
Abstract
Aboveground ecological impacts associated with agricultural land use change are evident as natural plant communities are replaced with managed production systems. These impacts have been extensively studied, unlike those belowground, which remain poorly understood. Soil bacteria are good candidates to monitor belowground ecological dynamics due to their prevalence within the soil system and ability to survive under harsh and changing conditions. Here, we use soil physicochemical assessment and 16S rRNA gene sequencing to investigate the soil physical and bacterial assemblage changes across a mixed-use agricultural landscape. We assess soil from remnant vegetation (Eucalyptus mallee), new and old vineyards, old pasture, and recently revegetated areas. Elevated concentrations of nitrogen (NO3−) and plant-available (Colwell) phosphorus were identified in the managed vineyard systems, highlighting the impact of agricultural inputs on soil nutrition. Alpha diversity comparison revealed a significant difference between the remnant mallee vegetation and the vineyard systems, with vineyards supporting highest bacterial diversity. Bacterial community composition of recently revegetated areas was similar to remnant vegetation systems, suggesting that bacterial communities can respond quickly to aboveground changes, and that actions taken to restore native plant communities may also act to recover natural microbial communities, with implications for soil and plant health. Findings here suggest that agriculture may disrupt the correlation between above- and belowground diversities by altering the natural processes that otherwise govern this relationship (e.g. disturbance, plant production, diversity of inputs), leading to the promotion of belowground microbial diversity in agricultural systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The intensification of agriculture that has occurred over the last 100 years, while increasing food production [1], has led to the degradation of many agricultural and natural landscapes [2, 3]. It is perhaps the conversion of natural ecosystems to production systems that is most profound, directly evident by a reduction in aboveground diversity. Production agriculture can also negatively influence soil condition, through depletion of soil organic carbon, acceleration of erosion, reduction of soil fertility, and through acidification and salinisation [2, 4, 5], affecting the productivity and sustainability of aboveground ecosystems [6,7,8,9]. Soil degradation leads to reduced agricultural output [10], as well as driving fundamental changes in soil biology [11,12,13,14], notably, the balance between component groups of microorganisms, many of which play a pivotal role in broader ecosystem function.
Bacteria perform a variety of functions critical to soil and plant health [15,16,17,18]. Bacteria assist in the conversion and uptake of plant available nutrients [19,20,21], act as phytostimulators promoting plant growth and resilience [22, 23], and biological control agents that protect plants against phytopathogens [24]. Secretions from soil bacteria help form microaggregates by binding soil particles that affect soil structure [25]. Soil microaggregates are increasingly recognised as a characteristic of healthy soil, improving gas exchange, water infiltration, and water holding capacity of the soil [26]. Given the range of functions performed by bacteria, the diversity and composition of their component communities can provide valuable insights into the health and function of associated environments [27,28,29,30,31], including agricultural systems and practices [11, 12, 32,33,34]. The investigation of soil bacterial communities has been suggested as a way to evaluate the condition of soil and productivity of correlating ecosystems [32, 35,36,37], with particular relevance to production systems where soil and plant health are intrinsically linked to productivity.
Different land systems and management practices can modify vegetation and soil physicochemical properties, which in turn influence aboveground biodiversity and ecological processes such as nutrient cycling and gas exchange [34, 38,39,40,41,42,43]. And while there is some evidence that land use practice can modify belowground microbial communities [11,12,13,14], it is still unclear how aboveground land systems influence the structure of soil bacterial communities, how much variation exists between and within these communities, and what is driving it [32, 35, 44]. The effect of land use change from natural to managed agriculture on soil bacterial communities is poorly understood, with both positive and negative correlations reported [36, 45,46,47,48,49,50,51], while an assumption of above- and belowground diversity linkage still exists. Further investigation across different environments is required to explore these assumptions [52, 53].
Much research has focused on the implications of aboveground land use on soil microbial communities across temperate and mesic biomes [51, 54,55,56,57] with considerably fewer studies investigating these interactions in less productive arid systems [36, 58], such as those found throughout much of Australia. Australia’s semi-arid zone occurs through the interior of the continent where average rainfall is between 250 and 500 mm per year. These systems support sclerophyllous vegetation of predominantly low-growing Eucalyptus species (commonly termed mallee or mallee scrub), drought-tolerant understory shrubs (e.g. Acacia and Chenapod species), and ephemeral grasses and herbs. Here, we investigated the conversion of these systems to production agriculture (vineyards) and the impact of this transition on soil physicochemical characteristics and bacterial community composition. Conversely, we investigate how the restoration of sites with a legacy of agriculture (ex-pastoral land) influences these critical soil components. Given the growing global interest in ecological restoration as a strategy to restore the flow of ecosystem services [6], such investigations are increasingly important in evaluating the success of restorative actions taken. A greater understanding of how the recovery of native plant communities (through active revegetation) influences soil microbial communities can help shed light on the significance of such actions on soil condition.
We hypothesised that managed agricultural systems would be associated with elevated concentrations of key nutrients (nitrate and phosphorus), and that distinct bacterial communities would be associated with different land use systems (i.e. vineyards, remnant mallee vegetation, revegetation, and ex agricultural land). Further, we expected to find a positive correlation between above and belowground diversities, and consequently that the conversion of diverse natural systems (remnant mallee vegetation) to monoculture agriculture (vineyards) would result in a reduction in soil bacterial diversity.
Materials and method
Study site
The study site was a mixed-use agriculture production landscape, encompassing agricultural and natural systems (Fig. 1). Located on the River Murray in the New South Wales Murray Darling Wine Region of Australia (34°37′47.8″S, 143°00′56.2″E), the site consisted of two commercial agricultural operations: wine vineyards and dried fruit vineyards. The region is highly productive producing high-value crops such as grapes, citrus, olives, nuts, stone fruit, cereal crops, and livestock. The area was classified as semi-arid with most of the annual average rainfall of ~ 300 mm falling during the Austral winter (i.e. June–August) [59]. Soil, aspect, and elevation were consistent across the site, with soils classified as calcarosols or mallee loam, ranging from brown to red-brown loamy sand, sandy loam, or loam [60].

Study system (NSW, Australia) showing the location of the 32 sample replicates owning to the five identified landscape units (ecological systems)
Much of the site was dominated by irrigated vineyards (roughly 50%), which has replaced the remnant native Eucalyptus mallee that would have occurred across most of the site and of the region prior to its conversion to agriculture (Fig. 1). The management practices of both vineyard systems were consistent, both being applied with two microbial inoculants (Supplementary 7). Along with the active vineyard operations and remaining remnant mallee vegetation, an ex-pastoral/cropping section existed along the north-eastern boarder of the site (~ 420 ha). This section was abandoned for agricultural use within the last 5 years (assessed as unsuitable for irrigated agriculture) although still possesses a legacy of past cropping and pastoralism via a system dominated by wheat, and mixed native and introduced grasslands.
Ecological systems
Five distinct land use/ecological systems were identified across the study site (landscape units, hereafter) (Figs. 1 and 2). Remnant mallee vegetation of mixed Eucalyptus (E. gracilis, E. brachycalyx, E. leptophylla, E. incrassata) (RemVeg, hereafter) was identified and used as the natural reference system in which the impact of land use change could be measured against. Three agricultural landscape units were identified: established vineyards (OldVineyard, hereafter) consisting of grape vines over 10 years old, new vineyards (NewVineyard, hereafter) consisting of grape vines under 2 years old, and a grassland section (old pasture) that had been abandoned for agricultural use (Excrop, hereafter). Finally, a native revegetation landscape unit (Reveg, hereafter) was identified, consisting of three plantings undertaken within 2 years of sampling (2019–2020); one seedling planted site, and two direct seeded revegetation sites which comprised a seed mix of approximately 15 local native plant species that had not yet emerged at time of sampling.

Sample images of landscape unit replicates; A remnant mallee vegetation (RemVeg), B new vineyard (NewVineyard), C established vineyard (OldVineyard), D pastoral land abandoned for agricultural use (ExCrop)
Replicates were identified for each of the five landscape units, totaling 32 individual sampling sites, broken down as follows; 15 replicates of the RemVeg landscape unit, six replicates of the OldVineyard landscape unit, five replicates of the NewVineyard landscape unit, three replicates of the Reveg landscape unit, and three replicates of the Excrop landscape unit (Fig. 1). The sampling design was developed with consideration to soil and landscape variation (aspect and elevation). Replicates were chosen based on their location across the study site, where possible landscape units were identified and sampled that were in close vicinity to one another and in consistent soil types, allowing for comparative analysis between each.
Soil sampling
Soils were sampled following the Biomes of Australian Soil Environments (BASE) project protocol [61], in December 2020 (i.e. Austral Summer). Briefly, one soil sample was taken from each of the landscape unit replicates (n = 32), which comprised three pooled sub-samples taken from a 30 m radius within the replicate. Soil was collected from the bulk soil surface horizon (0–10 cm depth), a portion (approx. 50 g) of which was stored in a sterile 50-mL tube to be used for DNA extraction, and another larger portion (~ 300 g) stored in a ziplock bag for physicochemical analysis. The top litter layer was carefully removed and a scoop taken to required depth (10 cm), with the three sub-samples thoroughly mixed prior to portioning. Samples to undergo physicochemical analysis were air dried in bag and stored at room temperature, while tubes where immediately stored at − 8 °C until microbial analysis was performed.
Additional metadata were also collected at each of the landscape unit replicates, consisting of photos of each sub-sampling location, GPS coordinates, and notes on vegetation community variables including a plant species list. Plant species lists were compiled via visual inventories during a 5-min search of the immediate area surrounding the sampling location (30 m soil sampling radius).
Soil analysis
Soil physicochemical analysis was undertaken by the Australian Precision Ag Laboratory (APAL, Adelaide, Australia). Specifically, ammonium (NH4+), nitrate (NO3−), plant-available (Colwell) phosphorus, potassium, sodium, magnesium, calcium, organic carbon, soil pH (CaCl2), and soil texture were quantified. The Colwell phosphorus test employed provides a measure of plant-available phosphorus, that being the bicarbonate-extractable phosphorus. The Colwell phosphorus method is considered to estimate phosphorus quantity and is the most common soil phosphorus test used in Australia [62]. Organic carbon was determined using the Walkley and Black wet oxidation method, providing an approximation of total soil organic carbon by measuring the readily oxidisable/decomposable carbon which is considered to account for roughly 80% of the total soil organic carbon pool [63]. DNA extraction and sequencing were undertaken by the Australian Genome Research Facility (AGRF, Adelaide, Australia) using the ‘DNeasy PowerSoil Pro Kit’ from Qiagen [64]. Briefly, soil samples were added to a bead beating tube for rapid and thorough homogenisation, cell lysis occurred by mechanical and chemical methods, total genomic DNA was captured on a silica membrane in a spin column format, and DNA was washed and eluted from the membrane ready for downstream analysis [64].
Bacterial 16S ribosomal rRNA was PCR amplified for each replicate using the forward 27f (AGAGTTTGATCMTGGCTCAG) and reverse 519r (GWATTACCGCGGCKGCTG) primers. Sequence data was analysed using the QIIME 2 (2019.7) platform [65]. The demultiplexed raw reads were primer trimmed using the cutadapt plugin, with a length cut-off of 240 bp for the forward primer (default –error-rate 0.1 –times 1 –overlap 3). DADA2 with default setting (–p-max-e 2, –p-chimera-method consensus) was used to denoise, dereplicate, and filter chimeras [66]. Taxonomy was assigned to amplicon sequence variants (ASVs) using the q2 feature classifier [67]. Sequences from the Greengenes databases (v13.8) were trimmed for the targeted regions (V1–V3) and used as a training dataset for the classifier resulting in an absolute abundance ASV table to be used in downstream analysis.
Statistical analysis
The vegetation data (plant species list) was used to determine the mean plant functional diversity associated with each of the landscape units (Supplementary 5 and 6). Observed species were categorised into one of five functional groups; perennial herbaceous groundcover (0–30 cm height, grasses and forbes); annual herbaceous groundcover (0–30 cm height, grasses and forbes); small shrub (< 1 m height, woody perennial); medium/large shrub (> 1 m height, woody perennial); and tree (woody plants with trunk and canopy over 3 m height). The observed plant functional groups were summed for each replicate, providing a plant functional diversity score for each sample replicate from which a mean plant function diversity score could be derived for each landscape unit.
The majority of statistical analysis was undertaken using R software (v4.03) [67, 68], employing the microbiome data analysis framework of the phyloseq package (v1.32.1) [69]. Both rarefied and non-rarefied data were analysed dependent on input and standardisation requirements of particular analysis. Firstly, rare sequence variants were removed (< 10 sequence reads) from the ASV table, using the ‘prune_taxa’ function of the phyloseq package. A linear model (LM) was used to identify significant relationships between soil variables and landscape units using the ‘lm’ function of the phyloseq package. To investigate differences in community composition between landscape units, ordination of ASV beta diversity was calculated with the ‘ordinate’ function in phyloseq using unrarefied data. Constrained analysis of principal coordinates (CAP) was performed on the Bray–Curtis dissimilarity matrix constrained by soil variables: organic carbon, nitrate, phosphorus, sodium, pH, calcium, magnesium, ammonium, and by plant functional diversity. Potassium was removed from ordination, as it was highly correlated with other variables (significance cut-off of > 0.7 or < − 0.7, Pearson’s product-moment correlation). Constraining variable significance was assessed non-parametrically via 999 permutations. The ‘betadisper’ function was used to test for homogeneity of group dispersions. A PERMANOVA (999 iterations) was run with the ‘pairwise_adonis2’ function of the pairwise.adonis package to test the significance of community compositional variation between landscape units [70].
To investigate diversity of landscape units, alpha diversity was calculated at ASV level using observed richness and Shannon and Simpson diversity indices, performed using the ‘estimate_richness’ function in the phyloseq package. Prior to alpha diversity calculations, ASV level data was rarefied to using the phyloseq packages ‘rarefy_even_depth’ function, and Shannon and Simpson index values were transformed to effective number of ASVs. A negative binomial generalised linear model (GLM) was used to test for differences in alpha diversity between landscape units, followed by ‘goodness of fit’ analysis using chi-squared distribution ‘pchisq’ function in the phyloseq package. A type II Wald chi2 test was run with the ‘ANOVA’ function of the car package to test main effects of the GLM’s (v3.0–10) [71]. Pairwise comparisons using Holm-Bonferroni P-adjustment were then made between landscape units using ‘pairwise’ function in the phyloseq package. A correlation matrix (Pearson product-moment) was used to identify significant relationships between soil and ecological (plant functional diversity) variables and diversity metrics, and to determine any correlating variables. Bacterial community composition was further investigated via a relative abundance stack plot, created by converting the rarefied family abundances to percentages. Rare families (< 2% of total rarefied sequences) were pooled into a single group named ‘pooled (< 2% relative abundance)’.
Results
Aboveground diversity
Remnant mallee vegetation (RemVeg) had the highest plant species richness and plant functional diversity, possessing all five functional groups across replicates (Table 1; Supplementary 5 and 6). Four plant functional groups were observed across replicates of the revegetation system (Reveg), which was also found to contain some large established and recruiting native vegetation. Seedlings of planted species were not included in the Reveg landscape unit species list, as they were not yet established. The managed vineyard systems (OldVineyard and NewVineyard) were found to have low plant functional diversity, typically consisting of a medium shrub layer (Vitis sp.) and an annual grassy groundcover. Likewise, the ex-pasture systems (ExCrop) predominantly consisted of two plant functional groups (perennial and annual herbaceous groundcover).
Soil physicochemical properties
Soil texture was consistent across the study site (Supplementary 1). Replicates of the remnant mallee vegetation landscape unit (RemVeg) ranged from sandy loam to silty loam. Similarly, both the vineyard landscape units (OldVineyard and NewVineyard), the revegetation (Reveg), and the ex-cropping landscape units (ExCrop) were identified as either silty loam, sandy loam, or loam.
Linear models (soil variable against landscape units) revealed a number of statistically significant correlations between physicochemical variables and landscape units, including a number of key nutrients. Nitrate (NO3−) was highest in both vineyard systems (NewVineyard, p < 0.001; OldVineyard, p = 0.004), plant-available (Colwell) phosphorus was elevated in the OldVineyard landscape unit (p < 0.001), while potassium was significantly lower in the RemVeg landscape unit (Fig. 3). Magnesium was elevated in the OldVineyard (p < 0.00) landscape units, calcium was elevated in the ExCrop (p = 0.007), NewVineyard (p = 0.003) and OldVineyard (p = 0.001) landscape units (Fig. 4), and sodium was elevated in the two vineyard systems (new, p = 0.015; old, p = 0.020).

Boxplot panel displaying organic carbon (mg/kg), nitrate (NO3.−; mg/kg), potassium (mg/kg) and phosphorus (Colwell; mg/kg) concentrations across landscape units, with linear model (soil variable by landscape unit) significance codes: ‘.’ P < 0.10; ‘*’ P < 0.05; ‘**’ P < 0.01; ‘***’ P < 0.001 (Holm-Bonferroni P-adjustment)

Boxplot panel displaying magnesium (mg/kg), calcium (mg/kg), sodium (mg/kg), and pH (CaCl2) concentrations across landscape units, with linear model (soil variable by landscape unit) significance codes: ‘.’ P < 0.10; ‘*’ P < 0.05; ‘**’ P < 0.01; ‘***’ P < 0.001 (Holm-Bonferroni P-adjustment)
Bacterial community composition
Landscape units were found to be associated with distinct bacterial communities (Fig. 5), with a significant PERMANOVA test on the dissimilarity matrix (Bray–Curtis, F = 2.86, p < 0.001). Pairwise community analysis revealed a significant difference in community composition between all but one of the 10 landscape unit pairwise comparisons, that being the ExCrop and Reveg landscape units (Supplementary 4). Constrained ordination (CAP) indicated a clear shift in community composition from the more natural landscape units (RemVeg and Reveg) to the highly modified systems (ExCrop, OldVineyard, and NewVineyard) (Fig. 5). Constraining gradients of soil physicochemistry and plant functional diversity explained 48% of variance in community composition, with organic carbon, nitrate, phosphorus, pH, and calcium found to be significant (Table 2). Beta dispersion test was significant (F = 17.839, p < 0.001), indicating that landscape units had variable species turnover among replicates.

First two axes of a constrained analysis of principal coordinates (CAP) using Bray–Curtis distance constrained by soil physicochemical and ecological (plant functional diversity) variables, which explained 48% of variance across all axes. Axes 1–2 (depicted) explain 18.1% (CAP1) and 9.8% (CAP2) of total variance, respectively. 95% confidence ellipses were applied post-hoc to landscape units
The more natural predominantly unmanaged native vegetation land systems (RemVeg and Reveg) separate out from the highly modified managed systems (OldVineyard, NewVineyard, and ExCrop) along the primary x axis (CAP1, 18.1% of variation explained) (Fig. 5). This partitioning appears to be strongly influenced by plant functional diversity and agricultural inputs, evident by vectors plant functional diversity, nitrate, phosphorus, calcium, and magnesium with soil pH also an influential variable affecting group partitioning along the x axis (CAP1). Bacterial community shift is also apparent within the highly modified and managed systems along the y axis (CAP2, 9.8% of variation explained), with the more natural ExCrop landscape unit (in comparison to the vineyard systems) clearly removed from the established vineyard system (OldVineyard), with the newly established vineyard system (NewVineyard) sitting between the two (Fig. 5).
Bacterial alpha diversity
Soil bacterial diversity was compared between landscape units at ASV level. Of the three diversity metrics calculated (observed richness, effective Shannon, and effective Simpson), Simpson diversity was found to be significantly different among landscape units, as determined by GLM (Simpson, chi2 = 0.225, p = 0.005; Shannon, chi2 = 0.225, p = 0.078) (Supplementary 2). The managed vineyard systems returned the highest bacterial diversity across all measured metrics, with both these systems returning significant Shannon diversity (NewVineyard, Z = 2.037, p = 0.042, OldVineyard, Z = 2.483, p = 0.013), while the OldVineyard returned significant Simpson diversity (Z = 3.496, p = 0.0004) (Fig. 6, Supplementary 2).

Boxplot of alpha diversity results across landscape units, displaying observed richness, Shannon (effective species), and Simpson (effective species) diversity metrics, with negative binomial GLM model (diversity metric by landscape unit) significance codes: ‘.’ P < 0.10; ‘*’ P < 0.05; ‘**’ P < 0.01; ‘***’ P < 0.001 (Holm-Bonferroni P-adjustment)
Alpha diversity pairwise comparisons revealed a significant difference (Simpson diversity) between the RemVeg and OldVineyard landscape units (Z = − 3.496, p = 0.004) (Supplementary 3). No significant correlations (cut-off of 0.7, Pearson’s product-moment correlation) were found between any of the soil physicochemical or ecological (plant functional diversity) variables measured and any of the calculated diversity metrics (observed richness, Shannon, Simpson) (Table 3). Several significant correlations were found between soil physicochemical variables (potassium/calcium, r = + 0.722; potassium/phosphorus, r = + 0.747; potassium/plant functional diversity, r = 0.675).
Taxa analysis
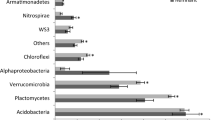
Bacterial taxa analysis was undertaken at the family taxonomic level, as it was expected that more specific and precise functional information could be sought because it was thought that similar functional groups (e.g. decomposers, parasites, mutualists) would be represented. Analysis revealed that rare taxa (< 2% relative abundance) dominate the system (34% relative abundance), with only 10 of the 210 families identified across the site found to be abundant (> 2% relative abundance), these being Rubrobacteraceae (17.4% relative abundance), Bacillaceae (9.2% relative abundance), Bradyrhizobiaceae (7.2% relative abundance), Pseudonocardiaceae (3.6% relative abundance), Micrococcaceae (3.1% relative abundance), Geodermatophilaceae (2.7% relative abundance), Rhodospirillaceae (5.1% relative abundance), Sphingomonadaceae (3.4% relative abundance), Sinobacteraceae (2.8% relative abundance), and Hyphomicrobiaceae (2.7% relative abundance).
The relative abundance of bacterial families within landscape units was further investigated, similarly finding that bacterial communities were dominated by rare taxa (< 2% relative abundance), ranging from 38% of community composition in the Reveg landscape unit to 30% in the ExCrop landscape unit (Fig. 7, Supplementary 8). Interestingly, the Rubrobacteraceae family was found to be the dominant taxa in all landscape unit communities except the established vineyard system (OldVineyard), where the Bacillaceae family was found to be most relatively abundant (Fig. 7, Supplementary 8).

Bacterial family stack plot (mean relative abundance). Displaying landscape unit bacterial community composition at family level
Discussion
Bacterial community composition and soil physicochemical characteristics of different land systems (landscape units) were investigated across a semi-arid production landscape to explore the impact of land use on soil bacterial communities. As hypothesised, the landscape units differed in their soil physicochemical characteristics. This was linked to a shift in the soil microbiome, such that distinct bacterial communities were associated with land use systems. Interestingly, highest bacterial diversity was observed in the managed vineyard system, highlighting that aboveground diversity does not necessarily correlate with belowground diversity. The restoration of native plant communities appears to be acting to recover native bacterial communities, suggesting that such actions have the capacity to not only influence aboveground species composition but also belowground bacterial community assemblage.
Managed vineyard systems associated with elevated levels of key nutrients
Elevated concentrations of nitrate (NO3−) and plant-available (Colwell) phosphorus were identified in the managed vineyard systems (OldVineyard and NewVineyard). The higher concentrations of these nutrients were not surprising, given the addition of soil microbial inoculants containing proportions of these nutrients (product A, nitrogen = 2.66% w/v, phosphorus = 1.2% w/v, potassium 0.25% w/v; product B, phosphorus = 2.09%), and the addition of other fertilisers that would also likely contain these nutrients.
This result serves to highlight the physiochemical changes in agricultural systems (mallee vegetation to vineyard agriculture) in relation to soil nutrition. Although increased concentrations of common agricultural inputs such as nitrogen and phosphorus could be viewed as positive in the context of agricultural productivity, the long-term sustainability of the system could be questioned given the well-recognised negative impacts associated with fertiliser use [2, 72].
Land systems/practices drive distinct bacterial communities
Our results indicate that bacterial community composition is strongly associated with land use, based on pairwise comparisons and constrained ordination (CAP) analysis of bacterial community composition (Fig. 5, Supplementary 4). Only one of the pairwise comparisons was not significant (ExCrop – Reveg). This is likely due to the fact that the revegetation systems have only recently (within the last 2 years) been converted from pastoral land (ExCrop), and as such the associated bacterial community still resembles the community associated with the ExCrop landscape units. The observed separation of natural systems (RemVeg and Reveg) from the modified agricultural systems (ExCrop, OldVineyard, and NewVineyard) in the CAP analysis indicates that agricultural land use change has modified bacterial community composition across the study system. The observed partition of natural and agricultural communities appears to be strongly correlated with, and potentially driven by, plant functional diversity and management practice.
Elevated concentrations of key nutrients in the vineyard systems may be the result of microbial inoculants and additional fertiliser inputs, indicating that these practices, and the associated change in soil nutrients, have influenced community composition, evident by statistically significant nitrate and phosphorus-constraining variables in CAP analysis (Fig. 5). Although not found to correlate with bacterial diversity (Table 3), soil pH also appears to influence community composition, in line with other studies returning similar results [73]. Constrained ordination (CAP) also revealed that plant functional diversity influenced community composition in the opposite direction to key nutrient vectors (nitrate and plant-available (Colwell) phosphorus) in the ordination space (Fig. 5). As expected, the conversion of remnant mallee vegetation to vineyard agriculture was found to reduce plant functional diversity, suggesting that land use change is also a key factor influencing bacterial community composition.
The Rubrobacteraceae family was the most abundant family across the study site (~ 17.4% relative abundance) and was most abundant in all landscape units except the established vineyards (OldVineyard). Recognised as one of the most radiation-resistant organisms [74], and halotolerant and desiccation tolerant [75], the Rubrobacteraceae has a selective advantage in extreme environments, including arid soils [74, 76], permafrost [77], and saline environments [78]. The greater relative abundance of Rubrobacteraceae across the study system is likely a legacy of the semi-arid soils that much of the site would have consisted of before its conversion to irrigated production agriculture. The reduced abundance of this family in vineyard systems highlights the ability of land use to alter the abundance of specific taxa. Indeed, revegetation of old pasture sites (Reveg landscape unit) has acted to shift the bacterial community back towards a reference state (Fig. 5)—community associated with remnant mallee vegetation (RemVeg). This finding suggests that the restoration of aboveground ecosystems can act to restore belowground bacterial communities, as reported in other studies [79], with potential implications for soil and wider ecosystem health. For instance, native microbial communities likely harbor a greater proportion of species possessing advantageous traits to local environmental conditions, providing a pool of well adapted (potentially plant beneficial) species that may disperse to adjacent production systems, such as vineyards.
To explore the efficacy of management practices employed to improve soil condition (inoculation), inoculated groups were investigated (where possible). The inoculated bacterial family Pseudomonadaceae was not found to be abundant (> 2% relative abundance) in any of the landscape units (Supplementary 8), including the landscape units in which it was applied (OldVineyard and NewVineyard), indicating that the addition of inoculants has not influenced the abundance of this family. No conclusion could be drawn regarding the efficacy of applied inoculants to proliferate members of the Actinomycetes group, as no information could be sourced regarding which specific taxon’s (e.g. species, genera, families) the inoculants contained.
Conversion of remnant mallee vegetation to vineyard agriculture increases bacterial diversity
Observed bacterial richness at amplicon sequence variant level was not significant among the seven landscape units assessed, while both effective Simpson and Shannon diversity metrics (which account for abundance and evenness) were found to be statistically significant (Supplementary 2). Shannon and Simpson diversity metrics are widely recommended and commonly used when analysing microbial diversity and have been shown to reduce the bias (richness over evenness) often associated with other diversity metrics [80, 81].
Results revealed that the managed vineyard landscape units had highest soil bacterial diversity, with both the established and new vineyard systems (NewVineyard and OldVineyard) returning statistically significant diversity results. This result was not in line with our expectations or with other studies that suggest a positive correlation between plant diversity/complexity and bacterial diversity [12, 51, 53, 81]. The conversion of the more diverse remnant mallee vegetation to monoculture agriculture has, in fact, increased belowground bacterial diversity, suggesting that agriculturally driven land use change has resulted in a decoupling of above- and belowground diversities.
Although a positive relationship between above- and belowground diversities has been observed in other studies [53], and could be considered a broadly accepted principle [82], we suggest that agriculture can act to disrupt this relationship via major modification of the natural soil system; modification occurring through the removal and replacement of natural plant communities (and their associated inputs), and ongoing management practices associated with production systems, such as the planting of crops, chemical/fertiliser application, and soil tillage. In this regard, we propose that agricultural land use (and associated practices) may be a stronger driver of soil bacterial diversity than aboveground plant diversity. In their analysis of experimental grasslands, Zak et al. (2003) found that plant diversity increased the biomass and composition of soil microbial communities, but attributed this to the increase in plant production associated with greater species diversity, rather than to plant diversity per se [83]. This goes some way to explaining findings here, given that plant production would likely be greatest in the agricultural systems (due to management practices such as fertiliser and water inputs) where bacterial diversity was also found to be highest (New and OldVineyards), adding support for our suggestion that agriculture fundamentally disrupts the natural processes that otherwise govern above- and belowground diversity linkage, such as plant production. Further support for this can be found in a global meta-analysis of more than 84 studies. Liu et al. (2020) found that microbial richness showed a moderate but positive correlation with plant diversity, and likewise suggested that plant communities with higher diversity may promote more diverse microbial communities through greater diversity of inputs (chemical, i.e. root exudates and physical, i.e. litter) and higher productivity, leading to increased niche space [53].
Another factor likely impacting diversity, and typical of agricultural systems, is disturbance. The intermediate disturbance hypothesis (where the diversity of competing species will be maximised at intermediate frequencies and/or intensities of disturbance or environmental change) [84] partly explains the diversity results found here. In stable-state environments, fewer well-adapted taxa outcompete and dominate those less adapted, reducing diversity. Conversely, intermittently disturbed environments can result in the persistence of a greater number of taxa due to increased environmental heterogeneity or habitats (niche space) in which different taxa are suitably adapted, and can exploit [16, 36]. Although it is recognised that the study here is not appropriately designed to test the IDH, results do point to disturbance as a significant factor driving bacterial diversity.
Land use change (from remnant mallee vegetation to managed vineyards) and associated management practices employed (e.g. cover cropping, chemical/fertiliser application, tillage) could be viewed as having a positive effect on soil health, given that there is some evidence to suggest that soil microbial diversity confers stability to stress and protection against soil-borne disease [84]. However, while microbial diversity is a valuable tool in evaluating change in soil condition, it is acknowledged that increased diversity does not necessarily indicate positive change. For instance, a bacterial community may have higher diversity than another while also consisting a greater proportion of parasitic taxa, potentially indicating poor soil condition (in the context of plant productivity). Thus, taxonomic community shifts should also be considered in evaluating the impacts of aboveground land use change, as this variable may be of greater significance to soil–plant systems than diversity per se. It is also recognised that increased bacterial diversity and soil fertility associated with the managed vineyard systems are likely the results of other inputs, such as microbial amendments, fertilizer, and water. Interestingly, nitrate and plant-available (Colwell) phosphorus were found to be elevated in both vineyard systems (Fig. 3), while no significant correlation was identified between these nutrients and bacterial diversity (Table 3). It could be assumed that the amendments (hydrolysed molasses, amino acids, fulvic acid, seaweed, and liquid fish) and/or the living bacteria within the inoculants are driving diversity in the inoculated systems (New and OldVineyards) [85]. However, this assumption lacks the appropriate statistical evidence to be validated here, and further work would be required to assess the impact of these soil amendments on bacterial diversity.
Conclusion
A reduction in aboveground plant diversity is inevitable when natural systems are converted to large-scale production monocultures, as was found here. It is also broadly assumed that this aboveground change results in a reduction in belowground diversity (above- and belowground diversity linkage). Our results stand in contrast to this assumption with the finding that agricultural systems (with reduced aboveground diversity) have increased soil bacterial diversity. We highlight that restoration of native plant communities can act to rapidly recover natural soil bacterial communities, which in turn could improve soil and plant health. However, the impact of shifts in bacterial community composition associated with land use systems detected here is not fully understood without a greater understanding of the functional significance of the key bacterial groups identified. Such shifts should be considered in future studies seeking to further our understanding of land use impacts on soil and plant health.
Data availability
The datasets generated during the current study are available in the Figshare repository, https://doi.org/10.6084/m9.figshare.19289450
Change history
23 February 2023
Missing Open Access funding information has been added in the Funding Note.
References
Foley JA, Ramankutty N, Brauman KA, Cassidy ES, Gerber JS, Johnston M, Mueller ND, O’Connell C, Ray DK, West PC, Balzer C, Bennett EM, Carpenter SR, Hill J, Monfreda C, Polasky S, Rockstrom J, Sheehan J, Siebert S, Tilman D, Zaks DP (2011) Solutions for a cultivated planet. Nature 478:337–342. https://doi.org/10.1038/nature10452
Foley JA, DeFries R, Asner GP, Barford C, Bonan G, Carpenter SR, Chapin FS, Coe MT, Daily GC, Gibbs HK, Helkowski JH, Holloway T, Howard EA, Kucharik CJ, Monfreda C, Patz JA, Prentice IC, Ramankutty N, Snyder PK (2005) Global consequences of land use. Science 309:570–574. https://doi.org/10.1126/science.1111772
Pingali PL (2012) Green revolution: impacts, limits, and the path ahead. Proc Natl Acad Sci U S A 109:12302–12308. https://doi.org/10.1073/pnas.0912953109
Louwagie G, Gay S, Sammeth F, Ratinger T (2010) The potential of European Union policies to address soil degradation in agriculture. Land Degrad Dev 22:5–17
Koch A, Chappell A, Eyres M, Scott E (2015) Monitor soil degradation or triage for soil security? An Australian challenge. Sustainability 7:4870–4892
Bongaarts J (2019) Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. Popul Dev Rev 45:680–681. https://doi.org/10.1111/padr.12283
Millennium Ecosystem Assessment (2005) Ecosystems and Human Well-Being: Synthesis. Island Press, Washington
Sutton PC, Anderson SJ, Costanza R, Kubiszewski I (2016) The ecological economics of land degradation: impacts on ecosystem service values. Ecol Econ 129:182–192. https://doi.org/10.1016/j.ecolecon.2016.06.016
Lal R (2004) Soil carbon sequestration impacts on global climate change and food security. Science 304:1623–1627. https://doi.org/10.1126/science.1097396
Jie C, Jing-zhang C, Man-zhi T, Zi-tong G (2002) Soil degradation: a global problem endangering sustainable development. J Geog Sci 12:243–252. https://doi.org/10.1007/BF02837480
Garcia-Orenes F, Morugan-Coronado A, Zornoza R, Cerda A, Scow K (2016) Correction: changes in soil microbial community structure influenced by agricultural management practices in a Mediterranean agro-ecosystem. PLoS One 11:e0152958. https://doi.org/10.1371/journal.pone.0152958
Holland TC, Bowen PA, Bogdanoff CP, Lowery TD, Shaposhnikova O, Smith S, Hart MM (2016) Evaluating the diversity of soil microbial communities in vineyards relative to adjacent native ecosystems. Appl Soil Ecol 100:91–103. https://doi.org/10.1016/j.apsoil.2015.12.001
Fierer N, Ladau J, Clemente JC, Leff JW, Owens SM, Pollard KS, Knight R, Gilbert JA, McCulley RL (2013) Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342:621–624. https://doi.org/10.1126/science.1243768
Wei G, Li M, Shi W, Tian R, Chang C, Wang Z, Wang N, Zhao G, Gao Z (2020) Similar drivers but different effects lead to distinct ecological patterns of soil bacterial and archaeal communities. Soil Biol Biochem 144:107759. https://doi.org/10.1016/j.soilbio.2020.107759
Hayat R, Ali S, Amara U, Khalid R, Ahmed I (2010) Soil beneficial bacteria and their role in plant growth promotion: a review. Ann Microbiol 60:579–598
Kennedy AC (1999) Bacterial diversity in agroecosystemsinvertebrate biodiversity as bioindicators of sustainable landscapes, pp. 65–76
Heijden VD (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310
Ramakrishna W, Yadav R, Li K (2019) Plant growth promoting bacteria in agriculture: two sides of a coin. Appl Soil Ecol 138:10–18. https://doi.org/10.1016/j.apsoil.2019.02.019
Saharan B, Nehra V (2011) Plant growth promoting rhizobacteria: a critical review. Life Sci Med Res 21:1–30
Kennedy I, Islam N (2001) The current and potential contribution of asymbiotic nitrogen fixation to nitrogen requirements on farms: a review. Aust J Exp Agric 41:447–457
Zlotnikov AK, Shapovalova YN, Makarov AA (2001) Association of Bacillus firmus E3 and Klebsiella terrigena E6 with increased ability for nitrogen fixation. Soil Biol Biochem 33:1525–1530. https://doi.org/10.1016/s0038-0717(01)00070-0
Glick BR, Todorovic B, Czarny J, Cheng Z, Duan J, McConkey B (2007) Promotion of plant growth by bacterial ACC deaminase. Crit Rev Plant Sci 26:227–242. https://doi.org/10.1080/07352680701572966
Spaepen S, Vanderleyden J, Remans R (2007) Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol Rev 31:425–448. https://doi.org/10.1111/j.1574-6976.2007.00072.x
Dawar S, Wahab S, Tariq M, Zaki MJ (2010) Application ofBacillusspecies in the control of root rot diseases of crop plants. Arch Phytopathol Plant Prot 43:412–418. https://doi.org/10.1080/03235400701850870
Bronick CJ, Lal R (2005) Soil structure and management: a review. Geoderma 124:3–22. https://doi.org/10.1016/j.geoderma.2004.03.005
Six J, Bossuyt H, Degryze S, Denef K (2004) A history of research on the link between (micro)aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res 79:7–31. https://doi.org/10.1016/j.still.2004.03.008
Borneman J, Skroch PW, O’Sullivan KM, Palus JA, Rumjanek NG, Jansen JL, Nienhuis J, Triplett EW (1996) Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol 62:1935–1943. https://doi.org/10.1128/aem.62.6.1935-1943.1996
Upchurch R, Chiu C-Y, Everett K, Dyszynski G, Coleman DC, Whitman WB (2008) Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol Biochem 40:1294–1305. https://doi.org/10.1016/j.soilbio.2007.06.027
Baker GC, Gaffar S, Cowan DA, Suharto AR (2001) Bacterial community analysis of Indonesian hot springs. FEMS Microbiol Lett 200:103–109. https://doi.org/10.1111/j.1574-6968.2001.tb10700.x
Miura Y, Hiraiwa MN, Ito T, Itonaga T, Watanabe Y, Okabe S (2007) Bacterial community structures in MBRs treating municipal wastewater: relationship between community stability and reactor performance. Water Res 41:627–637. https://doi.org/10.1016/j.watres.2006.11.005
Moissl C, Osman S, La Duc MT, Dekas A, Brodie E, DeSantis T, Venkateswaran K (2007) Molecular bacterial community analysis of clean rooms where spacecraft are assembled. FEMS Microbiol Ecol 61:509–521. https://doi.org/10.1111/j.1574-6941.2007.00360.x
Rampelotto P, de Siqueira Ferreira A, Barboza A, Fernando L, Roesch W (2013) Changes in diversity, abundance, and structure of soil bacterial communities in Brazilian savanna under different land use systems. Microb Ecol 66:593–607
Ondreičková K, Piliarová M, Bušo R, Hašana R, Schreiber Ľ, Gubiš J, Kraic J (2018) The structure and diversity of bacterial communities in differently managed soils studied by molecular fingerprinting methods. Sustainability 10:1095. https://doi.org/10.3390/su10041095
Jeanbille M, Buee M, Bach C, Cebron A, Frey-Klett P, Turpault MP, Uroz S (2016) Soil parameters drive the structure, diversity and metabolic potentials of the bacterial communities across temperate beech forest soil sequences. Microb Ecol 71:482–493. https://doi.org/10.1007/s00248-015-0669-5
Rodrigues JL, Pellizari VH, Mueller R, Baek K, Jesus Eda C, Paula FS, Mirza B, Hamaoui GS Jr, Tsai SM, Feigl B, Tiedje JM, Bohannan BJ, Nusslein K (2013) Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc Natl Acad Sci U S A 110:988–993. https://doi.org/10.1073/pnas.1220608110
Trivedi P, Delgado-Baquerizo M, Anderson IC, Singh BK (2016) Response of soil properties and microbial communities to agriculture: implications for primary productivity and soil health indicators. Front Plant Sci 7:990. https://doi.org/10.3389/fpls.2016.00990
Bartelt-Ryser J, Joshi J, Schmid B, Brandl H, Balser T (2005) Soil feedbacks of plant diversity on soil microbial communities and subsequent plant growth. Perspect Plant Ecol Evol Syst 7:27–49. https://doi.org/10.1016/j.ppees.2004.11.002
van de Koppel J, Rietkerk M, Weissing FJ (1997) Catastrophic vegetation shifts and soil degradation in terrestrial grazing systems. Trends Ecol Evol 12:352–356. https://doi.org/10.1016/s0169-5347(97)01133-6
Pulido M, Schnabel S, Francisco J, Contador L, Lozano-Parra J, González F (2016) The impact of heavy grazing on soil quality and pasture production in rangelands of SW Spain. Land Degrad Dev 29:219–230
Bacq-Labreuil A, Neal A, Crawford J, Mooney S, Akkari E, Zhang X, Clark I, Ritz K (2020) Significant structural evolution of a long-term fallow soil in response to agricultural management practices requires at least 10 years after conversion. Soil Sci 72:829–841
Bissett A, Richardson AE, Baker G, Thrall PH (2011) Long-term land use effects on soil microbial community structure and function. Appl Soil Ecol 51:66–78. https://doi.org/10.1016/j.apsoil.2011.08.010
Martínez ML, Pérez-Maqueo O, Vázquez G, Castillo-Campos G, García-Franco J, Mehltreter K, Equihua M, Landgrave R (2009) Effects of land use change on biodiversity and ecosystem services in tropical montane cloud forests of Mexico. For Ecol Manage 258:1856–1863. https://doi.org/10.1016/j.foreco.2009.02.023
Smith M, Conte P, Berns AE, Thomson JR, Cavagnaro TR (2012) Spatial patterns of, and environmental controls on, soil properties at a riparian–paddock interface. Soil Biol Biochem 49:38–45. https://doi.org/10.1016/j.soilbio.2012.02.007
Kennedy AC, Smith KL (1995) Soil microbial diversity and the sustainability of agricultural soils. Plant Soil 170:75–86. https://doi.org/10.1007/bf02183056
Hooper DU, Bignell DE, Brown VK, Brussard L, Mark Dangerfield J, Wall DH, Wardle DA, Coleman DC, Giller KE, Lavelle P, Van Der Putten WH, De Ruiter PC, Rusek J, Silver WL, Tiedje JM, Wolters V (2000) Interactions between aboveground and belowground biodiversity in terrestrial ecosystems: patterns, mechanisms, and feedbacks. Bioscience 50:1049. https://doi.org/10.1641/0006-3568(2000)050[1049:Ibaabb]2.0.Co;2
Kim HS, Lee SH, Jo HY, Finneran KT, Kwon MJ (2021) Diversity and composition of soil Acidobacteria and Proteobacteria communities as a bacterial indicator of past land-use change from forest to farmland. Sci Total Environ 797:148944. https://doi.org/10.1016/j.scitotenv.2021.148944
Lange M, Eisenhauer N, Sierra CA, Bessler H, Engels C, Griffiths RI, Mellado-Vazquez PG, Malik AA, Roy J, Scheu S, Steinbeiss S, Thomson BC, Trumbore SE, Gleixner G (2015) Plant diversity increases soil microbial activity and soil carbon storage. Nat Commun 6:6707. https://doi.org/10.1038/ncomms7707
Stephan A, Meyer AH, Schmid B (2000) Plant diversity affects culturable soil bacteria in experimental grassland communities. J Ecol 88:988–998. https://doi.org/10.1046/j.1365-2745.2000.00510.x
Wang H, Liu S, Li H, Tao X, Wang H, Qi J, Zhang Z (2022) Large-scale homogenization of soil bacterial communities in response to agricultural practices in paddy fields, China. Soil Biology and Biochemistry 164. https://doi.org/10.1016/j.soilbio.2021.108490
Wolinska A, Rekosz-Burlaga H, Goryluk-Salmonowicz A, Błaszczyk M, Stepniewska Z (2015) Bacterial abundance and dehydrogenase activity in selected agricultural soils from Lublin Region Polish. J Environ Stud 24:2677. https://doi.org/10.15244/pjoes/59323
Wen Z, Zheng H, Zhao H, Xie S, Liu L, Ouyang Z (2020) Land-use intensity indirectly affects soil multifunctionality via a cascade effect of plant diversity on soil bacterial diversity. Global Ecol Conserv 23:e01061. https://doi.org/10.1016/j.gecco.2020.e01061
Dedeyn G, Vanderputten W (2005) Linking aboveground and belowground diversity. Trends Ecol Evol 20:625–633. https://doi.org/10.1016/j.tree.2005.08.009
Liu L, Zhu K, Wurzburger N, Zhang J (2020) Relationships between plant diversity and soil microbial diversity vary across taxonomic groups and spatial scales. Ecosphere 11:e02999. https://doi.org/10.1002/ecs2.2999
Szoboszlay M, Dohrmann AB, Poeplau C, Don A, Tebbe CC (2017) Impact of land-use change and soil organic carbon quality on microbial diversity in soils across Europe. FEMS Microbiology Ecology 93. https://doi.org/10.1093/femsec/fix146
Romdhane S, Spor A, Banerjee S, Breuil M-C, Bru D, Chabbi A, Hallin S, van der Heijden MGA, Saghai A, Philippot L (2022) Land-use intensification differentially affects bacterial, fungal and protist communities and decreases microbiome network complexity. Environmental Microbiome 17. https://doi.org/10.1186/s40793-021-00396-9
Sui X, Zhang R, Frey B, Yang L, Li M-H, Ni H (2019) Land use change effects on diversity of soil bacterial, Acidobacterial and fungal communities in wetlands of the Sanjiang Plain, northeastern China. Sci Rep 9:18535. https://doi.org/10.1038/s41598-019-55063-4
Amoo AE, Babalola OO (2019) Impact of land use on bacterial diversity and community structure in temperate pine and indigenous forest soils. Diversity 11:217. https://doi.org/10.3390/d11110217
Lacerda-Júnior GV, Noronha MF, Cabral L, Delforno TP, de Sousa STP, Fernandes-Júnior PI, Melo IS, Oliveira VM (2019) Land use and seasonal effects on the soil microbiome of a Brazilian dry forest. Frontiers in Microbiology 10. https://doi.org/10.3389/fmicb.2019.00648
Meteorology AGBo (2021) Daily rainfall Robinvale. http://www.bom.gov.au/climate/data/. Accessed 11 June 2021
Isbell R (2002) The Australian soil classification. CSIRO Publishing, Melbourne
Bissett A, Fitzgerald A, Court L, Meintjes T, Mele PM, Reith F, Dennis PG, Breed MF, Brown B, Brown MV, Brugger J, Byrne M, Caddy-Retalic S, Carmody B, Coates DJ, Correa C, Ferrari BC, Gupta VV, Hamonts K, Haslem A, Hugenholtz P, Karan M, Koval J, Lowe AJ, Macdonald S, McGrath L, Martin D, Morgan M, North KI, Paungfoo-Lonhienne C, Pendall E, Phillips L, Pirzl R, Powell JR, Ragan MA, Schmidt S, Seymour N, Snape I, Stephen JR, Stevens M, Tinning M, Williams K, Yeoh YK, Zammit CM, Young A (2016) Introducing BASE: the biomes of Australian Soil Environments soil microbial diversity database. Gigascience 5:21. https://doi.org/10.1186/s13742-016-0126-5
Moody PW (2007) Interpretation of a single-point P buffering index for adjusting critical levels of the Colwell soil P test. Soil Res 45:55. https://doi.org/10.1071/sr06056
Chatterjee A, Lal R, Wielopolski L, Martin MZ, Ebinger MH (2009) Evaluation of different soil carbon determination methods. Crit Rev Plant Sci 28:164–178. https://doi.org/10.1080/07352680902776556
Qiagen (2021) DNeasy PowerSoil Pro Kit. Qiagen. https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/dna-purification/microbial-dna/dneasy-powersoil-pro-kit/.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Bokulich NA, Dillon MR, Zhang Y, Rideout JR, Bolyen E, Li H, Albert PS, Caporaso JG (2018) q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems 3. https://doi.org/10.1128/mSystems.00219-18
Foundation TR (2021) R: a language and environment for statistical computing’, R Foundation for Statistical Computing. The R Foundation. https://www.r-project.org/.
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. Plos One 8:e61217. https://doi.org/10.1371/journal.pone.0061217
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. https://doi.org/10.1111/j.1442-9993.2001.01070.pp.x
Fox J, Friendly M, Weisberg S (2013) Hypothesis tests for multivariate linear models using the car package. R J 5:39
Pahalvi HN, Rafiya L, Rashid S, Nisar B, Kamili AN (2021) Chemical fertilizers and their impact on soil healthmicrobiota and biofertilizers 2:1–20
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl Environ Microbiol 75:5111–5120. https://doi.org/10.1128/aem.00335-09
Albuquerque L, Johnson MM, Schumann P, Rainey FA, da Costa MS (2014) Description of two new thermophilic species of the genus Rubrobacter, Rubrobacter calidifluminis sp. nov. and Rubrobacter naiadicus sp. nov., and emended description of the genus Rubrobacter and the species Rubrobacter bracarensis. Syst Appl Microbiol 37:235–243. https://doi.org/10.1016/j.syapm.2014.03.001
Meier DV, Imminger S, Gillor O, Woebken D (2021) Distribution of mixotrophy and desiccation survival mechanisms across microbial genomes in an arid biological soil crust community. mSystems 6. https://doi.org/10.1128/mSystems.00786-20
Favet J, Lapanje A, Giongo A, Kennedy S, Aung YY, Cattaneo A, Davis-Richardson AG, Brown CT, Kort R, Brumsack HJ, Schnetger B, Chappell A, Kroijenga J, Beck A, Schwibbert K, Mohamed AH, Kirchner T, de Quadros PD, Triplett EW, Broughton WJ, Gorbushina AA (2013) Microbial hitchhikers on intercontinental dust: catching a lift in Chad. ISME J 7:850–867. https://doi.org/10.1038/ismej.2012.152
Jansson JK, Tas N (2014) The microbial ecology of permafrost. Nat Rev Microbiol 12:414–425. https://doi.org/10.1038/nrmicro3262
Kutovaya OV, Lebedeva MP, Tkhakakhova AK, Ivanova EA, Andronov EE (2015) Metagenomic characterization of biodiversity in the extremely arid desert soils of Kazakhstan. Eurasian Soil Sci 48:493–500. https://doi.org/10.1134/s106422931505004x
Gellie NJC, Mills JG, Breed MF, Lowe AJ (2017) Revegetation rewilds the soil bacterial microbiome of an old field. Mol Ecol 26:2895–2904. https://doi.org/10.1111/mec.14081
He Y, Zhou BJ, Deng GH, Jiang XT, Zhang H, Zhou HW (2013) Comparison of microbial diversity determined with the same variable tag sequence extracted from two different PCR amplicons. BMC Microbiol 13:208. https://doi.org/10.1186/1471-2180-13-208
Mills JG, Bissett A, Gellie NJC, Lowe AJ, Selway CA, Thomas T, Weinstein P, Weyrich LS, Breed MF (2020) Revegetation of urban green space rewilds soil microbiotas with implications for human health and urban design. Restor Ecol 28:S322–S334. https://doi.org/10.1111/rec.13175
Wardle DA, Bardgett RD, Klironomos JN, Setala H, van der Putten WH, Wall DH (2004) Ecological linkages between aboveground and belowground biota. Science 304:1629–1633. https://doi.org/10.1126/science.1094875
Zak DR, Holmes WE, White DC, Peacock AD, Tilman D (2003) Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84:2042–2050. https://doi.org/10.1890/02-0433
Brussaard L, de Ruiter PC, Brown GG (2007) Soil biodiversity for agricultural sustainability. Agr Ecosyst Environ 121:233–244. https://doi.org/10.1016/j.agee.2006.12.013
Santos MS, Nogueira MA, Hungria M (2019) Microbial inoculants: reviewing the past, discussing the present and previewing an outstanding future for the use of beneficial bacteria in agriculture. AMB Express 9:205. https://doi.org/10.1186/s13568-019-0932-0
Acknowledgements
We acknowledge and would like to thank Duxton Vineyards and Duxton Dried Fruits for providing the study site for this research and for their financial contributions to this research.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. Author ARGM and AJL received research support from Duxton Vineyards and Duxton Dried Fruit as part of an ongoing relationship between The University of Adelaide and Duxton Capital Australia.
Author information
Authors and Affiliations
Contributions
Authors ARGM, AJL, and TC contributed to the study conception and design. Material preparation, data collection, and analysis were performed by ARGM, AJL, and GG. The first draft of the manuscript was written by ARGM and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
ARGM commenced employment with Duxton Capital Australia (financial manager of Duxton Dried Fruits and Duxton Vineyards) in January 2021. Beyond assisting with on-ground soil sampling, Duxton Dried Fruit and Duxton Vineyards had no input in the design, and write-up of this research.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mason, A.R.G., Cavagnaro, T.R., Guerin, G.R. et al. Soil Bacterial Assemblage Across a Production Landscape: Agriculture Increases Diversity While Revegetation Recovers Community Composition. Microb Ecol 85, 1098–1112 (2023). https://doi.org/10.1007/s00248-023-02178-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-023-02178-x