
Abstract
Aphids harbor an array of symbionts that provide hosts with ecological benefits. Microbial community assembly generally varies with respect to aphid species, geography, and host plants. However, the influence of host genetics and ecological factors on shaping intraspecific microbial community structures has not been fully understood. In the present study, using Illumina sequencing of the V3 − V4 hypervariable region of the 16S rRNA gene, we characterized the microbial compositions associated with Mollitrichosiphum tenuicorpus from different regions and plants in China. The primary symbiont Buchnera aphidicola and the secondary symbiont Arsenophonus dominated the microbial flora in M. tenuicorpus. Ordination analyses and statistical tests suggested that geography and aphid genetics primarily contributed to the variation in the microbiota of M. tenuicorpus. We further confirmed the combined effect of aphid genetics and geography on shaping the structures of symbiont and secondary symbiont communities. Moreover, the significant correlation between aphid genetic divergence and symbiont community dissimilarity provides evidence for intraspecific phylosymbiosis in natural systems. Our study helped to elucidate the eco-evolutionary relationship between symbiont communities and aphids within one given species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The influence of ecological factors and host genetics on animal-associated microbial communities has been well documented [1,2,3]. Phylosymbiosis occurs when microbial community relationships significantly correlate with the evolutionary history of the host [4, 5]. This pattern does not necessarily presume the vertical inheritance of the entirety or some members of microbial communities. In addition to the codiversification of hosts and some microbes [6], phylosymbiosis may arise from ecological filtering by conserved host traits [7, 8]. Interspecific phylosymbiosis has been substantiated in certain insects, fishes, birds, and mammals [9,10,11,12]. At the intraspecific level, Kohl et al. [13] reported the phylosymbiotic relationship between gut microbiota and different geographical populations of American pikas. However, investigation of intraspecific phylosymbiosis has rarely been assessed in other animal groups. To completely understand the eco-evolutionary relationship of microbiota and individual host species, intraspecific phylosymbiosis analyses should be performed on more animal groups.
Aphids and their various symbionts provide an excellent model system to study the insect-microbe relationship from ecological and evolutionary perspectives. The microbial communities associated with aphids are typically dominated by symbionts [14,15,16,17]. The primary endosymbiont Buchnera aphidicola, which resides in specialized bacteriocytes of almost all aphids, provides hosts with essential nutrients lacking in phloem sap diets [18]. Buchnera is strictly vertically transmitted from mother to offspring [19] and co-speciates with aphids [20,21,22,23]. Aphids also host secondary symbionts that occupy secondary bacteriocytes, sheath cells, or hemocoel [24] and experience vertical and horizontal transmission [25,26,27]. Most attention has been given to nine secondary symbionts, namely, Arsenophonus, Fukatsuia symbiotica, Hamiltonella defensa, Regiella insecticola, Rickettsia, Rickettsiella viridis, Serratia symbiotica, Spiroplasma, and Wolbachia [25, 28,29,30,31,32,33,34,35]. Secondary symbionts exert diverse mutualistic effects in complex ecological environments, such as resistance to high temperature [36, 37], defense against parasitic wasps and fungal pathogens [38, 39], modification of body colors [34], and enhancement of host plant utilization [40,41,42].
The factors shaping the symbiont community structures primarily include aphid species [43], characteristics of aphids [17], geography [44], and host plants [16, 45]. The majority of surveys about intraspecific symbiont diversity have focused on the impact of ecological conditions on symbiont infection patterns [46,47,48,49,50,51]. For example, Tsuchida et al. [52] highlighted that the markedly different infection frequencies of the secondary symbionts in Acyrthosiphon pisum were associated with geographical distribution. Regarding the impact of aphid genetic divergence on symbionts, some studies have documented the associations between individual symbionts and hosts, such as the nonrandom presence of H. defensa across aphid genetic clusters in A. pisum [53] and the divergence of Buchnera in different geographical populations of Schlechtendalia chinensis [54]. Gauthier et al. [55] demonstrated that the variation in bacterial communities was not related to genetic divergence between biotypes of A. pisum. The intraspecific variation in the microbial community needs more exploration across both ecological and aphid genetic contexts.
Mollitrichosiphum tenuicorpus (Hemiptera: Aphididae: Greenideinae) is monocious with a holocyclic life cycle. This species feeds on young shoots of plants consisting of Alnus (Betulaceae), Castanospermum (Fabaceae), Litsea (Lauraceae), Meliosma (Sabiaceae), and several genera of Fagaceae, such as Castanea, Castanopsis, Lithocarpus, and Quercus [56]. M. tenuicorpus is distributed in eastern and southeast Asia [56, 57]. Previous studies have demonstrated that M. tenuicorpus is divided into three clades [58], which co-segregates with Buchnera at the intraspecific level [22]. Qin et al. [43] uncovered the microbial community composition of M. tenuicorpus. However, the intraspecific microbiota variation of this species has not been fully characterized to date. M. tenuicorpus provides an opportunity to explore the eco-evolutionary relationship between microbiota and insects within one species.
In this study, using Illumina sequencing of the 16S rRNA gene, we characterized the microbial community composition of M. tenuicorpus sampled from different plants and regions in China. Moreover, we assessed the effects of aphid genetic divergence, geography, host plant, and environmental conditions on the structures of bacterial, symbiont (incl. Buchnera and secondary symbionts), and secondary symbiont communities in the field. To investigate the pattern of intraspecific phylosymbiosis, the correlations between microbiota dissimilarities and aphid genetic divergences were also estimated.
Materials and Methods
Aphid Sampling and DNA Extraction
Aphid collection was carried out for seven genera of plants in 12 geographic regions of China. Collection information is shown in Table S1. The samples were frozen at − 20 °C until further processing. All samples were preserved in 75% and 95% ethanol for voucher specimen collections and molecular studies, respectively. Aphid identification was performed using morphological examination and DNA barcoding. All specimens and samples were deposited in the National Zoological Museum of China, Institute of Zoology, Chinese Academy of Sciences, Beijing, China.
A single adult from each sample was obtained for DNA extraction. Aphid individuals were washed with 70% ethanol for 5 min and rinsed with sterile water five times to remove body surface contaminants. Total DNA was extracted from the whole body of each aphid using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. A negative control substituting the DNA with sterile ultrapure water was prepared in the same way. DNA extracts were PCR-amplified targeting the cytochrome c oxidase subunit I (COI) gene with primers LCO1490 and HCO2198 [59] to verify aphid species and eliminate parasitized samples. Final DNA samples were kept at − 20 °C for further experiments.
16S rRNA Gene Amplification and Illumina Sequencing
Primers 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′) were employed to amplify the V3 − V4 hypervariable region of the 16S rRNA gene. Each 30-μL PCR reaction mixture comprised of 15 μL Phusion High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA, USA), 3 μL primers, and 10 μL template DNA. The PCR protocol was as follows: 1 min at 98 °C for initial denaturation; 30 cycles of 10 s at 98 °C for denaturing, 30 s at 50 °C for annealing, and 30 s at 72 °C for elongation; and 5 min at 72 °C for final extension. All samples including negative controls for DNA extraction and amplification were amplified in triplicate.
PCR products were observed on a 2% agarose gel and purified with GeneJET Gel Extraction Kit (Thermo Scientific, Wilmington, DE, USA). Amplicon libraries were prepared with NEBNext Ultra DNA Library Prep Kit (New England Biolabs). Library quality control was performed on Qubit 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Finally, sequencing was conducted on an Illumina HiSeq 2500 PE250 platform (Illumina, San Diego, CA, USA).
Bioinformatic Processing of Sequencing Data
Paired-end reads were merged using FLASH v1.2.7 [60] with a minimum overlap size of 10 bp and an error rate of 10% and assigned to each sample according to their unique barcodes. After filtering and removing chimeras by QIIME v1.9.1 [61], the remaining sequences with ≥ 97% similarity were clustered into the same operational taxonomic units (OTUs) using the UCLUST module. Representative sequences (i.e., the most abundant sequence in OTU clusters) were annotated against the SILVA 128 reference database [62] using the RDP classifier [63] with a 0.80 confidence threshold. Taxonomic classifications were also manually checked by BLAST against GenBank. OTUs with an abundance less than 0.005% were subsequently excluded according to Bokulich et al. [64]. For each sample, we averaged the sequence number across three technical PCR replicates to estimate the abundance of each OTU. To reduce the impact of the uneven sequencing depth on the downstream statistical analyses, the sum of sequence number per sample was rarefied to the minimum value across all samples (53,500 reads) in USEARCH v10.0 [65]. Then, we obtained an OTU count table containing taxonomic definitions and sequence number per sample. We converted OTU count data to relative abundance using the decostand function of the R package vegan [66]. Finally, the bacterial OTU table was prepared (Table S2a).
Microbial Community Analyses
To better explore the microbial community structures within M. tenuicorpus, OTUs classified as symbionts (incl. Buchnera and secondary symbionts) (Table S2b) and secondary symbionts (Table S2c) were screened out, and their relative abundances were calculated via division of the number of sequences assigned to each OTU by the sum of sequences in a given sample. All of the following statistical analyses were performed with bacterial, symbiont, and secondary symbiont data. We grouped all M. tenuicorpus samples according to aphid clades, geographic distribution, and host plant. Detailed grouping information is shown in Table S3. A heatmap visualizing the relative abundance of symbiont OTUs was generated using the pheatmap function in the R package pheatmap [67]. The maximum-likelihood trees showing the relatedness of symbionts and M. tenuicorpus aphids were constructed separately using RAxML v8.2.7 [68] (detailed methods are provided in the Electronic Supplementary Methods). Statistical inference was performed on all groups and groups with a sample size ≥ 3, except Mantel tests, Procrustes analyses, and redundancy analyses. Samples with ambiguous host plant information were excluded from analyses.
Based on OTU tables, Shannon and Simpson indices quantifying alpha diversity were assessed using the diversity function in vegan. Because the alpha diversity data were not normally distributed (Shapiro–Wilk test, P < 0.05), nonparametric Kruskal–Wallis tests were conducted to compare the microbiota variation across different groups of aphid clades, geographic distribution, and host plant.
Then, beta diversity, including the Jaccard presence/absence metric and Bray–Curtis relative abundance metric, was calculated using the vegdist function of vegan. We used unconstrained nonmetric multidimensional scaling (NMDS) and constrained principal coordinate analysis (cPCoA) to visualize the patterns from beta diversity data. NMDS was assessed with the metaMDS function of vegan (stress values < 0.05 indicate excellent representation). CPCoA was performed using the capscale and anova.cca functions in vegan. The significance of differences in microbial community structures was examined through analysis of similarities (ANOSIM; anosim function) and permutational multivariate analysis of variance (PERMANOVA; adonis function) with 9999 permutations in vegan. Both statistical tests were calculated based on Jaccard and Bray–Curtis distances, which generate a P value and a sample statistic (i.e., R of ANOSIM and R2 value of PERMANOVA). An R value between 0 and 1 represents the dissimilarity of community structures among groups, and the R2 value measures the degree of difference between two groups.
To further investigate the impact of aphid genetic divergence or geography on microbiota dissimilarities, partial Mantel test using matrices of aphid genetic divergence and beta diversity (Jaccard and Bray–Curtis distances) was performed. Aphid genetic divergences were evaluated by p distances between pairs of cytochrome c oxidase subunit I (COI) sequences in MEGA v7.0 [69]. Geographic distances were calculated in Geographic Distance Matrix Generator v1.2.3 [70]. The partial Mantel test is commonly used to examine the relationship between two matrices (e.g., microbial beta diversity and geographic distances) while holding another (e.g., genetic distances) constant. Analyses were implemented using the mantel function of the ecodist package with 9999 permutations [71]. We also performed multiple regression on distance matrices (MRM function of ecodist) to assess the combined effect of aphid genetic divergence and geography in shaping the microbial community structures [72]. Moreover, Mantel test (mantel function of vegan) and Procrustes (procrustes and protest function of vegan) analysis were conducted to investigate the intraspecific phylosymbiosis in M. tenuicorpus using the distances of aphid genetic and beta diversity. The Mantel test is a commonly used approach to estimate the relationship between two matrices. Procrustes analysis is more powerful [73], in which M2 varies from 0 (complete incongruence) to 1 (complete congruence).
Finally, to assess the importance of environmental factors in explaining the variation in microbial communities associated with M. tenuicorpus, redundancy analyses (RDA) were implemented on OTU tables. Environmental variables were generated from the “WorldClim” dataset using the getData function in the raster package and logarithmically transformed for normalization. We manually removed collinear environmental variables (function vif.cca of vegan) and obtained the maximum adjusted R2. The maximum temperature of the warmest month (Bio5) and annual precipitation (Bio12), latitude, and altitude were extracted as predictor variables. Next, the RDA model was applied using the rda function in vegan to study the relationship between the microbiota and these screened environmental variables.
Results
Microbial Community Profiling
We obtained 1,469,838 reads after quality control, with a mean of 56,532 reads per sample. The sequences were assigned into 106 OTUs, which belonged to 46 genera, 35 families, 25 orders, 14 classes, and 7 phyla of bacteria. Proteobacteria (average relative abundance across all samples, 98.85%) was the most dominant phylum of the microbial composition associated with M. tenuicorpus. At the class level, Gammaproteobacteria (96.66%) represented the most commonly classified bacteria. Enterobacteriales (96.28%) was the most abundant order, followed by Rickettsiales (1.84%). Enterobacteriaceae (96.26%) and Anaplasmataceae (1.41%) were common, and other bacterial families accounted for less than 0.50% (Table S4). Among the top 10 genera, the relative abundances of Buchnera aphidicola (83.62%), Arsenophonus (10.52%), and Wolbachia (1.41%) were greater than 1%. The alpha diversity estimates of bacterial communities across M. tenuicorpus samples ranged from 0.117 to 0.799 for the Shannon index and from 0.077 to 0.684 for the Simpson index (Table S5).
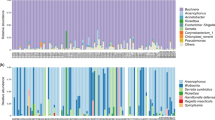
Each sample examined in this study simultaneously harbored 4–7 symbionts (Fig. 1). The primary endosymbiont Buchnera and secondary symbionts Arsenophonus and Wolbachia were detected in all samples (infection frequency, 26/26). In addition, M. tenuicorpus was infected with five other kinds of aphid secondary symbionts in which the relative abundances were low, including Hamiltonella defensa (11/26, 0.77%), Rickettsia (14/26, 0.41%), Serratia symbiotica (22/26, 0.13%), Spiroplasma (12/26, 0.04%), and Fukatsuia symbiotica (3/26, 0.02%). The alpha diversity estimates of symbiont communities across M. tenuicorpus samples ranged from 0.085 to 0.695 for the Shannon index and from 0.061 to 0.661 for the Simpson index (Table S5). After excluding the primary endosymbiont Buchnera, the alpha diversity estimates of secondary symbiont communities ranged from 0.184 to 0.923 for the Shannon index and from 0.143 to 0.808 for the Simpson index (Table S5).

Microbial community composition associated with Mollitrichosiphum tenuicorpus across all samples. The maximum-likelihood tree depicts the phylogenetic relationships between aphid samples
At the OTU level, OTU1 of Buchnera predominated in the most samples with a relative abundance of 81.66%, except for a sample from Tibet in which OTU1729 (48.70%) and OTU1 (25.45%) were most abundant (Fig. 2). Regarding the secondary symbionts, the predominant OTUs of Arsenophonus differed among the different aphid clades in M. tenuicorpus. The secondary symbiont composition of the sample collected from Tibet (sample ID: 15381) was dominated by OTU11 (9.49%) and OTU305 (5.61%) belonging to Arsenophonus. The most abundant secondary symbiont OTU of samples from northwestern Yunnan Province (sample ID: 13361, 24074 and 24067) was OTU3561 of Arsenophonus, in which the relative abundance ranged from 3.01 to 6.16%. OTU4 of Arsenophonus dominated the secondary symbiont communities of most samples widely distributed in southern China, with an average relative abundance of 7.92%.

Heatmap representing the distribution and relative abundances of symbiont OTUs among Mollitrichosiphum tenuicorpus. The maximum-likelihood trees display the phylogenetic relationships of Buchnera OTUs, secondary symbiont OTUs, and Mollitrichosiphum tenuicorpus, respectively. White groups indicate ambiguous information on the host plant
Microbial Community Signatures of Mollitrichosiphum tenuicorpus
Kruskal–Wallis tests based on alpha diversity indices did not reveal a significant effect of aphid genetic divergence (Shannon index, P = 0.213–0.616; Simpson index, P = 0.192–0.676) or host plants (Shannon, P = 0.155–0.624; Simpson, P = 0.163–0.566) on the bacterial, symbiont, and secondary symbiont communities. No significant variation in microbial communities was found among geographic regions (Shannon, P = 0.057–0.837; Simpson, P = 0.113–0.875), except for a significant variation in the secondary symbiont community based on the Simpson index (n ≥ 3, P = 0.036).
There was no recognizable clustering of samples structured by aphid clades, geographic regions, or host plants in unconstrained NMDS plots (Fig. S1–S3) using all types of beta diversity data. Conversely, constrained PCoA (cPCoA) analyses based on Jaccard and Bray–Curtis distances using all samples showed a significant beta-diversity pattern. CPCoA plots displayed significant clustering constrained by aphid clades (P = 0.001–0.013; Fig. 3a–c and Fig. S4a–c) and geographic regions (P = 0.001–0.011; Fig. 3d–f and Fig. S4d–f) in the bacterial, symbiont, and secondary symbiont communities. The overall variance in the data explained by geographic regions (57.1–62.2% of variance) was greater than that explained by aphid clades (18.2–26% of variance). Moreover, cPCoA analyses did not reveal a meaningful microbial community structure constrained by host plants (35.4–44.4% of variance, P = 0.3–0.87; Fig. 3g–i and Fig. S4g–i). Regarding beta diversity with a sample size ≥ 3, only cPCoA analyses of geographic groups were performed, as the data constrained by aphid clades and host plant were insufficient. Additionally, the cPCoA analyses merely uncovered a significant pattern of secondary symbiont community among geographic regions (P = 0.001; 40% of variance for Jaccard distances; 44.1% of variance for Bray–Curtis distances; Fig. S5c, f).

Constrained principal coordinate analysis (cPCoA) plots based on Bray–Curtis distances of bacterial (a, d, g), symbiont (b, e, h), and secondary symbiont (c, f, i) communities (n ≥ 1). Plots are structured by aphid clades (a–c), geographic region (d–f), and host plant (g–i). The abbreviations are given in Table S3
ANOSIM did not detect a significant impact of aphid genetic divergence (R = − 0.153 − 0.181, P = 0.180–0.741) or geography (R = − 0.163 − 0.045, P = 0.271 − 0.935) on shaping the structures of bacterial and symbiont communities (Table 1). Nonetheless, the variation in secondary symbiont communities was usually significant among samples grouped by aphid clades (R = 0.627 − 0.640, P = 0.003 − 0.008) and geographic regions (n ≥ 3, R = 0.388, P < 0.001). PERMANOVA corroborated that the secondary symbiont communities significantly differed among aphid clades (R = 0.196 − 0.303, P < 0.001) and geographic regions (R = 0.484 − 0.535, P < 0.001) (Table 1). The R2 values further suggested a greater contribution of geography than aphid genetic divergence. PERMANOVA also indicates a significant dissimilarity among samples in some datasets from different aphid clades (n ≥ 1, R2 = 0.323 − 0. 492, P = 0.008 − 0.009) and geographic regions (n ≥ 3, R2 = 0.262 − 0.286, P = 0.012 − 0.026) in bacterial and symbiont communities. Moreover, neither ANOSIM (R = − 0.019 − 0.038, P = 0.401 − 0.550) nor PERMANOVA (R = 0.374 − 0.630, P = 0.242 − 0.506) revealed a significant effect of host plant.
Based on partial Mantel tests, we did not detect a significant correlation between aphid genetic divergence and microbial profiles comprising the bacterial, symbiont, and secondary symbiont communities (r = 0.162 − 0.358, P = 0.844 − 0.993; Table S6) after removing the effect of geography. The microbial community structures were not significantly related to geography when controlling for the effect of aphid genetic divergence (r = 0.097 − 0.121, P = 0.894 − 0.947; Table S6). However, multiple regression on distance matrices revealed the significant combined effect of aphid genetic divergence and geography on symbiont (Bray–Curtis distance, R2 = 0.142, P = 0.048) and secondary symbiont communities (Jaccard distance, R2 = 0.229, P = 0.001; Bray–Curtis distance, R2 = 0. 250, P = 0.002) (Table 2). We further found a significant correlation between the distances of aphid genetic divergence and geography using the Mantel test (r = 0.6238, P < 0.001).
Based on Mantel tests, we observed a significant positive correlation between aphid genetic divergence and symbiont community dissimilarities (Bray–Curtis distances, r = 0.365, P = 0.049; Fig. 4e). The same linkage between aphid genetic divergence and secondary symbiont communities was also found (Jaccard distance, r = 0.469, P < 0.001; Bray–Curtis distance, r = 0.493, P = 0.002; Fig. 4c, f). Using Procrustes analyses, a significantly strong correlation between aphid genetic divergence and microbial communities was confirmed in the bacterial, symbiont, and secondary symbiont communities (M2 = 0.602 − 0.688, P = 0.001 − 0.018; Fig. 5).

Correlations between microbial beta diversity and aphid genetic distances estimated by Mantel tests in bacterial (a, d), symbiont (b, e), and secondary symbiont (c, f) communities. Microbial beta diversity was assessed by Jaccard (a–c) and Bray–Curtis (d–f) distances

Procrustean superimpositions for PCA-scaled aphid genetic distances vs. variations in bacterial (a, d), symbiont (b, e), and secondary symbiont (c, f) communities. Jaccard (a–c) and Bray–Curtis (d–f) distances were used to estimate the microbiota variations
Influence of Environmental Factors on Microbiota
As indicated by RDA, four environmental variables (Bio5, Bio12, latitude, and altitude) were significantly related to the bacterial community composition (R2 = 0.131, P = 0.045). In the RDA ordination plot, axis 1 and axis 2 explained 10.52% and 1.26%, respectively, of the variance in the relationship between the environmental variables and bacterial communities (Fig. S6). However, we did not detect a meaningful impact of the screened environmental factors on symbiont (R2 = 0.102, P = 0.077) and secondary symbiont communities (R2 = 0.110, P = 0.065).
Discussion
Symbiont Diversity of Mollitrichosiphum tenuicorpus
Aphid symbionts dominated the microbial community composition of M. tenuicorpus, among which Buchnera was the most abundant bacteria in all examined samples. This confirms the essential role of Buchnera in aphid survival and reproduction [74, 75]. In addition, M. tenuicorpus simultaneously harbored three to six types of secondary symbionts per sample. The frequent coinfection pattern in the present study substantiated the multiple infections of secondary symbionts within one aphid host reported in Mollitrichosiphum aphids [43].
Arsenophonus and Wolbachia were harbored by all examined M. tenuicorpus samples with high relative abundance, which suggested their ubiquity in aphids [14, 16, 17, 76]. Arsenophonus dominated the secondary symbiont community composition of M. tenuicorpus samples with the highest infection frequency and relative abundance. Moreover, we observed a high diversity of Arsenophonus, which was represented by ten types of OTUs in M. tenuicorpus. Previous studies have demonstrated that Arsenophonus can provide aphids with general fitness benefits [77] and facilitate specialization on a novel host plant [42, 78]. Wolbachia plays a role in manipulating the reproduction [79] of numerous terrestrial arthropods [80]. However, the precise impact of Wolbachia on aphids has not been explored. Further investigations are needed to illustrate the exact effects of Arsenophonus and Wolbachia on M. tenuicorpus aphids.
The other five secondary symbionts, namely, H. defensa, Rickettsia, S. symbiotica, Spiroplasma, and F. symbiotica, presented low relative abundances in M. tenuicorpus. Spiroplasma was detected for the first time in Mollitrichosiphum aphids, although its relative abundance was low. Spiroplasma has been reported in few aphid groups, including Aphis gossypii [16], Myzus persicae [81], Aphis citricidus [82], and some species in Eriosomatinae [45]. The prevalence of Spiroplasma may be underestimated in other aphids due to the low titers. In addition, we found an infection pattern of F. symbiotica with low prevalence and relative abundance in M. tenuicorpus, which is different from other aphid species in Mollitrichosiphum [43]. Only one sample harbored H. defensa with a relative abundance of 18.22%. After removing this sample, the relative abundance of H. defensa across all examined M. tenuicorpus aphids was low (0.07%). M. tenuicorpus aphids have extremely long siphunculi and can efficiently emit alarm pheromones [83] to reduce the risk of predation. Aphids usually do not carry secondary symbionts, providing the benefits that they have already conferred from ecological traits [49]. This may explain the low relative abundance of defensive secondary symbiont H. defensa in M. tenuicorpus.
Factors Determining Microbiota Variation within Mollitrichosiphum tenuicorpus
The influence of host plants on microbial communities has been reported within one aphid species [16, 55, 82]. Nevertheless, some studies have demonstrated that geographical distribution plays a more important role than host plant in determining the microbial flora within one aphid species [46, 84]. In the present study, the effect of host plant on microbial community structures associated with M. tenuicorpus was not significant. Our results revealed the greatest contribution of geography in shaping the structures of bacterial, symbiont, and secondary symbiont communities in M. tenuicorpus. The geographic variability of microbiota may arise from local environmental conditions [85]. However, we found only a weakly significant association between environmental factors and bacterial communities. The nonsignificant relationships between environmental variables and the structures of symbiont and secondary symbiont communities rule out the impact of abiotic features among different geographical regions.
Notably, we detected a significant combined effect of aphid genetic divergence and geography on the symbiont and secondary symbiont communities, while their separate effects were not significant using the partial Mantel test. The Mantel test confirmed the significant correlation between aphid genetic divergence and geographic distances in the present study. Overall, our results suggest that the structures of symbiont and secondary symbiont communities are determined by the combination of aphid genetic divergence and geography.
Intraspecific Phylosymbiosis in Mollitrichosiphum tenuicorpus
Procrustes analyses showed that the genetic divergence of M. tenuicorpus significantly related to the microbial profiles of bacterial, symbiont, and secondary symbiont communities using all types of data. We also detected the phylogenetic structures of symbiont and secondary symbiont communities with Mantel tests. Although there was little inconsistency in the results under different analysis methods and beta diversity distance metrics, the phylosymbiotic signals within one aphid species were uncovered in the symbiont and secondary symbiont communities.
Significant positive correlations between aphid relatedness and microbiota dissimilarities have been reported in aphids at the interspecies level [43, 86]. Various factors can lend to the phylogenetically structured microbiota, such as host filtering of environmental microbes [7], regulation of host immune system [87], host phylogeny-related diet preference [88], and host-microbe codiversification [6]. We detected a nonsignificant impact of environmental factors and host plants on the symbiont communities in M. tenuicorpus. Considering the similarities of host traits within M. tenuicorpus, it is less likely that the phylosymbiotic structures result from ecological filtering by aphid traits. Alternatively, some specific microbes serving as keystone or hub taxa can determine the composition of the whole microbiota via microbe-microbe interactions [2, 89]. Codiversification of hub microbes and hosts can lend to the pattern of phylosymbiosis. We suggest that the phylosymbiotic structures in symbiont communities associated with M. tenuicorpus are driven by the codiversification of aphids and predominant symbionts (i.e., the primary endosymbiont Buchnera and the secondary symbiont Arsenophonus).
The co-segregation between three clades of M. tenuicorpus and maternally inherited Buchnera has been substantiated by Liu et al. [22]. The divergence of the clade within M. tenuicorpus sampled from Tibet occurred earlier than that of other clades [90]. Despite the limitation of phylogenetic signals in the V3 − V4 hypervariable region of the 16S rRNA gene, the diversification of predominant OTUs belonging to Buchnera corresponded to host aphids in the present study. In addition, the variation in dominant OTUs of the secondary symbiont Arsenophonus was generally congruent with aphid genetic divergence, which indicates host-symbiont codiversification within M. tenuicorpus. However, further investigations with additional data are required to elucidate the shared diversification history between M. tenuicorpus and Arsenophonus.
Conclusions
Assessing the factors determining microbial communities is crucial for understanding of host-microbiome associations. We identified the microbial composition dominated by symbionts within one aphid species, Mollitrichosiphum tenuicorpus. The combined impact of aphid genetic divergence and geography was uncovered in the symbiont community profiles. Moreover, we provided evidence of intraspecific phylosymbiosis based on the significant correlation between M. tenuicorpus and symbiont flora. We highlighted the role of codiversification in shaping the phylosymbiotic pattern of M. tenuicorpus, paving the way for further investigations of aphid-symbiont interactions in natural populations.
Data Accessibility
All COI sequences were deposited in GenBank under accession numbers MZ073749–MZ073754. Raw 16S rRNA gene amplicon reads were submitted in the NCBI Sequence Read Archive under BioProject accession number PRJNA726742.
Change history
29 November 2021
The original version of this paper was updated to change of the presentation of Figure 1 and Table 1.
References
Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A, Palumbo AV, Chesler EJ, Podar M (2012) Host genetic and environmental effects on mouse intestinal microbiota. ISME J 6:2033–2044. https://doi.org/10.1038/ismej.2012.54
Kohl KD (2020) Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Phil Trans R Soc B 375:20190251. https://doi.org/10.1098/rstb.2019.0251
Parker ES, Newton IL, Moczek AP (2020) (My Microbiome) Would walk 10,000 miles: maintenance and turnover of microbial communities in introduced dung beetles. Microb Ecol 80:435–446. https://doi.org/10.1007/s00248-020-01514-9
Brucker RM, Bordenstein SR (2013) The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia. Science 341:667–669. https://doi.org/10.1126/science.1240659
Lim SJ, Bordenstein SR (2020) An introduction to phylosymbiosis. Proc R Soc Lond B Biol Sci 287:20192900. https://doi.org/10.1098/rspb.2019.2900
Sanders JG, Powell S, Kronauer DJ, Vasconcelos HL, Frederickson ME, Pierce NE (2014) Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol Ecol 23:1268–1283. https://doi.org/10.1111/mec.12611
Moran NA, Sloan DB (2015) The hologenome concept: helpful or hollow? PLoS Biol 13:e1002311. https://doi.org/10.1371/journal.pbio.1002311
Mazel F, Davis KM, Loudon A, Kwong WK, Groussin M, Parfrey LW (2018) Is host filtering the main driver of phylosymbiosis across the tree of life? mSystems 3:e00097-18. https://doi.org/10.1128/mSystems.00097-18
Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR (2016) Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol 14:e2000225. https://doi.org/10.1371/journal.pbio.2000225
Doane MP, Morris MM, Papudeshi B et al (2020) The skin microbiome of elasmobranchs follows phylosymbiosis, but in teleost fishes, the microbiomes converge. Microbiome 8:93. https://doi.org/10.1186/s40168-020-00840-x
Hammer TJ, Dickerson JC, McMillan WO, Fierer N (2020) Heliconius butterflies host characteristic and phylogenetically structured adult-stage microbiomes. Appl Environ Microbiol 86:e02007-e2020. https://doi.org/10.1128/AEM.02007-20
Tinker KA, Ottesen EA (2020) Phylosymbiosis across deeply diverging lineages of omnivorous cockroaches (order Blattodea). Appl Environ Microbiol 86:e02513-e2519. https://doi.org/10.1128/AEM.02513-19
Kohl KD, Varner J, Wilkening JL, Dearing MD (2018) Gut microbial communities of American pikas (Ochotona princeps): evidence for phylosymbiosis and adaptations to novel diets. J Anim Ecol 87:323–330. https://doi.org/10.1111/1365-2656.12692
Jousselin E, Clamens AL, Galan M, Bernard M, Maman S, Gschloessl B, Duport G, Meseguer AS, Calevro F, Coeur d’acierA (2016) Assessment of a 16S rRNA amplicon Illumina sequencing procedure for studying the microbiome of a symbiont-rich aphid genus. Mol Ecol Resour 16:628–640. https://doi.org/10.1111/1755-0998.12478
Guyomar C, Legeai F, Jousselin E, Mougel C, Lemaitre C, Simon J (2018) Multi-scale characterization of symbiont diversity in the pea aphid complex through metagenomic approaches. Microbiome 6:181. https://doi.org/10.1186/s40168-018-0562-9
Xu S, Jiang L, Qiao G, Chen J (2020) The bacterial flora associated with the polyphagous aphid Aphis gossypii Glover (Hemiptera: Aphididae) is strongly affected by host plants. Microb Ecol 79:971–984. https://doi.org/10.1007/s00248-019-01435-2
Xu TT, Chen J, Jiang LY, Qiao GX (2021) Diversity of bacteria associated with Hormaphidinae aphids (Hemiptera: Aphididae). Insect Sci 28:165–179. https://doi.org/10.1111/1744-7917.12746
Baumann P, Baumann L, Lai CY, Rouhbakhsh D, Moran NA, Clark MA (1995) Genetics, physiology, and evolutionary relationships of the genus Buchnera: intracellular symbionts of aphids. Annu Rev Microbiol 49:55–94. https://doi.org/10.1146/annurev.mi.49.100195.000415
Koga R, Meng X, Tsuchida T, Fukatsu T (2012) Cellular mechanism for selective vertical transmission of an obligate insect symbiont at the bacteriocyte-embryo interface. Proc Natl Acad Sci USA 109:E1230–E1237. https://doi.org/10.1073/pnas.1119212109
Munson MA, Baumann P, Clark MA, Baumann L, Moran NA, Voegtlin DJ, Campbell BC (1991) Evidence for the establishment of aphid-eubacterium endosymbiosis in an ancestor of four aphid families. J Bacteriol 173:6321–6324. https://doi.org/10.1128/jb.173.20.6321-6324.1991
Funk DJ, Helbling L, Wernegreen JJ, Moran NA (2000) Intraspecific phylogenetic congruence among multiple symbiont genomes. Proc R Soc Lond B Biol Sci 267:2517–2521. https://doi.org/10.1098/rspb.2000.1314
Liu L, Huang XL, Zhang RL, Jiang LY, Qiao GX (2013) Phylogenetic congruence between Mollitrichosiphum (Aphididae: Greenideinae) and Buchnera indicates insect-bacteria parallel evolution. Syst Entomol 38:81–92. https://doi.org/10.1111/j.1365-3113.2012.00647.x
Xu TT, Chen J, Jiang LY, Qiao GX (2018) Historical and cospeciating associations between Cerataphidini aphids (Hemiptera: Aphididae: Hormaphidinae) and their primary endosymbiont Buchnera aphidicola. Zool J Linn Soc-Lond 182:604–613. https://doi.org/10.1093/zoolinnean/zlx048
Fukatsu T, Nikoh N, Kawai R, Koga R (2000) The secondary endosymbiotic bacterium of the pea aphid Acyrthosiphon pisum (Insecta: Homoptera). Appl Environ Microbiol 66:2748–2758. https://doi.org/10.1128/AEM.66.7.2748-2758.2000
Russell JA, Latorre A, Sabater-Muñoz B, Moya A, Moran NA (2003) Side-stepping secondary symbionts: widespread horizontal transfer across and beyond the Aphidoidea. Mol Ecol 12:1061–1075. https://doi.org/10.1046/j.1365-294X.2003.01780.x
Michalik A, Szklarzewicz T, Jankowska W, Wieczorek K (2014) Endosymbiotic microorganisms of aphids (Hemiptera: Sternorrhyncha: Aphidoidea): ultrastructure, distribution and transovarial transmission. Eur J Entomol 111:91–104. https://doi.org/10.14411/eje.2014.011
Pons I, Renoz F, Noël C, Hance T (2019) Circulation of the cultivable symbiont Serratia symbiotica in aphids is mediated by plants. Front Microbiol 10:764. https://doi.org/10.3389/fmicb.2019.00764
Unterman BM, Baumann P, McLean DL (1989) Pea aphid symbiont relationships established by analysis of 16S rRNAs. J Bacteriol 171:2970–2974. https://doi.org/10.1128/jb.171.6.2970-2974.1989
Chen DQ, Campbell BC, Purcell AH (1996) A new Rickettsia from a herbivorous insect, the pea aphid Acyrthosiphon pisum (Harris). Curr Microbiol 33:123–128. https://doi.org/10.1007/s002849900086
Darby AC, Birkle LM, Turner SL, Douglas AE (2001) An aphid-borne bacterium allied to the secondary symbionts of whitefly. FEMS Microbiol Ecol 36:43–50. https://doi.org/10.1016/S0168-6496(01)00117-9
Fukatsu T, Tsuchida T, Nikoh N, Koga R (2001) Spiroplasma symbiont of the pea aphid, Acyrthosiphon pisum (Insecta: Homoptera). Appl Environ Microbiol 67:1284–1291. https://doi.org/10.1128/AEM.67.3.1284-1291.2001
Sandström JP, Russell JA, White JP, Moran NA (2001) Independent origins and horizontal transfer of bacterial symbionts of aphids. Mol Ecol 10:217–228. https://doi.org/10.1046/j.1365-294X.2001.01189.x
Guay JF, Boudreault S, Michaud D, Cloutier C (2009) Impact of environmental stress on aphid clonal resistance to parasitoids: role of Hamiltonella defensa bacterial symbiosis in association with a new facultative symbiont of the pea aphid. J Insect Physiol 55:919–926. https://doi.org/10.1016/j.jinsphys.2009.06.006
Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S, Simon JC, Fukatsu T (2010) Symbiotic bacterium modifies aphid body color. Science 330:1102–1104. https://doi.org/10.1126/science.1195463
Augustinos AA, Santos-Garcia D, Dionyssopoulou E, Moreira M, Papapanagiotou A, Scarvelakis M, Doudoumis V, Ramos S, Aguiar AF, Borges PAV, Khadem M, Latorre A, Tsiamis G, Bourtzis K (2011) Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS ONE 6:e28695. https://doi.org/10.1371/journal.pone.0028695
Chen DQ, Montllor CB, Purcell AH (2000) Fitness effects of two facultative endosymbiotic bacteria on the pea aphid, Acyrthosiphon pisum, and the blue alfalfa aphid A. kondoi. Entomol Exp Appl 95:315–323. https://doi.org/10.1046/j.1570-7458.2000.00670.x
Russell JA, Moran NA (2006) Costs and benefits of symbiont infection in aphids: variation among symbionts and across temperatures. Proc R Soc Lond B Biol Sci 273:603–610. https://doi.org/10.1098/rspb.2005.3348
Oliver KM, Moran NA, Hunter MS (2005) Variation in resistance to parasitism in aphids is due to symbionts not host genotype. Proc Natl Acad Sci USA 102:12795–12800. https://doi.org/10.1073/pnas.0506131102
Scarborough CL, Ferrari J, Godfray HCJ (2005) Aphid protected from pathogen by endosymbiont. Science 310:1781–1781. https://doi.org/10.1126/science.1120180
Tsuchida T, Koga R, Fukatsu T (2004) Host plant specialization governed by facultative symbiont. Science 303:1989–1989. https://doi.org/10.1126/science.1094611
Tsuchida T, Koga R, Matsumoto S, Fukatsu T (2011) Interspecific symbiont transfection confers a novel ecological trait to the recipient insect. Biol Letters 7:245–248. https://doi.org/10.1098/rsbl.2010.0699
Wagner SM, Martinez AJ, Ruan YM, Kim KL, Lenhart PA, Dehnel AC, Oliver KM, White JA (2015) Facultative endosymbionts mediate dietary breadth in a polyphagous herbivore. Funct Ecol 29:1402–1410. https://doi.org/10.1111/1365-2435.12459
Qin M, Chen J, Xu SF, Jiang LY, Qiao GX (2021) Microbiota associated with Mollitrichosiphum aphids (Hemiptera: Aphididae: Greenideinae): diversity, host species specificity and phylosymbiosis. Environ Microbiol 23:2184–2198. https://doi.org/10.1111/1462-2920.15391
Gallo-Franco JJ, Duque-Gamboa DN, Toro-Perea N (2019) Bacterial communities of Aphis gossypii and Myzus persicae (Hemiptera: Aphididae) from pepper crops (Capsicum sp.). Sci Rep 9:5766. https://doi.org/10.1038/s41598-019-42232-8
Xu TT, Jiang LY, Chen J, Qiao GX (2020) Host plants influence the symbiont diversity of Eriosomatinae (Hemiptera: Aphididae). Insects 11:217. https://doi.org/10.3390/insects11040217
Najar-Rodríguez AJ, McGraw EA, Mensah RK, Pittman GW, Walter GH (2009) The microbial flora of Aphis gossypii: patterns across host plants and geographical space. J Invertebr Pathol 100:123–126. https://doi.org/10.1016/j.jip.2008.10.005
Henry LM, Peccoud J, Simon JC, Hadfield JD, Maiden MJC, Ferrari J, Godfray HCJ (2013) Horizontally transmitted symbionts and host colonization of ecological niches. Curr Biol 23:1713–1717. https://doi.org/10.1016/j.cub.2013.07.029
Brady CM, Asplen MK, Desneux N, Heimpel GE, Hopper KR, Linnen CR, Oliver KM, Wulff JA, White JA (2014) Worldwide populations of the aphid Aphis craccivora are infected with diverse facultative bacterial symbionts. Microb Ecol 67:195–204. https://doi.org/10.1007/s00248-013-0314-0
Henry LM, Maiden MCJ, Ferrari J, Godfray HCJ (2015) Insect life history and the evolution of bacterial mutualism. Ecol Lett 18:516–525. https://doi.org/10.1111/ele.12425
Smith AH, Łukasik P, O’Connor MP, Lee A, Mayo G, Drott MT, Doll S, Tuttle R, Disciullo RA, Messina A, Oliver KM, Russell JA (2015) Patterns, causes and consequences of defensive microbiome dynamics across multiple scales. Mol Ecol 24:1135–1149. https://doi.org/10.1111/mec.13095
Liu XD, Lei HX, Chen FF (2019) Infection pattern and negative effects of a facultative endosymbiont on its insect host are environment-dependent. Sci Rep 9:4013. https://doi.org/10.1038/s41598-019-40607-5
Tsuchida T, Koga R, Shibao H, Matsumoto T, Fukatsu T (2002) Diversity and geographic distribution of secondary endosymbiotic bacteria in natural populations of the pea aphid, Acyrthosiphon pisum. Mol Ecol 11:2123–2135. https://doi.org/10.1046/j.1365-294X.2002.01606.x
Ferrari J, West JA, Via S, Godfray HCJ (2012) Population genetic structure and secondary symbionts in host-associated populations of the pea aphid complex. Evolution 66:375–390. https://doi.org/10.1111/j.1558-5646.2011.01436.x
Zhang Y, Su X, Harris AJ, Caraballo-Ortiz MA, Ren Z, Zhong Y (2018) Genetic structure of the bacterial endosymbiont Buchnera aphidicola from its host aphid Schlechtendalia chinensis and evolutionary implications. Curr Microbiol 75:309–315. https://doi.org/10.1007/s00284-017-1381-0
Gauthier JP, Outreman Y, Mieuzet L, Simon JC (2015) Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS ONE 10:e0120664. https://doi.org/10.1371/journal.pone.0120664
Blackman RL, Eastop VF (2021) Aphids on the world’s plants: an online identification and information guide. http://www.aphidsonworldsplants.info. Accessed 1 Mar 2021
Zhang D, Qiao G (2010) Mollitrichosiphum Suenaga from China (Hemiptera: Aphididae), with the description of one new species. Zootaxa 2608:1–24
Zhang R, Huang X, Jiang L, Qiao G (2011) Phylogeny and species differentiation of Mollitrichosiphum spp. (Aphididae, Greenideinae) based on mitochondrial COI and Cytb genes. Curr Zool 57:806–815. https://doi.org/10.1093/czoolo/57.6.806
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. https://doi.org/10.1128/Aem.00062-07
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. https://doi.org/10.1038/nmeth.2276
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Oksanen J, Blanchet FG, Friendly M et al (2018) Package ‘vegan’, community ecology package. Version 2.5–2. https://cran.r-project.org/src/contrib/Archive/vegan/. Accessed 9 Mar 2020
Kolde R, Kolde MR (2015) Package “pheatmap”. https://CRAN.R-project.org/package=pheatmap. Accessed 18 Mar 2020
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Ersts PJ (2020) Geographic Distance Matrix Generator v1.2.3. American Museum of Natural History, Center for Biodiversity and Conservation. http://biodiversityinformatics.amnh.org/open_source/gdmg. Accessed 25 Sept 2020
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19. https://doi.org/10.18637/jss.v022.i07
Legendre P (2008) Studying beta diversity: ecological variation partitioning by multiple regression and canonical analysis. J Plant Ecol 1:3–8. https://doi.org/10.1093/jpe/rtm001
Peres-Neto PR, Jackson DA (2001) How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia 129:169–178. https://doi.org/10.1007/s004420100720
Douglas AE (1998) Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol 43:17–37. https://doi.org/10.1146/annurev.ento.43.1.17
Wilson ACC, Ashton PD, Calevro F et al (2010) Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect Mol Biol 19:249–258. https://doi.org/10.1111/j.1365-2583.2009.00942.x
Wang Z, Su X, Wen J, Jiang L, Qiao G (2014) Widespread infection and diverse infection patterns of Wolbachia in Chinese aphids. Insect Sci 21:313–325. https://doi.org/10.1111/1744-7917.12102
Wulff JA, White JA (2015) The endosymbiont Arsenophonus provides a general benefit to soybean aphid (Hemiptera: Aphididae) regardless of host plant resistance (Rag). Environ Entomol 44:574–581. https://doi.org/10.1093/ee/nvv031
Lenhart PA, White JA (2020) Endosymbionts facilitate rapid evolution in a polyphagous herbivore. J Evol Biol 33:1507–1511. https://doi.org/10.1111/jeb.13697
Werren JH, Baldo L, Clark ME (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. https://doi.org/10.1038/nrmicro1969
Zug R, Hammerstein P (2012) Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS ONE 7:e38544. https://doi.org/10.1371/journal.pone.0038544
Xu S, Jiang L, Qiao G, Chen J (2020) Diversity of bacterial symbionts associated with Myzus persicae (Sulzer)(Hemiptera: Aphididae: Aphidinae) revealed by 16S rRNA Illumina sequencing. Microb Ecol 81:784–794. https://doi.org/10.1007/s00248-020-01622-6
Guidolin AS, Cônsoli FL (2018) Diversity of the most commonly reported facultative symbionts in two closely-related aphids with different host ranges. Neotrop Entomol 47:440–446. https://doi.org/10.1007/s13744-017-0532-0
Mondor EB, Roitberg BD, Stadler B (2002) Cornicle length in macrosiphini aphids: a comparison of ecological traits. Ecol Entomol 27:758–762
Jones RT, Bressan A, Greenwell AM, Fierer N (2011) Bacterial communities of two parthenogenetic aphid species cocolonizing two host plants across the Hawaiian Islands. Appl Environ Microbiol 77:8345–8349. https://doi.org/10.1128/Aem.05974-11
Sepúlveda DA, Zepeda-Paulo F, Ramírez CC, Lavandero B, Figueroa CC (2017) Diversity, frequency, and geographic distribution of facultative bacterial endosymbionts in introduced aphid pests. Insect Sci 24:511–521. https://doi.org/10.1111/1744-7917.12313
McLean AHC, Godfray HCJ, Ellers J, Henry LM (2019) Host relatedness influences the composition of aphid microbiomes. Environ Microbiol Rep 11:808–816. https://doi.org/10.1111/1758-2229.12795
Franzenburg S, Walter J, KuÈnzel S et al (2013) Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc Natl Acad Sci USA 110:E3730–E3738. https://doi.org/10.1073/pnas.1304960110
Miyake S, Ngugi DK, Stingl U (2015) Diet strongly influences the gut microbiota of surgeonfishes. Mol Ecol 24:656–672. https://doi.org/10.1111/mec.13050
Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, Kemen EM (2016) Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 14:e1002352. https://doi.org/10.1371/journal.pbio.1002352
Zhang R, Huang X, Jiang L, Lei F, Qiao G (2012) Species differentiation of Chinese Mollitrichosiphum (Aphididae: Greenideinae) driven by geographical isolation and host plant acquirement. Int J Mol Sci 13:10441–10460. https://doi.org/10.3390/ijms130810441
Acknowledgements
Thanks to all collectors for sample collections. Thanks to Fendi Yang for making voucher slides.
Funding
The work was supported by the National Natural Science Foundation of China (No. 31772492), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA19050303), the Key Collaborative Research Program of the Alliance of International Science Organizations (Grant No. ANSO-CR-KP-2020–04), and the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2020087).
Author information
Authors and Affiliations
Contributions
J.C. and G.Q. designed the project. G.Q. and L.J. identified voucher specimens. K.B.R. helps to collect samples. M.Q. conducted molecular experiments and all analyses. J.C. assisted data analyses. M.Q. wrote the manuscript and all authors contributed to revisions.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, M., Jiang, L., Kholmatov, B.R. et al. Phylosymbiotic Structures of the Microbiota in Mollitrichosiphum tenuicorpus (Hemiptera: Aphididae: Greenideinae). Microb Ecol 84, 227–239 (2022). https://doi.org/10.1007/s00248-021-01830-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-021-01830-8