
Abstract
The sources of fungal symbionts of insects are not well understood, yet the acquisition and assembly of fungal communities in mobile insect hosts have important implications for the ecology of migratory insects and their plant hosts. To determine potential sources of fungi associated with the fall armyworm (Spodoptera frugiperda), we characterized the fungal communities associated with four different ecological compartments (insects, infested leaves, uninfested leaves, and soil) and estimated the contributions of each of these potential sources to the insect’s fungal microbiome. Results show that insect fungal community composition was distinct from and more varied than the composition of fungal communities in the environment of those insects (plants and soil). Among the sources evaluated, on average we found a surprisingly large apparent contribution from other congeneric S. frugiperda insect larvae (ca. 25%) compared to the contribution from soil or plant sources (< 5%). However, a large proportion of the insect microbiome could not be attributed to the sampled sources and was instead attributed to unknown sources (ca. 50%). Surprisingly, we found little evidence for exchange of fungal taxa, with the exception of a Fusarium oxysporum and a Cladosporium sp. OTU, between larvae and the infested leaves on which they fed. Together, our results suggest that mobile insects such as S. frugiperda obtain their fungal symbionts from a variety of sources, not limited to plants and soil, but including conspecific insects and other unsampled environmental sources, and that transmission among insects may play an important role in acquisition of fungal symbionts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Unlike sessile plants, animals often move across different environments and exchange microbial symbionts through interactions with other host species and their environment [1]. However, relatively few studies have examined the sources of microbial symbionts of mobile hosts such as insects [2, 3] or determined the degree to which insects exchange fungal symbionts with their environment, such as the plants on which they feed [4,5,6]. The extent to which microbial communities are shared and exchanged across insect hosts, plant hosts, and the soil environment has important implications for transmission of pathogens from pest insects to crops, as well as the efficacy of microbial biocontrol applications for these pests.
Herbivorous insects may acquire their fungal symbionts from a variety of environmental sources (e.g., soil, air, and water) or from interactions with other organisms (e.g., plant hosts). Results from one of the few studies of fungal microbiomes of foliar feeding insects showed that these insects acquire some of their fungal symbionts from the plants on which they feed [7]. However, in other systems, bacteria that typically inhabit the soil are acquired each generation and form symbiotic associations with herbivorous insects even though those insects feed only above ground [8]. Similarly, a recent study demonstrated that a leaf-feeding caterpillar (Mamestra brassicae) obtained a larger portion of its microbiome from soil rather than from the host plant on which it feeds [2].
Foliar feeding insects may also impact the microbial communities in their plant hosts [9]. The damage caused by insect herbivory to host plants creates routes of entry for fungi [10], and insects can also directly transmit fungi to plants [6]. The transmission of microbial symbionts from insects to plants can have negative consequences for plant host productivity through transmission of plant pathogens [6, 11, 12]. Thus, understanding the extent to which insects transmit fungal symbionts to plant hosts is important for maintaining agricultural plant health.
The fall armyworm (Spodoptera frugiperda) is a significant agricultural pest that feeds upon a wide range of crop species but shows a preference for and causes the greatest damage in various grasses, including corn (Zea mays), sorghum (Sorghum bicolor), and rice (Oryza sativa) [13]. Although S. frugiperda lays its eggs on leaf surfaces, where larvae hatch and cause substantial damage to plants by consuming foliage as they mature, pupation occurs in soil prior to emergence as adults each spring [14]. In its native range in North America, S. frugiperda migrates annually from overwintering grounds in south Texas and Florida, moving up to 90 km per week to expand northward during summer months through the midwestern and eastern U.S.A., respectively [15]. Recently, S. frugiperda has invaded and become a widespread and serious pest on several other continents, including India, Africa, and Asia, with devastating impacts on economics and food security [16,17,18]. Estimated costs of up to $13 billion can be attributed to S. frugiperda in Africa alone [19]. Knowledge of the sources of its fungal microbiome will help advance the use of microbes for management of this invasive pest.
To better understand sources from which herbivorous insects such as S. frugiperda acquire their fungi and whether these insects may exchange fungal symbionts with their plant hosts, we characterized fungal symbiont communities associated with S. frugiperda and those of four different ecological compartments in the insects’ immediate environment: S. frugiperda larvae feeding on sorghum, both infested and uninfested leaves of sorghum (Sorghum bicolor), and the soil environment surrounding S. frugiperda-infested plants. Using these datasets, we addressed the following questions. (Q1) How are fungal communities structured across co-occurring insect, plant, and soil ecological compartments? (Q2) What are the relative contributions of the source communities in these different ecological compartments to the fungal communities of insects? (Q3) Does the insect, S. frugiperda, contribute fungal taxa to the communities of the plant host on which it feeds?
Methods
Sample Collection and Processing
Samples were collected from four S. frugiperda-infested sorghum fields in Kansas, U.S.A., in July 2018. The S. frugiperda larvae were identified using the characteristic inverted Y-shaped marking on the head and four raised spots on the eighth abdominal segment [20]. Sampled insects were at the 4 to 6 instar stages. While it is known that the gut microbiome of insects may change during developmental stages or in response to diet [21, 22], these larval stages represent more mature stages that share a similar morphology and have fed on leaves during early development, but not yet pupated [14]. All sampling points were at least 3 m apart, and at each sampling point, we obtained one set of samples from each of four ecological compartments: a single S. frugiperda larva (insect), the leaf on which it was feeding (infested leaf), a leaf from the same plant without indications of S. frugiperda damage (uninfested leaf), and the soil beneath the plant (soil). Soil was sampled from the surface to a depth of 15 cm [23, 24], as close to the base of the plant as possible using a trowel, which was wiped with 70% ethanol between samples. In each field, 6–10 sets were sampled, for a total of 32 sample sets. All samples were stored individually in sterile whirl bags on ice until being processed, up to 24 h after collection.
Insect and plant samples were split in half and surface sterilized by rinsing in water, washed in 75% ethanol for 1 min, washed in 50% commercial bleach for 1 min, washed in 75% ethanol for 1 min, and then rinsed in sterile distilled water for 1 min [20, 25], with half of each sample frozen in liquid nitrogen immediately after returning the samples from the field for NextGen amplicon sequencing and the other half used for culturing. The microbiome of the entire insect was characterized in order to capture both gut microbes and those circulating in hemolymph. Due to time constraints, fungal cultures were attempted for only 10 sets of samples across the four fields. To do so, after surface sterilization, plant leaf samples were sectioned into 20 4 × 4 mm sections and plated onto ½ strength corn meal agar (CMA) [26] in 1.5 mL Eppendorf tubes (mini slants). The corresponding insects from the same 10 sets were sectioned into quarters and plated on ½ strength CMA plates. As fungi emerged from insects, individual colonies were transferred to ½ CMA slants.
DNA Extraction, Amplification, and Sequencing
DNA was extracted from insect samples using a modified hexadecyltrimethylammonium bromide (CTAB) protocol [27, 28]. Frozen insect samples were lyophilized for 48 h and then ground to a fine powder using liquid nitrogen and sterile mortar and pestles. For each sample, homogenized insect tissue (10 mg) was added to 500 µL of a 2% CTAB solution [27] amended with 66 µL 70% sodium dodecyl sulfate (SDS) and 1.74 µL of proteinase K (60 mg/mL), incubated at 65 °C for 3 h [28], and then extracted first with 666 µL and a second time with 333 µL 24:1 chloroform isoamyl alcohol. The purified aqueous fraction was treated with 5 µL RNAse A (10 mg/mL) for two hours at 37 °C. DNA was precipitated with 460 µL of isopropanol incubated at − 20 °C for one hour, centrifuged at 14,000 rpm for 5 min, and the DNA pellet washed with cold 70% ethanol and resuspended in 50 µL 1% TE buffer. Total genomic DNA was extracted from plant tissue with the DNeasy Plant Mini Kit (QIAGEN, Valencia, CA, USA) using a volume of 200 µL of plant tissue ground to a fine powder using liquid nitrogen and sterile mortar and pestles. Individual soil samples were homogenized using a coffee grinder sterilized with 70% ethanol between samples [29] and total DNA extracted from 10 mg of ground soil using the PowerSoil DNA Isolation Kit (QIAGEN, Valencia, CA, USA).
To generate amplicon sequencing data, the concentrations of extracted DNA samples were determined via Qubit fluorometer (Invitrogen, Carlsbad, CA), diluted to ca. 20 ng/µL, and 20 µL per sample submitted to University of Minnesota Genomics Center (UMGC Microbiome Services, St. Paul, MN). The Internal Transcribed Spacer 1 region (ITS1F, ITS2 primers) of the rDNA SSU was amplified using a dual-index, two-step amplification method [30] and sequenced with a paired-end (2 × 250 bp) MiSeq 600 cycle kit in one lane (Illumina, San Diego, CA, USA).
To identify cultured fungi, total DNA was extracted from a small fungal tissue sample using the RED Extract’n’Amp Tissue PCR kit protocol (Sigma-Aldrich, St. Louis, MO, USA). The ca. 700 bp region of the rDNA locus was amplified by PCR using the ITS1F (SSU) and LR3R (LSU) primers [31, 32]. Successful amplification of DNA was confirmed via gel electrophoresis. DNA products were purified using the ExoSap-IT Product Cleanup Reagent protocol (Thermo Fisher Scientific, Waltham, MA, USA), and single-strand ITS1 sequences were obtained with Sanger sequencing using the ITS1F primer (GeneWiz, South Plainfield, NJ, USA).
Bioinformatic Data Processing
ITS1 sequences obtained from MiSeq amplicon sequencing were demultiplexed, and after removing primer sequences, contigs were assembled and trimmed to 225 bp. Trimmed sequences were clustered into operational taxonomic units (OTUs) at 97% sequence identity using the QIIME2 toolkit (https://qiime2.org) [33] at the Minnesota Supercomputing Institute (MSI, Minneapolis, MN, USA). The OTUs were assigned taxonomic identities using a consensus BLAST method in QIIME2 against the full UNITE + INSD 2017 dataset (https://unite.ut.ee/) [34]. OTU table and taxonomy files were exported, and all subsequent analyses were performed in R (https://www.r-project.org/) [35] unless otherwise specified. Mapping files with metadata for each sample were manually compiled in Excel.
DNA sequences obtained from cultures were processed in Geneious v. 5.5.6 [36]. Sequences with multiple overlapping chromatogram peaks were removed and the remaining sequences cleaned by trimming the ITS1F and LR3R primer sequences. Sequences were clustered into OTUs at 97% sequence identity using the QIIME2 toolkit and then assigned taxonomic identities using a consensus BLAST method against the full UNITE + INSD 2017 dataset [34].
Community Richness, Evenness, Composition, and Nestedness
To compare fungal taxa detected by culturing versus amplicon sequencing, all genera detected by culturing were compared to all genera detected by amplicon sequencing using the “get_taxa_unique” function of the “phyloseq” package in R [37]. The distribution and abundance of OTUs detected by both amplicon sequencing and culturing were plotted using the “plot_bar” function in the “phyloseq” R package [37].
The culture-independent amplicon sequencing results were used to characterize the fungal community structure of each ecological compartment (insect, infested and uninfested leaves, and soil). Sequencing depth for samples was compared using the “sample_sums” function in the “phyloseq” R package (https://joey711.github.io/phyloseq/index.html) [37] and visualized using the “rarecurve” function in the “vegan” R package (https://cran.r-project.org/web/packages/vegan/index.html) [38]. Sequence samples were not rarefied to a common number of sequences among samples, but rarefaction curves were constructed to estimate sampling sufficiency. Taxon richness in each fungal community was estimated as the observed number of OTUs using the “estimate_richness” function in the “phyloseq” R package [37]. Pielou’s evenness, which quantifies the skewness of taxon abundances within a community [39], was calculated using the “evenness” function in the “vegan” R package [38]. To assess differences among compartments for observed OTUs per sample and Pielou’s evenness, we performed separate linear mixed effect models using the “nmle” R package (https://cran.r-project.org/web/packages/nlme/index.html) [40] and performed subsequent post hoc Tukey tests in “base R” [35]. The full nlme mixed models included ecological compartment (compartment) as a fixed effect and field as a random effect (diversity ~ compartment, random = ~ 1|field). The significance of the random effect term was assessed by fitting a reduced model with only the fixed effect term (diversity ~ compartment) and then performing an ANOVA using the “aov” function in base R [35] to compare the full and reduced models.
Differences in fungal community composition between different ecological compartments were assessed as abundance-weighted Bray–Curtis (BC) distances [41] using the “distance” function in the “phyloseq” R package with the “Bray” method [37]. The “adonis” function in the “vegan” R package [38] was used to perform a nested permutational multivariate ANOVA (PERMANOVA, BC ~ compartment/field) to determine if there was significant variation in Bray–Curtis distances among fungal communities associated with ecological compartment (compartment) and spatial location (field). The “betadisper” function of the “vegan” R package [38] was used to test for variation in dispersion of fungal communities within compartments and among fields.
The extent to which the less diverse fungal communities (e.g., insects and plants) were subsets of, or nested within, the more diverse fungal communities in soil was estimated using the “nestednodf” function (Nestedness Ordered by Decreasing Fill (NODF)) of the “vegan” R package [38], which returns values between 1 (no nesting) and 100 (perfectly nested) [42]. The significance of the NODF statistic was evaluated using the “oecosimu” function with 99 permutations in R v4.0 [43]. The occurrence of OTUs ordered by marginal totals was visualized using the “ComplexHeatmap” R package (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html) [44]. Samples were ranked by total number of OTUs occurring in that sample (row totals), and average ranks of each ecological compartment were analyzed using a Kruskal–Wallis test [45] implemented with the “kruskal” function in the “agricolae” R package (https://cran.r-project.org/web/packages/agricolae/index.html) [46].
To determine if the occurrence of fungal taxa in insects was predicted by their abundance (as read counts) in soil, for each of the 10 most commonly observed OTUs shared between insects and soil, we conducted a logistic regression of soil abundance and insect occurrence for paired soil and insect samples within replicate sample sets (glm(OTU occurrence in insects ~ OTU abundance in soil), binomial distribution (base R)) [35]. We defined the core insect microbiome as those OTUs present in 50% or more of the sampled insects and showing a relative abundance of at least 0.05% in each sample in which they were detected [47].
FEAST Analyses
To estimate relative contributions of potential sources of symbionts to fungal communities in insect and plant compartments, we used the FEAST package (https://github.com/cozygene/FEAST) [48]. FEAST allows the user to designate microbial communities a priori as sinks or sources and then uses a maximization-estimation algorithm to attribute a percentage of taxa in the sink community to designated source communities based on similar patterns of occurrence and abundance of taxa between source and sink communities. Attribution of unknown sources occur in FEAST when a mismatch of taxon occurrence and abundance between the sink and source communities prevents assignment to a potential source, and thus, the sum of designated source contributions is less than 100%. The FEAST software currently does not allow identifying the specific taxa contributing to the unknown category or the designated sources.
Two types of FEAST analyses were performed: (1) a “leave-one-out” analysis to estimate average contribution from each potential source to the designated sink and (2) a “within-set” analysis, to estimate source contributions from the local environment associated with each sample set. For the “leave-one-out” analysis, all the sampled communities from the same ecological compartment were pooled as source communities. To consider the insect communities as sources for each individual insect sink, the target individual insect sink was left out of the pooled insect compartment source. For the “within-set” analysis, each sampled insect was designated as a sink, and the individual fungal communities in the infested leaf, uninfested leaf, and soil compartments within the same sample set were designated as potential sources. We also estimated the extent to which insects might contribute fungal symbionts to infested leaves by performing another “within-set” FEAST analyses treating each infested leaf as a sink and the uninfested leaf, insect, and soil compartments within the same sample set as potential sources.
Results
From a total of 128 samples (4 ecological compartments per set, 32 sample sets across four fields), amplicon sequencing results were excluded for five sets due to poor sequence recovery for the insect samples (< 100 reads). For the remaining 27 sets, we recovered 6,769,278 reads, of which 4,072,065 passed quality control filters. On average, soil samples yielded the most reads (41,997 (SD ± 12,168)), followed by uninfested leaf samples (34,363 (SD ± 8135)), infested leaf samples (32,779 (SD ± 10,824)), and the insect samples (9117 (SD ± 12,340)). Sequences were binned into 3007 OTUs with 97% similarity of the ITS1 region using QIIME2. Rarefaction curves for fungal communities in each ecological compartment leveled off, indicating sampling depth was adequate (Fig. S1). Almost all cultured fungal genera were also detected by amplicon sequencing (Fig. S2). Statistical evaluation of cultured fungal communities was not conducted because the average number of cultures obtained per sample was low (2.2 per insect, 4.6 per infested leaf, and 6.1 per uninfested leaf).
Fungal Community Structure and Composition in Different Ecological Compartments
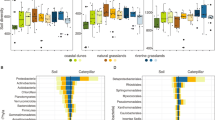
We compared fungal community richness (observed OTUs per sample), evenness (Pielou’s Evenness), and composition (Bray–Curtis distance) in the different ecological compartments. Variation in observed OTUs per sample was significantly affected by ecological compartment (p < 0.001, LME (observed ~ compartment, random = ~ 1|field)) and the spatial factor of field (p = 0.02) (Table 1). Subsequent post hoc Tukey pairwise tests revealed that soil fungal communities had significantly more (p < 0.001) observed OTUs per sample than did communities in other ecological compartments (Fig. 1a). Results for Pielou’s evenness (LME (evenness ~ compartment, random = ~ 1|field)) showed that ecological compartment, but not field, contributed significantly to variation in evenness of fungal communities (p < 0.001; Table 1). Subsequent post hoc Tukey tests showed that fungal communities in infested and uninfested leaves showed significantly lower evenness than fungal communities in soil (p = 4.96 × 10−14) and insects (p = 4.96 × 10−14) (Fig. 1b), likely due to the high abundance of one taxon, Cladosporium sp. 1 (OTU 2348) in both infested and uninfested leaves (Fig. S2a). The variation among insect fungal communities for Pielou’s evenness tended to be larger than for other ecological compartments (Fig. 1b). Consistent with high variation in evenness observed in insect communities, we found only five OTUs that might be considered core taxa, those present in 50% of more of insects sampled (prevalence) and with greater than 0.05% abundance per insect in which it occurred [45] (Table S1). Of these, only three taxa, Alternaria alternata (OTU 1549), Cladosporium chasmanthicola (OTU 1851), and Fusarium cuneirostrum (OTU 2481), showed an average relative abundance per insect greater than 10%.

Richness and evenness of fungal communities in different ecological compartments. (a) Box and whisker plot of the number of observed OTUs per sample in fungal communities of soil, insect, infested leaf, and uninfested leaf compartments. The number of observed OTUs per sample differed significantly among ecological compartments (LME, p < 0.001; Table 1). Post hoc Tukey pairwise analyses found soil fungal communities had greater numbers of observed OTUs per sample than the fungal communities of insects, infested leaves, and uninfested leaves (letters above boxplots). (b) Box and whisker plot of Pielou’s evenness of fungal communities in soil, insect, infested leaf, and uninfested leaf samples. Fungal community evenness differed significantly among ecological compartments (LME, p < 0.001; Table 1). Post hoc Tukey pairwise analyses found that evenness of fungal communities in infested and uninfested leaf samples was similar to each other but significantly lower (p < 0.001) than evenness in insect and soil fungal communities (letters above boxplots)
Analyses of community composition showed significant differences in Bray–Curtis distances among fungal communities in insect, soil, and plant compartments (p < 0.001, PERMANOVA (BC ~ compartment/field)), but little difference in Bray–Curtis distances among communities of infested and uninfested leaves (Table 2). Variation across fields also contributed significantly to variation in Bray–Curtis distances (p < 0.001; Table 2). The results of a Principal Coordinates Analysis (PCoA) of Bray–Curtis distances also showed that fungal communities from insects, plants, and soil were distinct, while those within plants (infested versus uninfested leaves) were overlapping and highly similar (Fig. 2).

PCoA of Bray–Curtis distances among fungal communities of different ecological compartments. Community composition in infested and uninfested plant leaves largely overlapped, whereas PERMANOVA of Bray–Curtis distances found significant differences in composition of fungal communities among ecological compartment (p < 0.001), likely due to the differences among plant, insect, and soil samples (Table 2)
As we observed that OTU (species) richness was greatest in soil communities, followed by those in insects, and finally those in plants, we used a nestedness analysis [42, 43] to evaluate the extent to which communities occupying living hosts (insects and plants) might be subsets of those residing in the soil. Results showed that the mean value of the nestedness statistic, NODF, was small but significantly different than 1 (NODF = 15.45 on scale of 1 to 100, p < 0.01), suggesting a low level of nestedness (Fig. 3). However, consistent with the Bray–Curtis results above, the columns (NODFcolumns, number of different taxa) significantly contributed to NODF (p < 0.01), whereas rows (NODFrows, total numbers of OTUs/individual sample) did not, suggesting that compositional turn-over, more than nestedness, contributes most to differences among these communities (Fig. 3) [49]. Consistent with the results for OTU richness, there were significant differences in rank order of richness (row totals of OTUs/individual sample) in samples representing the different ecological compartments (Kruskal Wallis test; Table S2). Soil fungal communities ranked as the most diverse, followed by fungal communities in insects, and then fungal communities in infested and uninfested leaves. Together, the results of the richness, Bray–Curtis, and nestedness analyses show that the less diverse fungal communities in insects were distinct from, but not strongly nested within, the taxa present in soil.

Nestedness of fungal communities from different ecological compartments. Columns represent the occurrence of individual OTUs (taxa) in each sample and are ordered by total occurrence across samples (column totals) from greatest to least (left to right). Rows represent the occurrence of observed OTUs in individual samples from different ecological compartments and are ordered by the total number of observed OTUs (row totals) from greatest to least (top to bottom). The ecological compartment from which samples were obtained is shown in the column to the right (blue = soil, red = insect, orange = infested leaf, green = uninfested leaf). The overall Nestedness Ordered by Decreasing Fill (NODF) statistic was small but significant (NODF = 15.45, p = 0.01). NODFcolumns (OTU occurrence in samples) contributed significantly to nestedness (p = 0.01), but NODFrows (numbers of OTUs in samples) did not (p = 0.39)
Nonetheless, we found that ca. 50% of fungal taxa were shared between soil and insect ecological compartments. We performed logistic regressions with the expectation that if insects obtain these taxa directly from soil, then taxa that are more abundant in soil samples should also occur more frequently in the associated insect samples. We did not find significant correlations for any taxon, suggesting that insects do not directly acquire many fungal symbionts from the soil (Table S3).
Estimated Source Contributions to Fungal Communities in Insects
We used the FEAST statistical package to estimate the relative contributions of fungal communities in each ecological compartment (designated as sources) to the fungal communities in insects (designated as sinks). We first conducted a “leave-one-out” analysis in which all samples for each potential source compartment were pooled, but the insect sample pool left out the individual insect sink under analysis. The results of the “leave-one-out” analyses showed that on average, FEAST attributed the pooled insect source communities as contributing a larger fraction of symbionts (30% SD ± 31.6) to the individual insect sinks, compared to the other designated source communities (soil, 3.1% SD ± 9.1; infested leaves, 6.3% SD ± 17; and uninfested leaves, 6.5% SD ± 15) (Fig. 4a). Interestingly, over half of the individual insect sink communities could not be attributed to one of the designated sources and were instead attributed to unknown sources (54% SD ± 32.6; Fig. 4a). There was substantial variation among individual insect sinks in the fraction of the fungal community attributed to each source (Fig. 4b). For example, fungal communities in four individual insects (f2040, f2055, f2130b, and f2131; Fig. 4b) were attributed almost completely to other insect sources, the fungal communities in two individual insects (f2139, f 2140; Fig. 4b) were mostly attributed to infested or uninfested leaf sources, and those in eleven of the individual insect sinks were primarily attributed to unknown sources (f2041, f2050, f2051, f2058, f2060, f2114T, f2116, f2118, f2119T, f2130, and f2132b; Fig. 4b).

Results of “leave-one-out” FEAST analyses. To estimate the relative contribution from potential source communities (ecological compartments) to fungal communities in insects, fungal communities in individual insects were designated as sinks and the fungal communities in soil, infested leaf, uninfested leaf, and remaining insects were pooled by ecological compartment and designated as sources. (a) Average source contributions to fungal communities in insects. Of the designated sources, other insects were attributed as a source for 30% (SD ± 31.6) of the fungal communities in insects, while soil (3.1% SD ± 9.1) and plant (infested 6.3% SD ± 17; uninfested 6.5% SD ± 15) sources contributed a much lower percentage to fungal communities in insects. A large proportion (54% SD ± 32.6) of the fungal communities in insects were attributed to unknown sources. (b) Estimated source contributions to fungal communities in individual insect samples. Each bar on the x-axis represents the fungal community in an individual insect, with the estimated relative contributions of designated pooled or unknown sources shown on the y-axis. Source contributions estimated by “leave-one-out” FEAST analyses were highly variable among individual insect fungal communities
To ask whether the large amount of variation in source contributions among individual insects that we note above might be due to variation across local environmental sources, we also performed FEAST analyses within each sample set at each sampling location, assigning the individual insect community as the sink and communities from other compartments within the same sample set as sources. Results of this “within-set” FEAST analyses showed that, on average, only small fractions of the fungal communities in insect sinks could be attributed to the fungal communities of the infested leaf (9.3% SD ± 20.2), uninfested leaf (9.5% SD ± 20.9), or soil compartment (10.3% SD ± 16.4) in the same set and sampling location, and most were instead attributed to unknown sources (70.8% SD ± 28.3; Fig. 5a). The unknown contribution estimated in the “within-set” FEAST analyses was likely inflated relative to the “leave-one-out” analyses because insects could not be included as a source. Surprisingly, fungal communities in infested leaves were not attributed as a source more frequently than were uninfested leaves even though the sampled insect was in contact with and consuming the infested leaf (Fig. 5a). Again, individual insect sinks exhibited substantial variation in source contributions, suggesting acquisition of symbionts from different sources encountered by these insects in their environment (Fig. 5c).

Results of “within-set” FEAST analyses. To estimate contributions from environmental sources in the immediate proximity of insects or to the infested leaf on which the insect fed, each individual insect or infested leaf was designated as a sink and the remaining ecological compartments within the same sample set (soil, infested leaf, and uninfested leaf for insects; insects, uninfested leaf, and soil for infested leaves) were designated as sources. (a) Average source contributions to fungal communities in insect samples. The “within-set” FEAST analyses attributed similar percent contributions from sources of soil (10.3%, SD ± 16.4), infested leaf (9.3%, SD ± 20.2), and uninfested leaf (9.5%, SD ± 20.9) to fungal communities in insects. A larger proportion were attributed to unknown sources (70.8%, SD ± 28.3) than in the results of “leave-one-out” analyses. (b) Average source contribution to fungal communities of infested leaf tissues. On average, a majority of fungal communities of infested leaves (86.3%, SD ± 24.4) were attributed to uninfested leaves, likely due to high similarity between these communities. In contrast, insect sources were rarely attributed (5%, SD ± 18.2) to the fungal communities of infested leaves. (c) Estimated source contributions to fungal communities of individual insects. Each bar on the x-axis represents a fungal community in an individual insect, with the estimated relative contributions of designated sources or unknown sources along the y-axis. Source contributions estimated by “within-set” FEAST analyses were variable among individual insect communities but dominated by unknown sources with negligible contribution attributed to infested leaves. (d) Estimated source contributions to individual fungal communities in infested leaf samples. Each bar on the x-axis represents a fungal community in an individual infested leaf sample with the estimated relative contributions of designated “within-set” and unknown sources along the y-axis. Insect sources were rarely attributed to the communities of infested leaf samples, consistent with the results above, and with the “leave-one-out” analysis
Estimated Contribution of Insect Sources to Fungal Communities in Infested Leaves
To assess the extent to which insects may contribute to the fungal communities of the infested leaves they reside on, we also performed “within-set” FEAST analyses using fungal communities of infested leaves as sinks and communities from other compartments within the same set as sources. Insect sources were not commonly attributed as a source for fungal communities in infested leaves (4.6% SD ± 18.2, Fig. 5b). Instead, most fungal communities of infested leaves were attributed to the uninfested leaf source (86.3% SD ± 24.4), which is likely due to the similarity of fungal communities in infested and uninfested leaves (Figs. 5b, d).
To identify specific taxa that might have been exchanged between insects and plants, we identified individual OTUs observed in both the insect and the paired infested leaf, but not in the uninfested leaf within the same sample set. We found only 15 cases fitting these criteria (Table S4). In 6 of these 15 cases, a Fusarium oxysporum OTU (OTU_671) was shared, and in 3 cases, Cladosporium sp. 2 (OTU_701) was shared. Interestingly, these two taxa were often found in insects (Table S1), suggesting that they are common in the environment of both the insects and the plants. In each of the remaining 6 cases, a different OTU was represented, suggesting that these were uncommon taxa not well sampled by our methods. Overall, these and the FEAST results above suggest that aside from these Fusarium and Cladosporium OTUs, insects exchange very few fungal symbionts with leaves on which they are feeding.
Discussion
We describe the fungal communities in four different ecological compartments (insects, infested leaf, uninfested leaf, and soil) and estimate the exchange of fungal symbionts among insects and these other compartments. We report three key findings. First, fungal community composition differed among soil, leaf, and insect samples, while the composition of infested and uninfested leaves of the same plant was very similar. Fungal communities in insects and plants harbored far fewer taxa than did soil communities but were not strongly nested within the richer soil communities. Second, the results of a “leave-one-out” FEAST analyses showed that other S. frugiperda insects were attributed as sources of the fungal communities in individual insects more commonly than the soil and plants sources. Along with our observation of considerable variation among individual insect sink communities in the sources attributed by FEAST, including substantial contributions attributed to unknown sources, our results suggest that S. frugiperda acquires fungi from a variety of environment sources, not only from plants and soil, but also from other S. frugiperda insects and environmental sources not sampled. Third, while few fungal taxa are apparently exchanged between the insect and the plant on which the insect feeds, Fusarium oxysporum and Cladosporium sp.2 OTUs apparently are and could be important to plant health. Our results provide insights into the sources that contribute most strongly to shaping the composition of fungal microbiomes in herbivorous insects and in the host plants on which they feed.
Ecological compartments of soil, plants, and insects harbored distinct communities that differed in diversity and evenness of fungal taxa. Compared to the fungal communities in plants and insects, those in soil showed the greatest OTU richness and were affected by spatial location (field), a pattern observed in other studies of microbial communities across different ecological compartments [29, 50]. Fungal communities in infested and uninfested S. bicolor leaves exhibited lower OTU richness and evenness than did those in other compartments, likely because these communities were dominated by a single Cladosporium taxon (OTU 2348). Previous characterization of endophyte communities of S. bicolor leaves also found similarly low diversity due to an abundance of Cladosporium taxa [51]. Fungal communities of insects showed intermediate richness and tended to have greater variation in evenness among individuals, compared to fungal communities in plants and soil. High variation among microbial communities of different individual insects is common among other lepidopteran microbial symbiont communities [3, 25, 52, 53], leading some to hypothesize these communities are more transient, strongly affected by environmental sources with which they interact, and composed of few “core” taxa [25]. However, recent research has identified a core bacterial microbiome in the gut of S. frugiperda across different plant hosts and locations in South America [47] and we identified five taxa shared across a majority of individual insects sampled (Table S1). While these taxa may be candidates for more stable symbiont associations with S. frugiperda, distinguishing between transient and stable symbionts is not possible from next-generation sequencing data sampled at a single time point and will require further experimental studies.
In contrast to previous studies, our results did not suggest that these insects acquire a majority of their symbionts directly from the soil or plant environment. Not only was there little correlation between the abundance of the most common taxa in soil and their occurrence in associated insects (Table S3), but there were relatively few shared taxa between plants and insects (Table S4). Similarly, the FEAST analyses did not attribute a large proportion of the microbiome to soil or plant sources, but instead to other insect and unknown sources (Figs. 4, 5). The large attribution to unknown sources could be due to sources we did not sample, for example, the epiphytic fungal communities on the surface of leaves [54] or fungi carried in the air, or could result from the inability of FEAST to match the composition (identity and abundance of taxa) of communities in an individual insect to that of the pooled, designated sources [48]. Such mismatches could occur if insects acquire fungal taxa from designated sources but subsequently filter those taxa, resulting in different abundances of these taxa in insects. Plant hosts are known to filter fungi from the soil environment, such that only a subset of taxa encountered in soil are able to survive, grow, and compete successfully in the interior of the root, resulting in a highly similar root microbial communities [29, 55]. In contrast, we observed the microbial communities in these insects to be highly variable with few taxa shared among individual insects. Additionally, insect symbiont communities were not strongly nested within those of soil or plants, indicating instead high turn-over between ecological compartments [49]. Overall, our data suggest that mobile insect hosts such as S. frugiperda encounter and acquire symbionts from a greater number of sources they may interact with in their environment, than do their non-mobile plant hosts.
Results from the “leave-one-out” FEAST analyses indicate that S. frugiperda may acquire a substantial fraction of its fungal symbionts from other conspecific insects rather than from environmental sources such as soil [2] or plant hosts [7]. We infer that the strong signal of insect sources in the FEAST analysis is not primarily due to vertical transmission, which should lead to more homogenous and less diverse communities than we observe here [56]. Although our understanding of vertical transmission from parent to offspring among insects has been primarily derived from studies of bacterial symbionts [57,58,59,60,61], vertical transmission of fungal symbionts in insects does occur [56, 62,63,64]. Transovarial transmission was recently demonstrated as a mechanism for Ophiocordyceps fungal symbionts of scale insects [63], as well as for the microsporidian pathogen Nosema bombycis in the related tobacco cutworm pest Spodoptera litura [65]. Although S. frugiperda may harbor a few taxa that are vertically transmitted, perhaps those we identify as core, and shared across a majority of insects sampled (Table S1), we infer that the strong source contribution of conspecific insects found by FEAST is more likely due to horizontal transmission, the acquisition of microbial symbionts from other insects or from shared environmental sources. Horizontal transmission is predicted to result in more variable and diverse microbial communities, with fewer taxa held in common among individuals [66], as we observe here. Close interactions between larvae inhabiting the same infested leaf, and in particular, cannibalism, could constitute mechanisms of horizontal transmission. Cannibalism is a documented behavior of S. frugiperda larvae both in the laboratory and the field and is known to transmit nuclear polyhedrosis virus among S. frugiperda larvae [67]. Further research using direct experimentation is needed to determine mechanisms by which this insect might acquire its fungal symbionts from other insects. Given the large attribution of unknown sources by FEAST to the fungal microbiome of S. frugiperda, as well as the high variability in both community evenness and source attributions by FEAST among individual insects, our results are best interpreted as showing that S. frugiperda acquires most of its fungal symbionts from a diversity of spatially varying sources, possibly including some that we did not sample.
In contrast to other studies demonstrating that phytophagous insects are competent vectors of fungal symbionts to plant hosts [4,5,6], we found surprisingly little evidence of exchange of fungal symbionts between the fall armyworm and infested sorghum plants. Additionally, we did not find fungal communities of uninfested plants to be nested within fungal communities of infested plants, as has been observed in other studies [9]. The only examples of potential exchange among insect and plant compartments included that of a Fusarium oxysporum taxon (OTU_617) and a Cladosporium sp. 2 taxon (OTU_701) found in both insects and their associated infested leaves, but not in the fungal communities of uninfested leaves of the same plant. Members of the F. oxysporum species complex are known to infect multiple hosts and have been detected as both endophytes and pathogens of plants [68], as well as pathogens of insects and animals [69, 70]. Additionally, sciarid (Bradysia spp., Lycoriella spp., and Sciara spp.) and shore flies (Scatella spp.) were previously shown to vector plant pathogenic strains of F. oxysporum among cucumber plants in a greenhouse study [71]. Cladosporium taxa are also documented as common endophytes of plants [72] and have also been found in the human gut [73] and as both beneficial symbionts and pathogens of insects [74, 75]. Consequently, while our results show that insect infestation does not substantially affect the fungal community structure of sorghum leaves and these insects likely do not gain many taxa from plants, the exchanges of Fusarium and Cladosporium taxa could affect plant and insect health.
We conclude that there are distinct fungal communities in S. frugiperda, its plant host S. bicolor, and the surrounding soil environment. Results suggest S. frugiperda likely acquires symbionts from a variety of sources in their environment, including a substantial contribution from conspecific insects, rather than primarily from the plants on which they feed or the soil. We found that the community structure of fungal symbionts of these migratory insects varied greatly from insect to insect, compared to variation across individuals of the sessile plant host. Surprisingly, there was relatively little contribution to the fungal microbiome of S. frugiperda from the plant hosts on which they fed and little evidence for transmission of fungal symbionts between the insect to its plant host. These results increase our understanding of the varied environmental sources of fungal taxa for the microbiome of this important insect pest and illustrate potential processes by which fungal microbiomes are structured across distinct, interacting host species.
Data Availability
Raw sequencing data will be deposited into SRA prior to publication and representative OTU sequences from cultured isolates will be deposited into GenBank. Metadata are to be deposited into SRA with the raw sequence data.
References
Robinson CD, Bohannan BJM, Britton RA (2019) Scales of persistence: transmission and the microbiome. Curr Opin Microbiol 50:42–49. https://doi.org/10.1016/j.mib.2019.09.009
Hannula SE, Zhu F, Heinen R, Bezemer TM (2019) Foliar-feeding insects acquire microbiomes from the soil rather than the host plant. Nat Commun 10:1254. https://doi.org/10.1038/s41467-019-09284-w
Jones AG, Mason CJ, Felton GW, Hoover K (2019) Host plant and population source drive diversity of microbial gut communities in two polyphagous insects. Sci Rep 9:11. https://doi.org/10.1038/s41598-019-39163-9
Kluth S, Kruess A, Tscharntke T (2002) Insects as vectors of plant pathogens: mutualistic and antagonistic interactions. Oecologia 133:193–199. https://doi.org/10.1007/s00442-002-1016-3
Mitchell PL (2004) Heteroptera as vectors of plant pathogens. Neotrop Entomol 33:519–545. https://doi.org/10.1590/s1519-566x2004000500001
Devarajan PT, Suryanarayanan TS (2006) Evidence for the role of phytophagous insects in dispersal of non-grass fungal endophytes. Fungal Divers 23:111–119
Harrison JG, Urruty DM, Forister ML (2016) An exploration of the fungal assemblage in each life history stage of the butterfly, Lycaeides melissa (Lycaenidae), as well as its host plant Astragalus canadensis (Fabaceae). Fungal Ecol 22:10–16. https://doi.org/10.1016/j.funeco.2016.02.001
Kikuchi Y, Hayatsu M, Hosokawa T, Nagayama A, Tago K, Fukatsu T (2012) Symbiont-mediated insecticide resistance. Proc Natl Acad Sci U S A 109:8618–8622. https://doi.org/10.1073/pnas.1200231109
David AS, Quiram GL, Sirota JI, Seabloom EW (2016) Quantifying the associations between fungal endophytes and biocontrol-induced herbivory of invasive purple loosestrife (Lythrum salicaria L.). Mycologia 108:625–637. https://doi.org/10.3852/15-207
Daleo P, Silliman BR, Alberti J, Escapa M, Canepuccia A, Pena N, Iribarne O (2009) Grazer facilitation of fungal infection and the control of plant growth in south-western Atlantic salt marshes. J Ecol 97:781–787. https://doi.org/10.1111/j.1365-2745.2009.01508.x
Mei CaF BS (2010) The use of beneficial microbial endophytes for plant biomass and stress tolerance improvement. Recent Pat Biotechnol 4:81–95
Frago E, Dicke M, Godfray HCJ (2012) Insect symbionts as hidden players in insect-plant interactions. Trends Ecol Evol 27:705–711. https://doi.org/10.1016/j.tree.2012.08.013
Montezano DG, Specht A, Sosa-Gomez DR, Roque-Specht VF, Sousa-Silva JC, Paula-Moraes SV, Peterson JA, Hunt TE (2018) Host plants of Spodoptera frugiperda (Lepidoptera: Noctuidae) in the Americas. Afr Entomol 26:286–300. https://doi.org/10.4001/003.026.0286
Sparks AN (1979) A review of the biology of the fall armyworm. Florida Entomologist 62:82–87
Nagoshi RN, Meagher RL, Hay-Roe M (2012) Inferring the annual migration patterns of fall armyworm (Lepidoptera: Noctuidae) in the United States from mitochondrial haplotypes. Ecol Evol 2:1458–1467. https://doi.org/10.1002/ece3.268
Suby SB, Soujanya PL, Yadava P, Patil J, Subaharan K, Prasad GS, Babu KS, Jat SL, Yathish KR, Vadassery J, Kalia VK, Bakthavatsalam N, Shekhar JC, Rakshit S (2020) Invasion of fall armyworm (Spodopterafrugiperda) in India nature, distribution, management and potential impact. Curr Sci 119:44–51. https://doi.org/10.18520/cs/v119/i1/44-51
Goergen G, Kumar PL, Sankung SB, Togola A, Tamo M (2016) First report of outbreaks of the fall armyworm Spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidae), a new alien invasive pest in west and central Africa. PLoS One 11:9. https://doi.org/10.1371/journal.pone.0165632
Jing W, Huang C, Li CY, Zhou HX, Ren YL, Li ZY, Xing LS, Zhang B, Qiao X, Liu B, Liu CH, Xi Y, Liu WX, Wang WK, Qian WQ, McKirdy S, Wan FH (2021) Biology, invasion, and management of the agricultural invader: Fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae). J Integr Agric 20:646–663. https://doi.org/10.1016/s2095-3119(20)63367-6
Day R, Bateman M, Beale T, Clottey V, Cock M, Colmenarez Y, Corniani N, Early R, Godwin J, Gomez J, Gonzalez MP, Murphy ST, Oppong-Mensah B, Phiri N, Pratt C, Silvestri S, Witt A (2017) Fall armyworm: implications and impacts for Africa. Outlooks Pest Manag 28:196–201. https://doi.org/10.1564/v28_oct_02
Arnold AE, Maynard Z, Gilbert GS (2001) Fungal endophytes in dicotyledonous neotropical trees: patterns of abundance and diversity. Mycol Res 105:1502–1507. https://doi.org/10.1017/s0953756201004956
Hammer TJ, Moran NA (2019) Links between metamorphosis and symbiosis in holometabolous insects. Philos Trans R Soc B-Biol Sci 374:10. https://doi.org/10.1098/rstb.2019.0068
Shukla SP, Sanders JG, Byrne MJ, Pierce NE (2016) Gut microbiota of dung beetles correspond to dietary specializations of adults and larvae. Mol Ecol 25:6092–6106. https://doi.org/10.1111/mec.13901
Hameed A, Asrar M, Hidayatullah AA (2016) Isolation of fungi from various agricultural fields as a function of soil depth and seasonal variability. Pak J Biotechnol 13:39–48
Luginbill P (1928) The fall army worm. USDA Technical Bulletin 34. U.S. Government Printing Office, United States Department Agriculture, Washington, D.C., pp 92
Hammer TJ, Janzen DH, Hallwachs W, Jaffe SP, Fierer N (2017) Caterpillars lack a resident gut microbiome. Proc Natl Acad Sci U S A 114:9641–9646. https://doi.org/10.1073/pnas.1707186114
Conant NF, Smith DT, Baker RD, Callaway JL (1971) Manual of clinical mycology. W.B. Saunders Co., Philadelphia, PA
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh plant tissue. Phytochem Bull 19:11–15
Detinger BTM, McLaughlin DJ (2006) Reconstructing the Clavariaceae using nuclear large subunit rDNA sequences, and a new genus segregated from Clavaria. Mycologia 98:746–762
Strom N, Hu WM, Haarith D, Chen SY, Bushley K (2020) Interactions between soil properties, fungal communities, the soybean cyst nematode, and crop yield under continuous corn and soybean monoculture. Appl Soil Ecol 147:14. https://doi.org/10.1016/j.apsoil.2019.103388
Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R, Knights D, Beckman KB (2016) Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol 34:942–949. https://doi.org/10.1038/nbt.3601
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. https://doi.org/10.1111/j.1365-294X.1993.tb00005.x
Hopple JS, Vilgalys R (1994) Phylogenetic relationships among coprinoid taxa and allies based on data from restriction site mapping of nuclear rDNA. Mycologia 86:96–107. https://doi.org/10.2307/3760723
Bolyen E, Rideout JR, Dillon MR, Bokulich N, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo JR, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang LJ, Kaehler BD, Bin Kang K, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, vander Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan YH, Wang MX, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang YL, Zhu QY, Knight R, Caporaso JG, (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
UNITE Community (2019) Full UNITE+INSD dataset for Fungi. Version 18.11.2018. UNITE Community. https://doi.org/10.15156/BIO/786347
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:11. https://doi.org/10.1371/journal.pone.0061217
Oksanen J, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens H, Szoecs E, Wagner H (2019) vegan: Community Ecology Package. R package version 2.5–6. https://cran.r-project.org/web/packages/vegan/index.html.
Pielou EC (1966) The measurement of diversity in different types of biological collections. J Theor Biol 13:131–144. https://doi.org/10.1016/0022-5193(66)90013-0
Pinhiero J, Bates D, DebRoy S, Sarkar D (2012) nmle: linear and nonlinear mixed effects models. R package version 3.1–152. https://CRAN.R-project.org/package=nmle
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27:326–349. https://doi.org/10.2307/1942268
Almeida-Neto M, Guimaraes P, Guimaraes PR, Loyola RD, Ulrich W (2008) A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos 117:1227–1239. https://doi.org/10.1111/j.0030-1299.2008.16644.x
Miklos I, Podani J (2004) Randomization of presence-absence matrices: comments and new algorithms. Ecology 85:86–92. https://doi.org/10.1890/03-0101
Gu ZG, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma 32:2847–2849. https://doi.org/10.1093/bioinformatics/btw313
Kruskal WH, Wallis WA (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47:583–621. https://doi.org/10.1080/01621459.1952.10483441
De Mendiburu F (2010) Agricolae: statistical procedures for agricultural research. R package version 1. https://CRAN.R-project.org/package=agricolae
Oliveira NC, Rodrigues PAP, Consoli FL (2022) Host-adapted strains of Spodoptera frugiperda hold and share a core microbial community across the western hemisphere. Microb Ecol. https://doi.org/10.1007/s00248-022-02008-6
Shenhav L, Thompson M, Joseph TA, Briscoe L, Furman O, Bogumil D, Mizrahi I, Pe’er I, Halperin E (2019) FEAST: fast expectation-maximization for microbial source tracking. Nat Methods 16:627–632. https://doi.org/10.1038/s41592-019-0431-x
Ulrich W, Almeida-Neto M (2012) On the meanings of nestedness: back to the basics. Ecography 35:865–871. https://doi.org/10.1111/j.1600-0587.2012.07671.x
Amend AS, Cobian GM, Laruson AJ, Remple K, Tucker SJ, Poff KE, Antaky C, Boraks A, Jones CA, Kuehu D, Lensing BR, Pejhanmehr M, Richardson DT, Riley PP (2019) Phytobiomes are compositionally nested from the ground up. PeerJ 7:18. https://doi.org/10.7717/peerj.6609
Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Singan V, Vogel J, Hutmacher RB, Dahlberg JA, Coleman-Derr D, Lemaux PG, Taylor JW (2020) Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat Commun 11:14. https://doi.org/10.1038/s41467-019-13913-9
Priya NG, Ojha A, Kajla MK, Raj A, Rajagopal R (2012) Host plant induced variation in gut bacteria of Helicoverpaarmigera. PLoS One 7:10. https://doi.org/10.1371/journal.pone.0030768
Staudacher H, Kaltenpoth M, Breeuwer JAJ, Menken SBJ, Heckel DG, Groot AT (2016) Variability of bacterial communities in the moth Heliothis virescens indicates transient association with the host. PLoS One 11:21. https://doi.org/10.1371/journal.pone.0154514
Xiong C, He JZ, Singh BK, Zhu YG, Wang JT, Li PP, Zhang QB, Han LL, Shen JP, Ge AH, Wu CF, Zhang LM (2021) Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ Microbiol 23:1907–1924. https://doi.org/10.1111/1462-2920.15262
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90. https://doi.org/10.1038/nature11237
van de Peppel LJJ, Aanen DK, Biedermann PHW (2018) Low intraspecific genetic diversity indicates asexuality and vertical transmission in the fungal cultivars of ambrosia beetles. Fungal Ecol 32:57–64. https://doi.org/10.1016/j.funeco.2017.11.010
Douglas AE (1998) Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol 43:17–37. https://doi.org/10.1146/annurev.ento.43.1.17
Engel P, Moran NA (2013) The gut microbiota of insects - diversity in structure and function. Fems Microbiol Rev 37:699–735. https://doi.org/10.1111/1574-6976.12025
Salem H, Florez L, Gerardo N, Kaltenpoth M (2015) An out-of-body experience: the extracellular dimension for the transmission of mutualistic bacteria in insects. Proc R Soc B-Biol Sci 282:10. https://doi.org/10.1098/rspb.2014.2957
Moran NA, Ochman H, Hammer TJ (2019) Evolutionary and ecological consequences of gut microbial communities. In: Futuyma, DJ (ed.) Annual Review of Ecology, Evolution, and Systematics, Vol 50. Annual Reviews, Palo Alto, pp. 451–475
Yeoman CJ, Brutscher LM, Esen OC, Ibaoglu F, Fowler C, Eren AM, Wanner K, Weaver DK (2019) Genome-resolved insights into a novel Spiroplasma symbiont of the Wheat Stem Sawfly (Cephus cinctus). PeerJ 7:23. https://doi.org/10.7717/peerj.7548
Cheng DJ, Hou RF (2001) Histological observations on transovarial transmission of a yeast-like symbiote in Nilaparvata lugens Stal (Homoptera, Delphacidae). Tissue Cell 33:273–279. https://doi.org/10.1054/tice.2001.0173
Szklarzewicz T, Michalik K, Grzywacz B, Kalandyk-Kolodziejczyk M, Michalik A (2021) Fungal associates of soft scale insects (Coccomorpha: Coccidae). Cells 10:13. https://doi.org/10.3390/cells10081922
Matsuura Y, Moriyama M, Lukasik P, Vanderpool D, Tanahashi M, Meng XY, McCutcheon JP, Fukatsu T (2018) Recurrent symbiont recruitment from fungal parasites in cicadas. Proc Natl Acad Sci U S A 115:E5970–E5979. https://doi.org/10.1073/pnas.1803245115
Pei BY, Wang CX, Yu B, Xia D, Li T, Zhou ZY (2021) The first report on the transovarial transmission of microsporidian Nosema bombycis in lepidopteran crop pests Spodoptera litura and Helicoverpa armigera. Microorganisms 9:11. https://doi.org/10.3390/microorganisms9071442
Bruijning M, Henry LP, Forsberg SKG, Metcalf CJE, Ayroles JF (2022) Natural selection for imprecise vertical transmission in host-microbiota systems. Nat Ecol Evol 6:77–87. https://doi.org/10.1038/s41559-021-01593-y
Chapman JW, Williams T, Escribano A, Caballero P, Cave RD, Goulson D (1999) Fitness consequences of cannibalism in the fall armyworm, Spodoptera frugiperda. Behav Ecol 10:298–303. https://doi.org/10.1093/beheco/10.3.298
de Lamo FJ, Takken FLW (2020) Biocontrol by Fusarium oxysporum using endophyte-mediated resistance. Front Plant Sci 11:15. https://doi.org/10.3389/fpls.2020.00037
O’Donnell K, Sutton DA, Rinaldi MG, Magnon KC, Cox PA, Revankar SG, Sanche S, Geiser DM, Juba JH, van Burik JAH, Padhye A, Anaissie EJ, Francesconi A, Walsh TJ, Robinson JS (2004) Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J Clin Microbiol 42:5109–5120. https://doi.org/10.1128/jcm.42.11.5109-5120.2004
Ortoneda M, Guarro J, Madrid MP, Caracuel Z, Roncero MIG, Mayayo E, Di Pietro A (2004) Fusarium oxysporum as a multihost model for the genetic dissection of fungal virulence in plants and mammals. Infect Immun 72:1760–1766. https://doi.org/10.1128/iai.72.3.1760-1766.2004
Scarlett K, Tesoriero L, Daniel R, Guest D (2014) Sciarid and shore flies as aerial vectors of Fusarium oxysporum f. sp. cucumerinum in greenhouse cucumbers. J Appl Entomol 138:368–377. https://doi.org/10.1111/jen.12098
Busby PE, Ridout M, Newcombe G (2016) Fungal endophytes: modifiers of plant disease. Plant MolBiol 90:645–655. https://doi.org/10.1007/s11103-015-0412-0
Hallen-Adams HE, Suhr MJ (2017) Fungi in the healthy human gastrointestinal tract. Virulence 8:352–358. https://doi.org/10.1080/21505594.2016.1247140
Liu W, Yu SH, Zhang HP, Fu ZY, An JQ, Zhang JY, Yang P (2022) Two Cladosporium fungi with opposite functions to the chinese white wax scale insect have different genome characters. J Fungi 8:21. https://doi.org/10.3390/jof8030286
Brentassi ME, Medina R, de la Fuente D, Franco ME, Toledo AV, Saparrat MCN, Balatti PA (2020) Endomycobiome associated with females of the planthopper Delphacodes kuscheli (Hemiptera: Delphacidae): a metabarcoding approach. Heliyon 6:9. https://doi.org/10.1016/j.heliyon.2020.e04634
Funding
This work was funded in part by the Natural History Award from the Dayton Bell Museum Fund and through startup funds at the University of Minnesota to KEB.
Author information
Authors and Affiliations
Contributions
MW and GM conceived of the study design. MW conducted field sampling, isolation of DNA and fungal cultures from samples, and analyzed sequence data. GM and KEB co-mentored MW and provided guidance, feedback, and suggestions for analysis and co-wrote the manuscript with MW. KEB provided funding for next-generation sequencing.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Watson, M., May, G. & Bushley, K.E. Sources of Fungal Symbionts in the Microbiome of a Mobile Insect Host, Spodoptera frugiperda. Microb Ecol 86, 900–913 (2023). https://doi.org/10.1007/s00248-022-02140-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-022-02140-3