durand S, Evangelista H, FitzJohn R, Friendly M, Furneaux B, Hannigan G, Hill M, Lahti L, McGlinn D, Ouellette M, Ribeiro Cunha E, Smith T, Stier A, Ter Braak C, Weedon J (2022) _Vegan: community ecology package_. R package version 2.6–2, https://CRAN.R.project.org/package=vegan.
Hansson S, Larsson U, Johansson S (1990) Selective predation by herring and mysids, and zooplankton community structure in a Baltic Sea coastal area. J Plankton Res 12:1099–1116. https://doi.org/10.1093/plankt/12.5.1099
Hansen FC, Möllmann C, Schütz U, Neumann T (2006) Spatio-temporal distribution and production of calanoid copepods in the central Baltic Sea. J Plankton Res 28:39–54. https://doi.org/10.1093/plankt/fbi097
Bunse C, Pinhassi J (2017) Marine bacterioplankton seasonal succession dynamics. Trends Microbiol 25:494–505. https://doi.org/10.1016/j.tim.2016.12.013
Winder M, Varpe Ø (2020) Interactions in plankton food web: seasonal succession and phenology of Baltic Sea zooplankton. Pp 162–191. In: Teodósio MA, Barbosa AB [eds.] Zooplankton Ecology. CRC Press
Serandour B, Jan KMG, Novotny A, Winder M (2023) Opportunistic vs selective feeding strategies of zooplankton under changing environmental conditions. J Plankton Res 45:389–403. https://doi.org/10.1093/plankt/fbad007
Allison SD, Martiny JBH (2008) Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci 105:11512–11519. https://doi.org/10.1073/pnas.0801925105
Shade A, Peter H, Allison SD et al (2012) Fundamentals of microbial community resistance and resilience. Front Microbiol 3:. https://doi.org/10.3389/fmicb.2012.00417
Callens M, De Meester L, Muylaert K et al (2020) The bacterioplankton community composition and a host genotype dependent occurrence of taxa shape the Daphnia magna gut bacterial community. FEMS Microbiol Ecol 96:. https://doi.org/10.1093/femsec/fiaa128
Ge S-X, Shi F-M, Pei J-H et al (2021) Gut bacteria associated with Monochamus saltuarius (Coleoptera: Cerambycidae) and their possible roles in host plant adaptations. Front Microbiol 12:. https://doi.org/10.3389/fmicb.2021.687211
Sallinger E, Robeson MS, Haselkorn TS (2021) Characterization of the bacterial microbiomes of social amoebae and exploration of the roles of host and environment on microbiome composition. Environ Microbiol 23:126–142. https://doi.org/10.1111/1462-2920.15279
Acknowledgements
We thank the pelagic monitoring group at Stockholm University, especially Stefan Svensson and Jakob Walve for their support during sampling.
Funding
Open access funding provided by Stockholm University. We acknowledge support from the National Genomics Infrastructure in Stockholm and Uppsala, funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, the Swedish Research Council, and SNIC/Uppsala Multidisciplinary Centre for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure. The research was funded by the Swedish Research Council (project number 2016–04685).
Author information
Authors and Affiliations
Contributions
TX: conceptualization, data curation, formal analysis, investigation, visualization, and writing original draft. AN: sample collection, methodology, biostatistical analysis, and data curation. SZT: sample collection and methodology. PH: supervision and writing review and editing. MW: conceptualization, visualization, funding acquisition, supervision, and writing review and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, T., Novotny, A., Zamora-Terol, S. et al. Dynamics of Gut Bacteria Across Different Zooplankton Genera in the Baltic Sea. Microb Ecol 87, 48 (2024). https://doi.org/10.1007/s00248-024-02362-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00248-024-02362-7