
Abstract
Bats are flying mammals distributed worldwide known to host several types of Coronavirus (CoV). Since they were reported as the probable source of spillover of highly pathogenic CoV into the human population, investigating the circulation of this virus in bats around the world became of great importance. We analyzed samples from 103 bats from two distinct regions in Brazil. Coronavirus from the Alphacoronavirus genus was detected in 12 animals, 11 from São José do Rio Preto—SP region and 1 from Barreiras—BA region, resulting in a prevalence of 17.18% and 2.56% respectively. The virus was detected not only in intestines but also in lungs and liver. Phylogenetic analysis based on nsP12 genomic region suggests that the sequences group according to host family and sampling location. Studies on the circulation of these viruses in bats remain important to understand the ecology and evolutionary relationship of these pathogens.
Similar content being viewed by others


Introduction
Coronaviruses (CoVs) are single-stranded positive sense RNA viruses that belong to the Coronaviridae family. The family comprises four genera: Alphacoronavirus (α-CoV), Betacoronavirus (β-CoV), Gammacoronavirus (γ-CoV), and Deltacoronavirus (δ-CoV) [1]. These viruses infect several species of animals and can cause hepatic, enteric, neurological, and acute and chronic respiratory infection [2]. In humans, they generally cause mild upper respiratory tract disease with low mortality [3]. In 2003 and 2012 respectively, the emergence of highly pathogenic β-CoV SARS-CoV (severe acute respiratory syndrome) and MERS-CoV (Middle East respiratory syndrome) revealed that this group of viruses can also cause severe respiratory illness in humans [4, 5].
Bats are widespread flying mammals that are distributed worldwide. About 1150 bat species have been described and 179 were reported in Brazil [6,7,8]. Numerous viruses have been reported to infect bats, and these animals are being studied as potential reservoir for several zoonotic diseases, including Coronaviruses [3, 9, 10]. The majority of Bat Coronavirus (BtCoV) have been identified in bats from Asia, Africa, and Europe [11,12,13,14,15,16,17,18,19]; however, CoV has also been reported in bats from South American countries including Brazil, Trinidad, Costa Rica, Ecuador, Panama, and Mexico [20,21,22,23,24,25].
Although there is no report of CoV pathogenesis in bats, studies suggest that SARS-CoV and MERS-CoV have a zoonotic origin in bats [26]. Therefore, studying the circulation of BtCoV in bats around the world is of great importance to detect potential sources of spillover of pathogenic viruses into the human population. Here, we investigate the presence of coronavirus in bats from two distinct regions in Brazil.
Methods
Samples
Bats were collected in São José do Rio Preto region located in southeastern Brazil at the state of São Paulo (SP) and in Barreiras region, located in the northeastern part of the country at the state of Bahia (BA). The first is a transition zone between the Atlantic Forrest biome, composed by an ecosystem of semi-deciduous forests, and the Cerrado biome characterized by savannahs. While the second region is situated within the Cerrado biome. Both regions are under tropical climates with dry winters (Tropical Aw according to Köppen-Geiger). The animals were collected in urban and peri-urban locations, using mist nets and euthanized according to the Guide for the Care and Use of Laboratory Animals of the Institute for Laboratory Animal Resources Commission on Life Sciences National Research Council [27]. Also, collections were made under environmental licenses from the Brazilian Institute of Environment and Natural Renewable Resources (IBAMA), SISBIO no.: 21707-1 (Process: 02027.001957/2006-02); 46190-1 and the project was approved by the Ethics Committee on the Use of Animals of the Institute of Biosciences, Letters and Exact Sciences (CEUA/IBILCE—Protocol 135/2016). Sex was determined based on Vizotto and Tadei, Miranda et al., and Laval [28,29,30]. Intestines, liver, and lungs were removed from bats from São José do Rio Preto—SP and intestines and liver from bats collected in Barreiras—BA. All samples were stored at − 150 °C until further processing.
RNA Extraction and cDNA Synthesis
Samples were thawed, immersed in TRIzol (Thermo Fisher Scientific), and tissues were homogenized mechanically by a polytron homogenizer (Marconi, Piracicaba, SP BR). Extraction was carried out according to manufacturer’s instructions. RNA pellet was resuspended in water treated with DEPC (Sigma Aldrich), quantified by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and stored at − 150 °C. The cDNA synthesis was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems).
Coronavirus Detection and Sequencing
The presence of Coronavirus in the organs was tested by a pan-Coronavirus NESTED-PCR as described by Chu et al. targeting the RNA-dependent RNA polymerase (RdRp), nsP12 region [31]. Positive samples were purified using DNA Clean & Concentrator (Zymo Research) and sequenced by Sanger method [32], using the set of primers from the NESTED reaction, in a 3130 XL Genetic Analyzer (Applied Biosystems). Sequences were submitted to Electropherogram Quality Analysis tool from Embrapa Genetic Resources and Biotechnology website (http://asparagin.cenargen.embrapa.br/phph/). The platform performed the quality analysis using Phred [33] and contig assembly by CAP3 [34].
Sequence ID
Sequences are identified by BtCoV, followed by the region where the host was collected (BA—Barreiras; SJRP—São José do Rio Preto), the reference of the animal (e.g., 03, 04 …) and by the identification of the organ where the viral RNA was detected (I—intestines; Li—liver; Lu—lungs). Nucleotide sequences were submitted to GenBank under the accession numbers MH974764 to MH974780, and specific identification is available in Online Resource 1.
Datasets and Phylogenetic Analysis
Two datasets were assembled to perform the phylogenetic analysis. The first one (dataset 1) was composed by all the sequences generated in this study plus sequences representing all four Coronavirus genera (Online Resource 1): α-CoV, β-CoV, γ-CoV and δ-CoV. The second dataset (Dataset 2) is composed by our sequences and sequences available in GenBank of the nsP12 region of BtCoV from the Alphacoronavirus genus (Online Resource 2). Sequences were aligned using Muscle [35, 36], nested in the Seaview 4.6.1 package [37]. The datasets were edited using BioEdit 7.2.5 package [38].
A Bayesian Markov Chain Monte Carlo (MCMC) estimation of the phylogenetic trees based on the sequences in both datasets was done using BEAST v1.8 [39]. The analysis was carried out using General Time Reversible model plus gamma distribution (GTR + G) for dataset 1 and GTR + G + invariable sites (GTR + I + G) for dataset 2, as selected by jModelTest [40], and both runs under a lognormal relaxed clock and constant population size. For dataset 1, the MCMC chain ran for 20,000,000 steps sampling every 2000 steps and for dataset 2 for 100,000,000 steps sampling every 5000 steps. The phylogenetic trees were summarized using Tree Annotator 1.8 and edited using iTOL 4.2.3 [41,42,43].
Association between host traits and tree topology was statistically tested based on the posterior set of trees (PST) generated by BEAST, using the software BaTS 0.9 [44]. The Parsimony Score (PS), Association Index (AI), and Monophyletic Clade (MC) metrics were analyzed, and values of p ≤ 0.01 were considered significant.
Images Design and Attribution
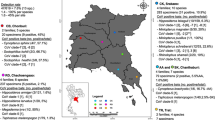
The map presented in Fig. 1 was designed by the authors using QGIS 3.2.3 based on maps of cities and states provided by Brazilian Institute of Geography and Statistics (IBGE) [45, 46]. Vectorial images of animals used in Figs. 1 and 2 were provided by vecteezy.com, www.freepik.es, and pixabay.com.

Map of Brazil showing the locations where infected bats where collected. The host families of infected animals are represented in the respective sampling location (see legend). São Paulo state—dark blue; São José do Rio Preto region—light blue; Bahia state—orange; Barreiras region—light orange

Bayesian phylogenetic tree based on a dataset of 52 nucleotide sequences of 426 bp from the nsP12 genomic region of CoV. Nodes display posterior probabilities higher than 0.7. The analysis includes 17 sequences from this study along with representatives of the four CoV genera, represented by different colors according to the legend. Images beside the tips of the tree are a graphic representation of the host. Accession numbers of each sequence is presented in Online Resource 1. Sequences generated in this study
Results
Samples and Coronavirus Detection
A total of 103 bats were collected, 64 from São José do Rio Preto region and 39 from Barreiras region. In both locations, males were more sampled than females, 60.9% and 66.7% respectively. In São José do Rio Preto, the species that was most sampled was Molossus rufus (21.9%) followed by Molossus molossus and Artibeus literatus (20.3% each). In Barreiras, Molossus molossus (28.2%) was most sampled followed by Artibeus literatus and Artibeus planirostris (23.1% each). Online Resource 3 present the information on sex, family, species, and organs of all animals collected.
The presence of CoV RNA was tested by reverse transcription NESTED-PCR specific for the nsP12 genomic region. From bats collected in São José do Rio Preto region intestines, liver, and lungs were tested. As for the animals from Barreiras region, only intestines and liver were tested, due to availability. Positive samples were sequenced and the resulting sequences were submitted to BLASTn (https://www.ncbi.nlm.nih.gov/BLAST/) in order to confirm CoV identification. The results revealed that 12 animals were infected with coronavirus, 11 from São José do Rio Preto and 1 from Barreiras. Viral RNA was detected in more than one organ in some animals totalizing 17 positive samples. Only one animal, SJRP03, tested positive in all organs. Coronavirus was mostly present in intestine samples, except for SJRP06 and SJRP10. Table 1 displays information on family, species, gender, and the organs where the virus was detected.
Considering the regions separately, the prevalence of CoV in bats from São José do Rio Preto region was 17.18% (11/64) and 2.56% (1/39) in bats from Barreiras region. Also, males were more infected than females since in São José do Rio Preto, CoV was detected in 23.1% (9/39) of males and in only 8% (2/25) of females. In Barreiras—BA, the only bat infected was male. The locations where infected bats were collected are presented in Fig. 1. It also show the families of bats that hosted CoV found in each location.
Phylogenetic Analysis
A total of 17 sequences, from 12 different animals, were generated in this study. The first analysis we performed was to determine the phylogenetic relationship of the sequences identified in both regions with representatives of the four Coronavirus genera. Based on dataset 1, composed by 52 nucleotide sequences of 426 bp (see “Methods”), we ran a Bayesian phylogenetic analysis. The phylogenetic tree indicates that the 17 sequences from this study belong to the α-CoV genus since they group in a monophyletic branch together with all sequences from this genus, with strong branch support (1.00) (Fig. 2).
We decided to analyze our sequences in the context of Bat Coronaviruses from the α-CoV genus. To this end, we assembled dataset 2 composed by 139 nucleotide sequences of 338 bp of the nsP12 region, which included all sequences from this group available in GenBank, plus our sequences. A Bayesian MCMC analysis using BEAST 1.8 was performed. The summarized tree is presented in Fig. 3. The analysis of the phylogenetic tree revealed a tendency of viruses isolated from the same host family to group in monophyletic branches with strong branch support, although there were some exceptions. In order to test for this association, we performed a statistical analysis, based on the posterior set of trees using BaTS software. According to the results, there is a positive association between the clustering in the phylogenetic tree and bat families with a p value ≤ 0.01 for PS and AI. As for MC values, all families displayed significant association except for Mormoopidae, since there is only one virus identified in this family in our analysis (Table 2) (Online Resource 4A).

Bayesian phylogenetic tree based on a dataset of 139 nucleotide sequences of 338 bp from the nsP12 genomic region of CoV . The analysis includes sequences of BtCoV from the α-CoV genus. Nodes display posterior probabilities higher than 0.7. Tips are colored according to the host family (see legend). Sequences generated in this study
We also tested for an association between phylogeny and the location where the virus was sampled. The results revealed a positive association between this trait and phylogenetic clustering based on AI and PS metrics. The MC metrics was significant for all locations with more than two representatives (Table 3) (Online Resource 4B). (Sampling site of each sequence used in the analysis is provided in Online Resource 2).
Discussion
The outbreaks of the β-CoV SARS-CoV and MERS-CoV evidenced the importance of bats as natural reservoir and as a source of spillover of Coronaviruses into the human population [26]. Understanding the diversity and the ecological aspects of these viruses in bats is important to prevent new outbreaks. Here, we investigated the presence of Coronavirus RNA in bats, living in urban and peri-urban areas, from two geographically distant regions in Brazil, São José do Rio Preto—SP and Barreiras—BA. Our results revealed the presence of Bat Coronavirus from the α-CoV genus in specimens from both regions. None of them are closely related to pathogenic viruses to humans. A prevalence of 17.2% was observed among bats sampled from the São José do Rio Preto region which is similar to the rate of 16.7% reported in bats from Hong Kong [47]. Although one study in the UK reported a comparably high BtCoV prevalence of 23% [48], reported prevalence rates usually range from 3% to 10% [13, 16, 17, 22, 24, 49,50,51,52,53], similar to our current findings in the Barreiras region (2.56%). A global study with over 12,000 samples reported a BtCoV prevalence of 8.6% [54]. The CoV prevalence among bats from the São José do Rio Preto region is therefore higher than estimated for Barreiras as well as compared to other studies. Several factors might influence the estimates, like the number of specimens sampled, host species diversity, number of sites of collection, geographic regions, among others [54]. In this study, 54.5% (6/11) of the infected specimens from São José do Rio Preto region were collected in the same site. The location is a private property known to harbor about six different species of bats in the roof of the house. The fact that these animals were collected in the same location might have influenced the high CoV prevalence in this region, since they live in close contact aiding the transmission. Another factor that might have influenced is that in addition to intestines, we also analyzed lungs and liver for the presence of CoV, while most prevalence studies only test fecal or intestine samples. Although most of the infected animals tested positive in the intestines (81.8%—9/11), in two animals, CoV was only detected in other organs (lungs and liver), raising the chances of viral detection. Other authors have also reported the presence of CoV RNA in tissues and secretions other than intestines or fecal samples, such as kidney, heart, lungs, and oropharyngeal swabs [25, 55, 56]. However, this is the first report of BtCoV detection in the liver. Since the animals presented no clinical symptoms, the significance of the detection of virus in lungs and liver remains unclear. Experimental infection of bats with CoV from the Betacoronavirus genus has been tried by Watanabe and colleagues [57]. Results revealed that the virus was only detected in the intestines. In our study, the infection of lungs and liver was not restricted to one host species or family since it was found in Molossus rufus (Molossidae), Eptesicus sp. (Vespertilionidae), and Glossophaga soricina (Phyllostomidae).
We analyzed the phylogenetic relationship of the virus identified in this study with other BtCoV from the α-CoV genus (Fig. 3). The sequences generated in our study are distributed in three distinct monophyletic branches. Most sequences, 13 out of 16, of CoV identified in bats from São José do Rio Preto grouped into a monophyletic branch with strong posterior probability (0.77). In the same branch, there are also three sequences of CoV identified in bats from the cities of Araçatuba (KC886321) and Penapolis (KT717388, KT717389), about 100 km distant from São José do Rio Preto. This cluster shares a most recent common ancestor, also with high branch support, with the virus we identified in a Molossus molossus bat in Barreiras (BtCoV_BA19I). The sequence BtCoV_SJRP44I groups apart from all other in a monophyletic branch (0.99) together with sequence KT717382. Both viruses were detected in Artibeus literatus bats. It is interesting that KT717382 was identified in Iguaçu National Park—PR, about 850 km from São José do Rio Preto—SP, in 2012 [22]. Sequences BtCoV_SJRP08I and BtCoV_SJRP22I, both from Phyllostomus discolor bats, group together in another monophyletic branch sharing a most recent common ancestor with a CoV from a P. discolor (JQ731783) bat collected in Panama [23]. Both clusters include all strains, included in the analysis, identified in the respective species. This indicates that the BtCoV identified in these hosts could be species-specific, being more closely related to the viruses identified in the same host, even if they are geographically distant. In fact, other sequences from our study, from hosts collected in the same sites, were infected with BtCoV that grouped apart from the P. discolor and A. literatus clusters (Fig. 3; Online Resource 2). An alternative explanation is that the occurrence of related strains in distant locations is a consequence of transmission during bat migration. Given the genetic distance between the strains sampled, this would obviously not be due to direct transmission but would presumably involve unsampled intermediate strains. Several bat species are known to migrate seasonally following resources, such as food. Migration studies report that small- and medium-sized bats can migrate long distances, with a maximum migration distance of 1905 km reported for a Vespertilionidae bat [58]. However, there are no studies of migration patterns for P. discolor and few for A. literatus, none of which report such long distance [59, 60]. It is therefore more likely that the close relatedness of BtCoV from P. discolor and from A. literatus is a reflection of the virus “host specificity.”
Overall, the BtCoV phylogenetic tree shows highly supported branches clustering primarily according to host family. Some families have two different clusters like Molossidae, Phyllostomidae, and Rhinolophidae while Miniopteridae and Hipposideridae present only one each. Since there are some exceptions and CoV from Vespertilionidae bats does not seem to group together, we statistically tested the hypothesis of a positive relationship between host family and phylogeny and it was confirmed. However, due to the exceptions, we decided to test for a possible association with location where the hosts were collected. For example, as previously described, most CoV from São José do Rio Preto grouped into a monophyletic and most of them belong to the Molossidae family. Still, among them there are sequences from virus identified in two animals from other families, Vespertilionidae (BtCoV_SJRP06) and Phyllostomidae (BtCoV_SJRP10). So, we statistically tested for an association between the location where the host was collected and phylogeny and there is also a positive association. Our results indicate that despite the association between BtCoV and host family seen here, the close contact between animals from different families may result in virus transmission. We conclude that both traits, host family and location, influence BtCoV phylogenetic relationship.
Here, we report the detection of Alphacoronavirus in bats from Brazil, not only in intestines but also in lungs and liver. We found a higher prevalence of BtCoV in animals collected in São José do Rio Preto—SP region, when compared to Barreiras—BA. The investigation of CoV in bats throughout the world remains important to characterize and understand the circulation of these viruses, identifying potential sources of spillover of pathogenic strains into the human population.
References
Adams MJ, Carstens EB (2012) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch Virol 157:1411–1422. https://doi.org/10.1007/s00705-012-1299-6
Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev: MMBR 69:635–664. https://doi.org/10.1128/MMBR.69.4.635-664.2005
Coleman CM, Frieman MB (2014) Coronaviruses: important emerging human pathogens. J Virol 88:5209–5212. https://doi.org/10.1128/JVI.03488-13
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. https://doi.org/10.1056/NEJMoa1211721
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. https://doi.org/10.1056/NEJMoa030747
Feldhamer GA, Drickamer LC, Vessey SH, Merritt JF, Krajewski C (2005) Mammalogy: adaptation, diversity, ecology. Johns Hopkins University Press, Baltimore
Nogueira MR, IPd L, Moratelli R, Tavares VC, Gregorin R, Peracchi AL (2014) Checklist of Brazilian bats, with comments on original records. Check List 10:808–821. https://doi.org/10.15560/10.4.808
Wilson DERD (2005) Mammal species of the world: a taxonomic and geographic reference. Johns Hopkins University Press, Baltimore
Luis AD, Hayman DT, O'Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA, Fooks AR, Rupprecht CE, Wood JL, Webb CT (2013) A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc Biol Sci 280:20122753. https://doi.org/10.1098/rspb.2012.2753rspb.2012
Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T (2006) Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev 19:531–545. https://doi.org/10.1128/CMR.00017-06
Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, Seebens A, Niedrig M, Pfefferle S, Yordanov S, Zhelyazkov L, Hermanns U, Vallo P, Lukashev A, Muller MA, Deng H, Herrler G, Drosten C (2010) Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol 84:11336–11349. https://doi.org/10.1128/JVI.00650-10JVI.00650-10
Drexler JF, Corman VM, Wegner T, Tateno AF, Zerbinati RM, Gloza-Rausch F, Seebens A, Muller MA, Drosten C (2011) Amplification of emerging viruses in a bat colony. Emerg Infect Dis 17:449–456. https://doi.org/10.3201/eid1703.100526
Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Drexler JF, Petersen N, Annan A, Grywna K, Muller M, Pfefferle S, Drosten C (2008) Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis 14:626–631. https://doi.org/10.3201/eid1404.071439
Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, Muller MA, Annan A, Vallo P, Adu-Sarkodie Y, Kruppa TF, Drosten C (2009) Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis 15:1377–1384. https://doi.org/10.3201/eid1509.090224
Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Lau SK, Woo PC, Suen KY, Yuen KY, Guan Y, Peiris JS (2005) Identification of a novel coronavirus in bats. J Virol 79:2001–2009. https://doi.org/10.1128/JVI.79.4.2001-2009.2005
Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, Peiris JS (2006) Coronaviruses in bent-winged bats (Miniopterus spp.). J Gen Virol 87:2461–2466. https://doi.org/10.1099/vir.0.82203-0
Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Li G, Dong BQ, Liu W, Cheung CL, Xu KM, Song WJ, Vijaykrishna D, Poon LL, Peiris JS, Smith GJ, Chen H, Guan Y (2006) Prevalence and genetic diversity of coronaviruses in bats from China. J Virol 80:7481–7490. https://doi.org/10.1128/JVI.00697-06
Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE (2009) Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis 15:482–485. https://doi.org/10.3201/eid1503.081013
Quan PL, Firth C, Street C, Henriquez JA, Petrosov A, Tashmukhamedova A, Hutchison SK, Egholm M, Osinubi MO, Niezgoda M, Ogunkoya AB, Briese T, Rupprecht CE, Lipkin WI (2010) Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio 1. https://doi.org/10.1128/mBio.00208-10e00208
Anthony SJ, Ojeda-Flores R, Rico-Chavez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez-Leon M, Sotomayor-Bonilla J, Aguirre AA, Avila-Flores R, Medellin RA, Goldstein T, Suzan G, Daszak P, Lipkin WI (2013) Coronaviruses in bats from Mexico. J Gen Virol 94:1028–1038. https://doi.org/10.1099/vir.0.049759-0
Goes LG, Ruvalcaba SG, Campos AA, Queiroz LH, de Carvalho C, Jerez JA, Durigon EL, Davalos LI, Dominguez SR (2013) Novel bat coronaviruses, Brazil and Mexico. Emerg Infect Dis 19:1711–1713. https://doi.org/10.3201/eid1910.130525
Goes LGB, Campos ACA, Carvalho C, Ambar G, Queiroz LH, Cruz-Neto AP, Munir M, Durigon EL (2016) Genetic diversity of bats coronaviruses in the Atlantic Forest hotspot biome, Brazil. Infect Genet Evol 44:510–513. https://doi.org/10.1016/j.meegid.2016.07.034
Corman VM, Rasche A, Diallo TD, Cottontail VM, Stocker A, Souza BF, Correa JI, Carneiro AJ, Franke CR, Nagy M, Metz M, Knornschild M, Kalko EK, Ghanem SJ, Morales KD, Salsamendi E, Spinola M, Herrler G, Voigt CC, Tschapka M, Drosten C, Drexler JF (2013) Highly diversified coronaviruses in neotropical bats. J Gen Virol 94:1984–1994. https://doi.org/10.1099/vir.0.054841-0
Asano KM, Hora AS, Scheffer KC, Fahl WO, Iamamoto K, Mori E, Brandao PE (2016) Alphacoronavirus in urban Molossidae and Phyllostomidae bats, Brazil. Virol J 13:110. https://doi.org/10.1186/s12985-016-0569-4
Carrington CV, Foster JE, Zhu HC, Zhang JX, Smith GJ, Thompson N, Auguste AJ, Ramkissoon V, Adesiyun AA, Guan Y (2008) Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerg Infect Dis 14:1890–1893. https://doi.org/10.3201/eid1412.080642
Hu B, Ge X, Wang LF, Shi Z (2015) Bat origin of human coronaviruses. Virol J 12:221. https://doi.org/10.1186/s12985-015-0422-1
NIH (2011) Guide for the care and use of laboratory animals. In: CftUotGftCaUoL (ed) Animals. National Academies Press (US), Washington
Vizotto LD, Taddei VA (1973) Chave para determinação de quirópteros brasileiros. Gráfica Francal, São José do Rio Preto
Miranda JMD, Bernardi IP, Passos FC (2006) A new species of Eptesicus (Mammalia: Chiroptera: Vespertilionidae) from the Atlantic Forest, Brazil. Zootaxa:57–68
LaVal RK (1973) A revision of the Neotropical bats of the genus Myotis. Natural History Museum, Los Angeles County , Los Angeles
Chu DK, Leung CY, Gilbert M, Joyner PH, Ng EM, Tse TM, Guan Y, Peiris JS, Poon LL (2011) Avian coronavirus in wild aquatic birds. J Virol 85:12815–12820. https://doi.org/10.1128/JVI.05838-11JVI.05838-11
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics 5:113. https://doi.org/10.1186/1471-2105-5-113
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. https://doi.org/10.1093/molbev/msp259
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium Oxford University Press
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973 doi: 10.1093/molbev/mss075mss075 [pii]
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. https://doi.org/10.1038/nmeth.2109nmeth
Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128. https://doi.org/10.1093/bioinformatics/btl529
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. https://doi.org/10.1093/nar/gkw290
Letunic I, Bork P (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. https://doi.org/10.1093/nar/gkr201
Parker J, Rambaut A, Pybus OG (2008) Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect Genet Evol 8:239–246. https://doi.org/10.1016/j.meegid.2007.08.001
QGIS (2018) QGIS Development Team - QGIS Geographic Information System. Open Source Geospatial Foundation Project
IBGE (2007) IBGE - Instituto Brasileiro de Geografia e Estatística - bases cartográficas. https://mapas.ibge.gov.br/bases-e-referenciais/bases-cartograficas.html. Accessed 2018
Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, Xu H, Guo R, Chan KH, Zheng BJ, Yuen KY (2007) Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 367:428–439. https://doi.org/10.1016/j.virol.2007.06.009
August TA, Mathews F, Nunn MA (2012) Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis 12:530–533. https://doi.org/10.1089/vbz.2011.0829
Goffard A, Demanche C, Arthur L, Pincon C, Michaux J, Dubuisson J (2015) Alphacoronaviruses detected in French bats are phylogeographically linked to coronaviruses of European bats. Viruses 7:6279–6290 doi: 10.3390/v7122937v7122937 [pii]
Osborne C, Cryan PM, O'Shea TJ, Oko LM, Ndaluka C, Calisher CH, Berglund AD, Klavetter ML, Bowen RA, Holmes KV, Dominguez SR (2011) Alphacoronaviruses in New World bats: prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS One 6:e19156. https://doi.org/10.1371/journal.pone.0019156PONE-D-11-00052
Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB (2013) Detection of coronaviruses in bats of various species in Italy. Viruses 5:2679–2689. https://doi.org/10.3390/v5112679v5112679
Dominguez SR, O'Shea TJ, Oko LM, Holmes KV (2007) Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis 13:1295–1300. https://doi.org/10.3201/eid1309.070491
Wacharapluesadee S, Duengkae P, Rodpan A, Kaewpom T, Maneeorn P, Kanchanasaka B, Yingsakmongkon S, Sittidetboripat N, Chareesaen C, Khlangsap N, Pidthong A, Leadprathom K, Ghai S, Epstein JH, Daszak P, Olival KJ, Blair PJ, Callahan MV, Hemachudha T (2015) Diversity of coronavirus in bats from Eastern Thailand. Virol J 12:57. https://doi.org/10.1186/s12985-015-0289-110.1186/s12985-015-0289-1
Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, Hicks AL, Joly DO, Wolfe ND, Daszak P, Karesh W, Lipkin WI, Morse SS, Mazet JAK, Goldstein T (2017) Global patterns in coronavirus diversity. Virus Evol 3:vex012. https://doi.org/10.1093/ve/
Luo Y, Li B, Jiang RD, Hu BJ, Luo DS, Zhu GJ, Hu B, Liu HZ, Zhang YZ, Yang XL, Shi ZL (2018) Longitudinal surveillance of Betacoronaviruses in fruit bats in Yunnan Province, China during 2009-2016. Virol Sin 33:87–95. https://doi.org/10.1007/s12250-018-0017-2
Hu D, Zhu C, Wang Y, Ai L, Yang L, Ye F, Ding C, Chen J, He B, Zhu J, Qian H, Xu W, Feng Y, Tan W, Wang C (2017) Virome analysis for identification of novel mammalian viruses in bats from Southeast China. Sci Rep 7:10917. https://doi.org/10.1038/s41598-017-11384-w
Watanabe S, Masangkay JS, Nagata N, Morikawa S, Mizutani T, Fukushi S, Alviola P, Omatsu T, Ueda N, Iha K, Taniguchi S, Fujii H, Tsuda S, Endoh M, Kato K, Tohya Y, Kyuwa S, Yoshikawa Y, Akashi H (2010) Bat coronaviruses and experimental infection of bats, the Philippines. Emerg Infect Dis 16:1217–1223. https://doi.org/10.3201/eid1608.100208
Hutterer RIT, Meyer-Cords C, Rodrigues L (2005) Bat migrations in Europe: a review of banding data and literature. Federal Agency for Nature Conservation, Bonn
Adams RA, Pedersen SC 2013 Bat evolution, ecology, and conservation
Arnone IS, Trajano E, Pulchério-Leite A, Passos FC (2016) Long-distance movement by a great fruit-eating bat, Artibeus lituratus (Olfers, 1818), in southeastern Brazil (Chiroptera, Phyllostomidae): evidence for migration in Neotropical bats? Biota Neotropica 16. https://doi.org/10.1590/1676-0611-BN-2015-0026
Funding
This work was funded by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico). Grant numbers: FAPESP 2015/09704-6 and CNPq 165802/2015-4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This research project was approved by the Ethics Committee on the Use of Animals of the Institute of Biosciences, Letters and Exact Sciences (CEUA/IBILCE-Protocol 135/2016) (Information stated in the Methods section under the headline Samples).
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Sequence data
GenBank Accession Numbers from MH974764 to MH974780
Electronic supplementary material
Online Resource 1:
GenBank accession number of the sequences from Dataset 1, including the sequences from this study. (XLSX 9 kb)
Online Resource 2:
GenBank accession number, host family and species, date and place of host sampling of the sequences from Dataset 2. (XLSX 16 kb)
Online Resource 3:
Information on sex, family, species and organs of all animals collected in this study. (XLSX 13 kb)
Online Resource 4:
Results of statistical test of association between traits and phylogeny done on BaTS software. A. Statistical analysis of the association of host family and phylogenetic clustering; B. Statistical analysis of the association of location and phylogenetic clustering. Parsimony Score (PS); Association Index (AI); Monophyletic Clade (MC). Values of p ≤ 0.01 were considered significant. (PDF 976 kb)
Rights and permissions
About this article

Cite this article
Bittar, C., Machado, R.R.G., Comelis, M.T. et al. Alphacoronavirus Detection in Lungs, Liver, and Intestines of Bats from Brazil. Microb Ecol 79, 203–212 (2020). https://doi.org/10.1007/s00248-019-01391-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-019-01391-x