
Abstract
Dilated cardiomyopathy (DCM) is characterized by left or bilateral ventricular dilation and systolic dysfunction without rational conditions, which can lead to progressive heart failure and sudden cardiac death. Most of the pathogenic genes have been reported in adult population by locus mapping in familial cases and animal model studies. However, it still remains challenging to decipher the role of genetics in the etiology of pediatric DCM. We applied whole-exome sequencing (WES) for 30 sporadic pediatric DCM subjects and 100 non-DCM local controls. We identified the pathogenic mutations using bioinformatics tools based on genomic strategies synergistically and confirmed mutations by Sanger sequencing. We identified compound heterozygous nonsense mutations in DSP (c.3799C > T, p.R1267X; c.4444G > T, p.E1482X). In sporadic cases, the two heterozygous mutations in XIRP2 were identified. Then we performed an exome-wide association study with 30 case and 100 control subjects. Interestingly, we could not identify TTN truncating variants in all cases. Collectively, we observed a significant risk signal between carriers of TTN deleterious missense variants and DCM risk (odds ratio 4.0, 95% confidence interval 1.1–22.2, p = 3.12 × 10−2). Our observations expanded the spectrum of mutations and were valuable in the pre- and postnatal screening and genetic diagnosis for DCM.



Similar content being viewed by others


References
Taylor MR, Carniel E, Mestroni L (2006) Cardiomyopathy, familial dilated. Orphanet J Rare Dis 1:27. https://doi.org/10.1186/1750-1172-1-27
Hershberger RE, Siegfried JD (2011) Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 57(16):1641–1649. https://doi.org/10.1016/j.jacc.2011.01.015
Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD (1992) The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 326(2):77–82. https://doi.org/10.1056/NEJM199201093260201
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S, Alastalo TP, Koskenvuo JW, Helio T (2015) Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 36(34):2327–2337. https://doi.org/10.1093/eurheartj/ehv253
Merlo M, Pivetta A, Pinamonti B, Stolfo D, Zecchin M, Barbati G, Di Lenarda A, Sinagra G (2014) Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: changing mortality over the last 30 years. Eur J Heart Fail 16(3):317–324. https://doi.org/10.1002/ejhf.16
Jefferies JL, Towbin JA (2010) Dilated cardiomyopathy. Lancet 375(9716):752–762. https://doi.org/10.1016/S0140-6736(09)62023-7
Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, Ramani F, Di Lenarda A, Mestroni L, Sinagra G (2016) Natural history of dilated cardiomyopathy in children. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.003450
Weintraub RG, Alexander PMA (2017) Outcomes in pediatric dilated cardiomyopathy: Quo Vadis? J Am Coll Cardiol 70(21):2674–2676. https://doi.org/10.1016/j.jacc.2017.09.1100
Long PA, Larsen BT, Evans JM, Olson TM (2015) Exome sequencing identifies pathogenic and modifier mutations in a child with sporadic dilated cardiomyopathy. J Am Heart Assoc. https://doi.org/10.1161/JAHA.115.002443
Long PA, Zimmermann MT, Kim M, Evans JM, Xu X, Olson TM (2016) De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum Genet 135(8):909–917. https://doi.org/10.1007/s00439-016-1685-3
Simpson S, Edwards J, Ferguson-Mignan TF, Cobb M, Mongan NP, Rutland CS (2015) genetics of human and canine dilated cardiomyopathy. Int J Genomics 2015:204823. https://doi.org/10.1155/2015/204823
Almomani R, Verhagen JM, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, van der Zwaag PA, Frohn-Mulder IM, Bertoli-Avella AM, Boven LG, van Slegtenhorst MA, van der Smagt JJ, van IJcken WF, Timmer B, van Stuijvenberg M, Verdijk RM, Saffitz JE, du Plessis FA, Michels M, Hofstra RM, Sinke RJ, van Tintelen JP, Wessels MW, Jongbloed JD, van de Laar IM (2016) Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol 67(5):515–525. https://doi.org/10.1016/j.jacc.2015.10.093
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29(2):270–276. https://doi.org/10.1093/eurheartj/ehm342
Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MR (2014) Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol 37(1–2):13–18. https://doi.org/10.1016/j.ppedcard.2014.10.003
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padron-Barthe L, Duro-Aguado I, Jimenez-Jaimez J, Hidalgo-Olivares VM, Garcia-Campo E, Lanzillo C, Suarez-Mier MP, Yonath H, Marcos-Alonso S, Ochoa JP, Santome JL, Garcia-Giustiniani D, Rodriguez-Garrido JL, Dominguez F, Merlo M, Palomino J, Pena ML, Trujillo JP, Martin-Vila A, Stolfo D, Molina P, Lara-Pezzi E, Calvo-Iglesias FE, Nof E, Calo L, Barriales-Villa R, Gimeno-Blanes JR, Arad M, Garcia-Pavia P, Monserrat L (2016) Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 68(22):2440–2451. https://doi.org/10.1016/j.jacc.2016.09.927
Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, Sweet ME, Slavov DB, Stafford N, Zeller MJ, Alnefaie R, Rowland TJ, Brun F, Jones KL, Gowan K, Mestroni L, Garrity DM, Taylor MR (2016) FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Trans Sci 1(5):344–359. https://doi.org/10.1016/j.jacbts.2016.05.004
Al-Hassnan ZN, Shinwari ZM, Wakil SM, Tulbah S, Mohammed S, Rahbeeni Z, Alghamdi M, Rababh M, Colak D, Kaya N, Al-Fayyadh M, Alburaiki J (2016) A substitution mutation in cardiac ubiquitin ligase, FBXO32, is associated with an autosomal recessive form of dilated cardiomyopathy. BMC Med Genet 17:3. https://doi.org/10.1186/s12881-016-0267-5
Li J, Yang S, Pu Z, Dai J, Jiang T, Du F, Jiang Z, Cheng Y, Dai G, Wang J, Qi J, Cao L, Cheng X, Ren C, Li X, Qin Y (2017) Whole-exome sequencing identifies SGCD and ACVRL1 mutations associated with total anomalous pulmonary venous return (TAPVR) in Chinese population. Oncotarget 8(17):27812–27819. https://doi.org/10.18632/oncotarget.15434
Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H, Sahin SE, Dupont E, Severs NJ, Leigh IM, Yuksel-Apak M, Kelsell DP, Wollnik B (2006) Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet 43(2):e5. https://doi.org/10.1136/jmg.2005.032904
Sanna-Cherchi S, Khan K, Westland R, Krithivasan P, Fievet L, Rasouly HM, Ionita-Laza I, Capone VP, Fasel DA, Kiryluk K, Kamalakaran S, Bodria M, Otto EA, Sampson MG, Gillies CE, Vega-Warner V, Vukojevic K, Pediaditakis I, Makar GS, Mitrotti A, Verbitsky M, Martino J, Liu Q, Na YJ, Goj V, Ardissino G, Gigante M, Gesualdo L, Janezcko M, Zaniew M, Mendelsohn CL, Shril S, Hildebrandt F, van Wijk JAE, Arapovic A, Saraga M, Allegri L, Izzi C, Scolari F, Tasic V, Ghiggeri GM, Latos-Bielenska A, Materna-Kiryluk A, Mane S, Goldstein DB, Lifton RP, Katsanis N, Davis EE, Gharavi AG (2017) Exome-wide association study identifies GREB1L mutations in congenital kidney malformations. Am J Hum Genet 101(6):1034. https://doi.org/10.1016/j.ajhg.2017.11.003
Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH (2014) The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 16(8):601–608. https://doi.org/10.1038/gim.2013.204
Kazerounian S, Uitto J, Aho S (2002) Unique role for the periplakin tail in intermediate filament association: specific binding to keratin 8 and vimentin. Exp Dermatol 11(5):428–438
Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA (2002) Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 118(2):232–238. https://doi.org/10.1046/j.0022-202x.2001.01664.x
Herbert Pratt C, Potter CS, Fairfield H, Reinholdt LG, Bergstrom DE, Harris BS, Greenstein I, Dadras SS, Liang BT, Schofield PN, Sundberg JP (2015) Dsp rul: a spontaneous mouse mutation in desmoplakin as a model of Carvajal-Huerta syndrome. Exp Mol Pathol 98(2):164–172. https://doi.org/10.1016/j.yexmp.2015.01.015
Wang DZ, Hu X, Lin JL, Kitten GT, Solursh M, Lin JJ (1996) Differential displaying of mRNAs from the atrioventricular region of developing chicken hearts at stages 15 and 21. Front Biosci 1:a1–15
McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, Ohashi K, Kielbasa OM, Mathew M, Ewen EP, Walsh K, Gavras H, Naya FJ (2010) Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res 106(5):952–960. https://doi.org/10.1161/CIRCRESAHA.109.209007
Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson-Wagner EA, Scholz TD, Yang B, Lin JJ (2010) Essential roles of an intercalated disc protein, mXinbeta, in postnatal heart growth and survival. Circ Res 106(9):1468–1478. https://doi.org/10.1161/CIRCRESAHA.109.212787
Akinrinade O, Alastalo TP, Koskenvuo JW (2016) Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet 90(1):49–54. https://doi.org/10.1111/cge.12741
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366(7):619–628. https://doi.org/10.1056/NEJMoa1110186
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O’Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Trans Med 7(270):27. https://doi.org/10.1126/scitranslmed.3010134
Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H, Mestroni L (2016) A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med 3:21. https://doi.org/10.3389/fcvm.2016.00021
Xu X, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, Fishman MC (2002) Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet 30(2):205–209. https://doi.org/10.1038/ng816
Levitas A, Muhammad E, Harel G, Saada A, Caspi VC, Manor E, Beck JC, Sheffield V, Parvari R (2010) Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Human Genet 18(10):1160–1165. https://doi.org/10.1038/ejhg.2010.83
Acknowledgements
The authors thank all the participating families and patients for their contributions.
Funding
This work was supported by the National Natural Science Foundation of China (81670284), Nanjing Science and Technology Project (201715057), Nanjing Medical Science and technique Development Foundation, Nanjing Department of Health (QRX17024, ZKX13037). The authors confirm that the funders had no influence over the study design, content of the article, or selection of this journal.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics Approval and Consent to Participate
The ethical committee of the Children’s Hospital of Nanjing Medical University approved of this study for human research, and written informed consents were obtained from all DCM patients and legal guardians.
Consent for Publication
Permissions were received from the patients whose photographs are to be published.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
246_2019_2096_MOESM1_ESM.docx
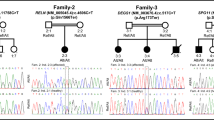
Supplementary material 1 (DOCX 323 kb). Fig. A1: The workflow of DCM genetic analysis. Abbreviations: MAF, minor allele frequency; UTR, untranslated region; Inheritance models: Autosomal recessive, De novo and Compound heterozygotes manner. Fig. A2: Sanger chromatograms of XIRP2 mutations
Rights and permissions
About this article

Cite this article
Dai, G., Pu, Z., Cheng, X. et al. Whole-Exome Sequencing Reveals Novel Genetic Variation for Dilated Cardiomyopathy in Pediatric Chinese Patients. Pediatr Cardiol 40, 950–957 (2019). https://doi.org/10.1007/s00246-019-02096-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-019-02096-1