
Abstract
Purpose
The aim of this Phase 1, open-label, positron emission tomography (PET) study was to determine the degree of striatal D2/D3 receptor occupancy induced by the serotonin–dopamine activity modulator, brexpiprazole, at different single dose levels in the range 0.25–6 mg.
Methods
Occupancy was measured at 4 and 23.5 h post-dose using the D2/D3 receptor antagonist [11C]raclopride. The pharmacokinetics, safety and tolerability of brexpiprazole were assessed in parallel.
Results
Fifteen healthy participants were enrolled (mean age 33.9 years; 93.3% male). Mean D2/D3 receptor occupancy in the putamen and caudate nucleus increased with brexpiprazole dose, leveled out at 77–88% with brexpiprazole 5 mg and 6 mg at 4 h post-dose, and remained at a similar level at 23.5 h post-dose (74–83%). Estimates of maximum obtainable receptor occupancy (Omax) were 89.2% for the putamen and 95.4% for the caudate nucleus; plasma concentrations predicted to provide 50% of Omax (EC50) were 8.13 ng/mL and 7.75 ng/mL, respectively. Brexpiprazole area under the concentration–time curve (AUC∞) and maximum plasma concentration (Cmax) increased approximately proportional to dose. No notable subjective or objective adverse effects were observed in this cohort.
Conclusion
By extrapolating the observed single-dose D2/D3 receptor occupancy data in healthy participants, multiple doses of brexpiprazole 2 mg/day and above are expected to result in an efficacious brexpiprazole concentration, consistent with clinically active doses in schizophrenia and major depressive disorder.
Trial registration
ClinicalTrials.gov NCT00805454 December 9, 2008.
Similar content being viewed by others


Introduction
Brexpiprazole (7-{4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy}quinolin-2(1H)-one; chemical structure shown in Fig. S1 in the Online Resource) is a serotonin–dopamine activity modulator that acts as a partial agonist at serotonin 5-HT1A (Ki = 0.12 nM) and dopamine D2 (Ki = 0.30 nM) receptors, and as an antagonist at serotonin 5-HT2A (Ki = 0.47 nM) and noradrenaline α1B/α2C (Ki = 0.17/0.59 nM) receptors, all with subnanomolar affinity [1]. Brexpiprazole also has nanomolar affinity (Ki < 5 nM) for dopamine D3 receptors (as a partial agonist; Ki = 1.1 nM), serotonin 5-HT2B/5-HT7 receptors (as an antagonist; Ki = 1.9/3.7 nM), and noradrenaline α1A/α1D receptors (Ki = 3.8/2.6 nM) [1]. In comparison to brexpiprazole, the dopamine receptor partial agonist, aripiprazole, has similar affinity at D2 (Ki = 0.87 nM) and D3 (Ki = 1.6 nM) receptors, but ten times lower affinity for 5-HT1A (Ki = 1.3 nM) and 5-HT2A (Ki = 4.7 nM) receptors [1].
The efficacy and safety of brexpiprazole as monotherapy in schizophrenia and as adjunctive therapy to antidepressant treatment in major depressive disorder (MDD) have been demonstrated in several randomized, double-blind, placebo-controlled studies [2,3,4,5,6,7,8]. Brexpiprazole is approved in various countries and regions for the treatment of schizophrenia and the adjunctive treatment of MDD in adults.
Positron emission tomography (PET) studies of typical and atypical antipsychotics that act as D2 receptor antagonists have shown that the degree of striatal D2/D3 receptor occupancy provides evidence of target engagement and the window for clinical improvement in schizophrenia, and also the minimal threshold for predicting side effects such as akathisia, hyperprolactinemia, and extrapyramidal symptoms [9,10,11]. According to PET studies of multiple different antipsychotic drugs at conventional doses, the optimal striatal D2/D3 receptor occupancy by an antagonist to achieve a clinical effect in schizophrenia with a low incidence of side effects is in the region of 65–80% [9, 11, 12]. Aripiprazole exhibits a higher D2/D3 receptor occupancy at clinically relevant doses (closer to 90% or 95%), without increasing the risk of extrapyramidal symptoms, because it acts as a partial agonist at these dopamine receptors [12, 13]; in other words, even though the occupancy with aripiprazole is greater than that with antagonists, the functional antagonism may be less due to its partial agonist activity [14].
The aim of this open-label PET study – the first human D2/D3 receptor target engagement study to be conducted with brexpiprazole – was to determine the degree of striatal D2/D3 receptor occupancy induced by brexpiprazole at different single dose levels, using the D2/D3 receptor antagonist [11C]raclopride. Pharmacokinetics, safety, and tolerability were also assessed.
Materials and methods
This study (ClinicalTrials.gov identifier: NCT00805454) was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice Guideline [15] and the Declaration of Helsinki [16]. The study protocol was approved by the Johns Hopkins School of Medicine Institutional Review Board, and written informed consent was obtained from all individual participants included in the study. The study started on November 25, 2008, and was completed on July 17, 2009.
Participants
Participants were enrolled at two centers in the United States: Johns Hopkins Hospital (Baltimore) and a local contract research organization (SNBL Clinical Pharmacology Center, Baltimore). The key inclusion criteria were that participants (male or female of non-child-bearing potential) must be healthy, aged 18–45 years, and have a body mass index (BMI) of 19–32 kg/m2. Key exclusion criteria were a positive alcohol or drug screen, having smoked within 2 months, having used any medication or vitamin supplement in the 14 days prior to dosing (30 days for antibiotics), and any previous exposure to antipsychotics. Participants were also excluded if they had a history of serious medical, neurological, and mental disorders. Participants were screened for the presence of abnormalities on a comprehensive metabolic panel, complete blood counts, liver and renal function tests, an electrocardiogram, urinalysis, and a structural magnetic resonance imaging (MRI) scan of the brain (described below). As an exploratory study, no formal sample size calculation was performed; up to 25 participants were planned for enrollment.
Study design
This was a Phase 1, open-label, single-dose, PET and pharmacokinetic study (study design shown in Fig. S2 in the Online Resource). The primary objective was to determine the degree of striatal D2/D3 receptor occupancy induced by brexpiprazole at different single dose levels. Secondary objectives were to determine the pharmacokinetics of brexpiprazole and its main metabolite following a single oral dose, and to determine the safety and tolerability of brexpiprazole following a single oral dose. Using observed data from various single-dose administrations, predictions of D2/D3 receptor occupancy at expected multiple-dose plasma concentrations were also made.
Participants were screened from day − 29 to day − 2, and eligible participants were checked in to the clinic on day − 1, where they remained until day 7 (except when transported to the PET center for post-dose PET scans, described below).
Participants received a single dose of brexpiprazole, in the range of 0.25–6 mg, on day 1. The first two participants received 0.5 mg; the dose for each subsequent two participants could be increased, repeated, or decreased based on results from the previous two participants. Brexpiprazole was taken with water following a minimum 2 h fast. Participants took no medications other than brexpiprazole during the course of the study. Participants were prohibited to consume grapefruit, grapefruit juice, Seville oranges, or Seville orange juice in the 72 h prior to dosing. In addition, participants were not allowed to consume alcohol, or food and beverages containing methylxanthines (caffeinated coffee, caffeinated tea, caffeinated soda, and chocolate), in the 72 h prior to dosing.
On day 7 (or earlier in the event of early termination), after collecting blood samples and completing standard safety assessments, participants were discharged from the clinic. On day 10, participants returned to the clinic for a follow-up safety evaluation. Participants were contacted by telephone 30 days post-dose for further safety follow-up.
PET and MRI procedures
PET and MRI procedures were carried out at the Johns Hopkins Hospital. PET was obtained with a high-resolution research tomography (HRRT) scanner [17, 18], with 207 image slices of approximately 1.2 mm thickness in 2.4–2.8 mm transaxial spatial resolution. Three PET scans were performed per participant: at baseline (i.e., during the period from day − 29 to day − 1), at 4 h post-dose (day 1), and at 23.5 h post-dose (day 2). These timings were to reflect baseline, maximum plasma concentration (Cmax), and post-Cmax time points, respectively. In each PET session, the participant had an indwelling intravenous (IV) catheter inserted, and was positioned on the scanner bed using a thermoplastic mask to reduce head motion. A transmission scan was performed using a 137Cs source to correct for the effects of attenuation. Dynamic PET started with a bolus IV injection of the selective dopamine D2/D3 receptor antagonist, [11C]raclopride, and lasted for 90 min. The PET radiopharmaceutical, [11C]raclopride, was produced in the Johns Hopkins radiochemistry facility in high specific activity using methods previously published [19]. The dynamic PET scans were reconstructed into 30 frames (four 15-s, four 30-s, three 1-min, two 2-min, five 4-min, and twelve 5-min frames), correcting for attenuation, scatter, and dead time. Each PET frame consisted of 256 by 256 by 207 (axial) voxels (1.2 mm cubic), decay corrected to the tracer injection time.
Structural spoiled gradient recalled (SPGR) MRI was performed at screening, using a 3.0 T SIGNA scanner (GE Healthcare, Chicago, IL, USA), to rule out cerebral lesions, to define volumes of interest and to perform PET-to-MRI coregistration and spatial normalization.
PET analysis
Volumes of interest (VOIs) were defined on individual participant MRI images for the putamen, caudate nucleus, and cerebellum gray matter, and transferred to the PET space using PET-to-MRI coregistration parameters from SPM5’s coregistration module [20]. Then, VOIs were applied to PET frames to generate time–activity curves. The primary outcome variable, binding potential relative to non-displaceable binding (BPND) [21], was determined by the multilinear reference method with two parameters (MRTM2) [22], with the cerebellum as the reference region. Estimates of dopamine D2/D3 receptor occupancy by brexpiprazole were obtained as follows: occupancy (%) = (BPNDB − BPNDD) / BPNDB × 100, where superscripts B and D indicate BPND at baseline and post-dose scans, respectively. Plots of occupancy (pooled over participants separately for the putamen and caudate nucleus) versus brexpiprazole plasma concentration (Cp) were fitted by the following saturation equation: occupancy = Omax × Cp / (Cp + EC50), where Omax is the maximum obtainable D2/D3 receptor occupancy, and EC50 is the plasma concentration predicted to provide 50% of Omax. In the fitting procedures, Omax was 1) fixed at the theoretical maximum (100%), and 2) estimated in addition to EC50; the Omax associated with a lower Akaike information criterion [23] was taken as optimal. Functional maps of BPND were generated by voxel-wise MRTM2 to visually confirm dose-dependent changes of BPND from the baseline to post-dose scans. For this purpose, functional maps were spatially normalized to a standard brain by applying parameters of PET-to-MRI coregistration and spatial normalization of MRI in one step [24], and smoothed by a Gaussian kernel of 8 mm full-width at half-maximum to compensate for insufficiency of spatial normalization.
Pharmacokinetic procedures and analysis
Full details of the pharmacokinetic procedures and analysis are presented in the Online Resource. In summary, the plasma concentration of brexpiprazole and its main metabolite, DM-3411, were determined using high-performance liquid chromatography with tandem mass spectrometric detection (HPLC-MS/MS). The following pharmacokinetic parameters were determined: Cmax, time to Cmax (tmax), area under the concentration–time curve to the last observable concentration at time t (AUCt) and to infinity (AUC∞), terminal-phase elimination half-life (t½,z), and apparent clearance of drug from plasma after extravascular administration (CL/F).
Safety assessments and analysis
The safety of brexpiprazole was evaluated at both centers by the reporting of adverse events (AEs) and by standard safety assessments at various time points, including physical examination, electrocardiograms, vital signs, and clinical laboratory tests. All participants who took a dose of brexpiprazole were included in the safety analysis.
Results
Disposition and demographics
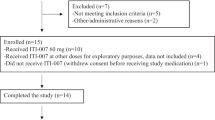
A total of 15 healthy volunteer participants were enrolled, all of whom took a single dose of oral brexpiprazole (study flow shown in Fig. S3 in the Online Resource). Two participants each received brexpiprazole 0.25 mg, 0.5 mg, 1 mg, 2 mg, 4 mg, and 6 mg; three participants received brexpiprazole 5 mg. Fourteen participants (93.3%) completed the trial; one participant (5 mg dose) was withdrawn by the investigator due to problems with the PET scanner.
In the enrolled sample, the baseline mean (standard deviation) age was 33.9 (6.8) years, weight was 81.7 (13.4) kg, and BMI was 26.6 (4.2) kg/m2; 93.3% of participants (14/15) were male, 86.7% (13/15) were Black/African American, and 13.3% (2/15) were Asian.
Dopamine D2/D3 receptor occupancy
Trans-axial BPND images at a level showing the putamen and caudate nucleus are shown in Fig. 1, at baseline and for low and high brexpiprazole plasma concentrations. At 4 h post-dose, the mean D2/D3 receptor occupancy in the putamen and caudate nucleus increased with brexpiprazole dose (from < 20% with brexpiprazole 0.25 mg), leveling out at 77–88% with brexpiprazole 5 mg and 6 mg (Fig. 2). A similar pattern of D2/D3 receptor occupancy was observed at 23.5 h post-dose (Fig. 2), leveling out at 74–83% with brexpiprazole 5 mg and 6 mg. Single doses of 2–4 mg resulted in D2/D3 receptor occupancies of 59–75% at 4 h and 53–74% at 23.5 h post-dose.

Mean trans-axial a MRI (n = 13) and b–d PET BPND images with [11C]raclopride, at a level showing the putamen and caudate nucleus. Panel b shows mean BPND at baseline (n = 13). Panel c shows mean BPND for arbitrarily selected ‘low’ brexpiprazole plasma concentration (< 20 ng/mL; n = 13). Panel d shows mean BPND for arbitrarily selected ‘high’ brexpiprazole plasma concentration (> 20 ng/mL; n = 12). BPND binding potential relative to non-displaceable binding, C caudate nucleus, MRI magnetic resonance imaging, P putamen, PET positron emission tomography

Mean dopamine D2/D3 receptor occupancy at 4 h and 23.5 h following a single oral dose of brexpiprazole (n = 2 per dose group except where there is no error bar, n = 1), as a function of dose. Error bars represent standard deviation
Estimates of Omax and EC50 and model prediction curves were similar at 4 and 23.5 h post-dose, and so the datasets were combined. Plots of D2/D3 receptor occupancy as a function of brexpiprazole plasma concentration are shown in Fig. 3a. Plasma concentrations of 60 ng/mL corresponded to approximately 80–90% D2/D3 receptor occupancy in the putamen and caudate nucleus. Based on the combined data, estimates of Omax and EC50 were 89.2% and 8.13 ng/mL, respectively, in the putamen, and 95.4% and 7.75 ng/mL, respectively, in the caudate nucleus. Fixing Omax at 100%, estimates of EC50 were 11.5 ng/mL in the putamen and 8.99 ng/mL in the caudate nucleus. Akaike information criterion values were lower for (and thus supported using) estimated Omax in the putamen, and fixed Omax (100%) in the caudate nucleus.

Dopamine D2/D3 receptor occupancy versus brexpiprazole plasma concentration a following a single oral dose (n = 12), and b extrapolated to expected multiple-dose concentrations. The mean plasma concentration across four time points spanning the duration of PET data acquisition was used for each data point. Omax maximum obtainable receptor occupancy, PET positron emission tomography
Extrapolations of the predicted D2/D3 receptor occupancy beyond a brexpiprazole plasma concentration of 60 ng/mL based on estimates of Omax and EC50 are shown in Fig. 3b.
Pharmacokinetic profile
Mean brexpiprazole and DM-3411 plasma concentrations versus time following administration of a single, oral dose of brexpiprazole are shown in Fig. 4. A summary of the pharmacokinetic parameters of brexpiprazole and DM-3411 are shown in Table 1. Brexpiprazole AUC∞ and Cmax increased approximately proportional to dose.

Mean a brexpiprazole and b its main metabolite, DM-3411, plasma concentration over time following a single oral dose of brexpiprazole (n = 2 per dose group)
Safety
Overall, 11/15 participants (73.3%) reported at least one treatment-emergent adverse event (TEAE). The most commonly reported TEAEs were postural orthostatic tachycardia syndrome (4 participants, 26.7%), nausea (3 participants, 20.0%), and headache (3 participants, 20.0%). A list of TEAEs occurring in ≥ 2 participants by dose is presented in Table S1 (Online Resource). The majority of TEAEs occurred at the higher brexpiprazole doses (4 mg, 5 mg, and 6 mg). All TEAEs were mild or moderate in severity. There were no discontinuations from the study due to TEAEs or due to electrocardiogram, vital sign, or clinical laboratory abnormalities.
Discussion
In this open-label study in healthy participants, single-dose administration of oral brexpiprazole in the dose range of 0.25–6 mg resulted in dose-dependent increases in striatal D2/D3 receptor occupancy, and dose-dependent increases in Cmax and AUC∞. This result aligns with a Japanese pharmacokinetics study in patients with schizophrenia, in which multiple-dose administration of oral brexpiprazole (1, 4, and 6 mg/day) increased Cmax and AUC in a dose-dependent manner; steady state, based on mean brexpiprazole plasma concentrations in pre-dosing samples, was estimated to be reached after 10 days [25]. Furthermore, a recent PET study by Girgis et al. in the United States demonstrated robust dose-dependent occupancy at D2 receptors in 12 patients with schizophrenia following 10 days’ administration of brexpiprazole (1 or 4 mg/day) [26].
The recommended dose range for brexpiprazole in schizophrenia is 2–4 mg/day [27]. In the present study, single doses of 2–4 mg resulted in D2/D3 receptor occupancies of 59–75% at 4 h in the putamen and caudate nucleus, and 53–74% at 23.5 h. By extrapolating the observed single-dose data, multiple doses of 2 mg/day and above are expected to result in D2/D3 receptor occupancies of above 80%. The Girgis et al. study found a slightly lower steady-state D2 receptor occupancy (62–80% with the 4 mg dose), attributed to the choice of radioligand: [11C]-(+)-PHNO, a D3-preferring D2/D3 receptor agonist, rather than [11C]raclopride, a D2-preferring D2/D3 receptor antagonist (Kd = 1.43 nM for D2A receptors and 1.58 nM for D3 receptors) [26, 28]. Whereas the brexpiprazole 2 mg and 4 mg doses have similar efficacy on schizophrenia symptoms when averaged across clinical trial samples [29], individual patients may benefit from different doses, and this individual response will be influenced by receptor occupancy.
In the present study, estimates of Omax and EC50 were 89.2% and 8.13 ng/mL, respectively, for the putamen, and 95.4% and 7.75 ng/mL for the caudate nucleus. These values are similar to those of aripiprazole in patients with schizophrenia, as observed using the D2/D3 receptor antagonist, [18F]fallypride: 92% and 10 ng/mL for the putamen, and 92% and 9 ng/mL for the caudate nucleus [30].
Brexpiprazole’s D2/D3 receptor occupancy was comparable at 4 h and at 23.5 h post-dose (of note, the half-life of brexpiprazole is approximately 91 h [27], and the half-life of aripiprazole is approximately 75 h [31]). This stable occupancy over 24 h supports the observed efficacy of once-daily dosing of brexpiprazole in schizophrenia and MDD [2,3,4,5,6,7,8]. The time to maximum plasma concentration for brexpiprazole 2–4 mg was 5–6 h in this study, and is reported as within 4 h in the prescribing information [27], comparable to that of oral aripiprazole tablets (3–5 h) [31].
A single dose of brexpiprazole was generally safe and well tolerated at levels up to approximately 90% striatal D2/D3 receptor occupancy in this sample of healthy participants. Overall in the schizophrenia clinical development program, brexpiprazole 2–4 mg/day was well tolerated, and no TEAEs had an incidence of ≥ 5% and twice that of placebo in short-term clinical efficacy studies [32]. Similarly, adjunctive brexpiprazole 1–3 mg/day was well tolerated in the MDD clinical development program, with only increased weight and akathisia occurring with an incidence of ≥ 5% and twice that of placebo in short-term clinical efficacy studies; these AEs generally did not lead to discontinuation [33]. The recommended dose of adjunctive brexpiprazole in MDD (2–3 mg/day) factors in CYP considerations; concomitant use of brexpiprazole with strong CYP2D6 inhibitors (e.g., paroxetine, fluoxetine) increases the exposure of brexpiprazole compared to the use of brexpiprazole alone [27].
There are two major contributions of this study to the academic understanding of the efficacy of dopamine partial agonists. Firstly, the study supports and further confirms the rationale for targeting > 80% occupancy of D2/D3 receptors by dopamine partial agonists for clinical effect. Indeed, in a previous collaboration, Johns Hopkins University and Otsuka carried out the first human D2/D3 receptor target engagement study with the partial agonist, aripiprazole [13]. Using the same PET radiotracer as the present study ([11C]raclopride), thereby allowing direct comparison between studies, aripiprazole had a steady-state occupancy of up to 95% at efficacious doses, with no increase in extrapyramidal symptoms [13]. (Of note, compared with aripiprazole, brexpiprazole has lower intrinsic activity at D2/D3 receptors, meaning that its intrinsic activity falls between that of aripiprazole and those of ‘pure’ antagonists [1].) In contrast, ‘pure’ D2 receptor antagonists are associated with dose-limiting side effects (such as extrapyramidal symptoms) above the therapeutic window of 65–80% occupancy [9,10,11,12]. Thus, the present results suggest that >80% occupancy of D2/D3 receptors is a ‘benchmark’ for dopamine partial agonists for the treatment of schizophrenia. The results of this single-dose, Phase 1, translational study in healthy volunteers were used to determine target dose ranges for subsequent registrational efficacy and safety studies of brexpiprazole in schizophrenia.
Secondly, since D3 receptor availability was not appreciably reduced by brexpiprazole treatment in the Girgis et al. study (as measured using PHNO) [26], the efficacious doses indicated by the present results may correspond to D2 receptor occupancy, rather than D3 receptor occupancy. Furthermore, given the known greater sensitivity to endogenous dopamine of agonist PET ligands (i.e., PHNO) versus antagonist PET ligands (i.e., raclopride) [34, 35], brexpiprazole’s lower occupancy with PHNO could allude to greater endogenous dopamine as a result of the partial dopamine agonism. Hence, by direct comparison of the raclopride results with the PHNO results, it can be hypothesized that endogenous dopamine may be a feature of brexpiprazole at D2 receptor sites. This may be a more straightforward explanation of the lower occupancy in the Girgis et al. study, as opposed to the rather complex mechanism of G protein affinity changes described by the authors [26].
The conclusions of this PET study are limited by its small sample size (N = 15), and the chance finding of a predominantly male, Black/African American sample (similarly, the Girgis et al. study had a small [N = 12] and predominantly Black/African American sample [26]). Nonetheless, the data were sufficient to fully describe brexpiprazole’s D2/D3 receptor dose–occupancy curve, and, in combination with pharmacokinetic outcomes, were sufficient to reliably select doses for registrational efficacy and safety studies in schizophrenia and MDD. With regard to dosing, based on prior tolerability data, the highest dose in the present study was 6 mg, limiting observations up to approximately 90% receptor occupancy. Finally, the study used single dosing rather than multiple dosing, which does not reflect use in clinical practice, although modeling was used to predict occupancies after multiple dosing.
Conclusions
This was the first PET imaging study with brexpiprazole, and the second PET imaging study (after a study with aripiprazole) of target engagement by a dopamine partial agonist in healthy participants. In conclusion, the occupancy of dopamine D2/D3 receptors by brexpiprazole correlated with dose in healthy participants, and was consistent with the approved, clinically active, doses of 2–4 mg/day for schizophrenia and 2–3 mg/day for the adjunctive treatment of MDD [27]. The results of this study guided the dose selection in subsequent clinical trials, which ultimately resulted in the approval of brexpiprazole for the treatment of schizophrenia and the adjunctive treatment of MDD. These data, together with the earlier PET findings with aripiprazole, highlight the translational value of D2/D3 receptor occupancy assessment in the development of future dopamine modulators for the treatment of patients with schizophrenia.
Data availability
To submit inquiries related to Otsuka Clinical Research, or to request access to individual participant data (IPD) associated with any Otsuka clinical trial, please visit https://clinical-trials.otsuka.com/. For all approved IPD access requests, Otsuka will share anonymized IPD on a remotely accessible data sharing platform.
Change history
04 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00228-020-03071-z
References
Maeda K, Sugino H, Akazawa H, Amada N, Shimada J, Futamura T, Yamashita H, Ito N, McQuade RD, Mørk A, Pehrson AL, Hentzer M, Nielsen V, Bundgaard C, Arnt J, Stensbøl TB, Kikuchi T (2014) Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin–dopamine activity modulator. J Pharmacol Exp Ther 350:589–604
Correll CU, Skuban A, Ouyang J, Hobart M, Pfister S, McQuade RD, Nyilas M, Carson WH, Sanchez R, Eriksson H (2015) Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry 172:870–880
Kane JM, Skuban A, Ouyang J, Hobart M, Pfister S, McQuade RD, Nyilas M, Carson WH, Sanchez R, Eriksson H (2015) A multicenter, randomized, double-blind, controlled Phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res 164:127–135
Fleischhacker WW, Hobart M, Ouyang J, Forbes A, Pfister S, McQuade R, Carson WH, Sanchez R, Nyilas M, Weiller E (2017) Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol 20:11–21
Thase ME, Youakim JM, Skuban A, Hobart M, Augustine C, Zhang P, McQuade RD, Carson WH, Nyilas M, Sanchez R, Eriksson H (2015) Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a Phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry 76:1224–1231
Thase ME, Youakim JM, Skuban A, Hobart M, Zhang P, McQuade RD, Nyilas M, Carson WH, Sanchez R, Eriksson H (2015) Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a Phase 3, randomized, double-blind study. J Clin Psychiatry 76:1232–1240
Hobart M, Skuban A, Zhang P, Augustine C, Brewer C, Hefting N, Sanchez R, McQuade RD (2018) A randomized, placebo-controlled study of the efficacy and safety of fixed-dose brexpiprazole 2 mg/d as adjunctive treatment of adults with major depressive disorder. J Clin Psychiatry 79:17m12058
Hobart M, Skuban A, Zhang P, Josiassen MK, Hefting N, Augustine C, Brewer C, Sanchez R, McQuade RD (2018) Efficacy and safety of flexibly dosed brexpiprazole for the adjunctive treatment of major depressive disorder: a randomized, active-referenced, placebo-controlled study. Curr Med Res Opin 34:633–642
Farde L, Wiesel FA, Halldin C, Sedvall G (1988) Central D2-dopamine receptor occupancy in schizophrenic patients treated with antipsychotic drugs. Arch Gen Psychiatry 45:71–76
Nordström AL, Farde L, Wiesel FA, Forslund K, Pauli S, Halldin C, Uppfeldt G (1993) Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects: a double-blind PET study of schizophrenic patients. Biol Psychiatry 33:227–235
Kapur S, Zipursky R, Jones C, Remington G, Houle S (2000) Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 157:514–520
Wong DF, Tauscher J, Gründer G (2009) The role of imaging in proof of concept for CNS drug discovery and development. Neuropsychopharmacol 34:187–203
Yokoi F, Gründer G, Biziere K, Stephane M, Dogan AS, Dannals RF, Ravert H, Suri A, Bramer S, Wong DF (2002) Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacol 27:248–259
Gründer G, Carlsson A, Wong DF (2003) Mechanism of new antipsychotic medications: occupancy is not just antagonism. Arch Gen Psychiatry 60:974–977
International Conference on Harmonisation (1997) Good clinical practice: consolidated guideline; availability. Fed Regist 62:25692–25709
World Medical Association (2008) Declaration of Helsinki–ethical principles for medical research involving human subjects. https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/doh-oct2008/. Accessed 4 May 2020
Rahmim A, Cheng JC, Blinder S, Camborde ML, Sossi V (2005) Statistical dynamic image reconstruction in state-of-the-art high-resolution PET. Phys Med Biol 50:4887–4912
Sossi V, de Jong HWAM, Barker WC, Bloomfield P, Burbar Z, Camborde M-L, Comtat C, Eriksson LA, Houle S, Keator D, Knob C, Krais R, Lammertsma AA, Rahmim A, Sibomana M, Teras M, Thompson CJ, Trebossen R, Votaw J, Walker MD, Wienhard K, Wong DF (2005) The second generation HRRT – a multi-Centre scanner performance investigation. IEEE Nuclear Science Symposium Conference Record. https://doi.org/10.1109/NSSMIC.2005.1596770
Ehrin E, Gawell L, Högberg T, de Paulis T, Ström P (1987) Synthesis of [methoxy-3H]- and [methoxy-11C]-labelled raclopride. Specific dopamine-D2 receptor ligands. J Label Compd Radiopharm 24:931–940
Ashburner J, Friston KJ (2004) Rigid body registration. In: Frackowiak RSJ, Ashburner J, Penny WD et al (eds) Human brain function, 2nd edn. Academic Press, San Diego, pp 635–654
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27:1533–1539
Ichise M, Liow JS, Lu JQ, Takano A, Model K, Toyama H, Suhara T, Suzuki K, Innis RB, Carson RE (2003) Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab 23:1096–1112
Akaike H (1974) A new look at statistical model identification. IEEE Trans Autom Control 19:716–723
Ashburner J, Friston KJ (2004) High-dimensional image warping. In: Frackowiak RSJ, Ashburner J, Penny WD et al (eds) Human brain function, 2nd edn. Academic Press, San Diego, pp 673–694
Ishigooka J, Iwashita S, Higashi K, Liew EL, Tadori Y (2018) Pharmacokinetics and safety of brexpiprazole following multiple-dose administration to Japanese patients with schizophrenia. J Clin Pharmacol 58:74–80
Girgis RR, Forbes A, Abi-Dargham A, Slifstein M (2020) A positron emission tomography occupancy study of brexpiprazole at dopamine D2 and D3 and serotonin 5-HT1A and 5-HT2A receptors, and serotonin reuptake transporters in subjects with schizophrenia. Neuropsychopharmacol 45:786–792
Rexulti® (brexpiprazole) tablets, for oral use (2020) Prescribing information. https://www.otsuka-us.com/media/static/Rexulti-PI.pdf. Accessed 4 May 2020
Malmberg A, Nordvall G, Johansson AM, Mohell N, Hacksell U (1994) Molecular basis for the binding of 2-aminotetralins to human dopamine D2A and D3 receptors. Mol Pharmacol 46:299–312
Correll CU, Skuban A, Hobart M, Ouyang J, Weiller E, Weiss C, Kane JM (2016) Efficacy of brexpiprazole in patients with acute schizophrenia: review of three randomized, double-blind, placebo-controlled studies. Schizophr Res 174:82–92
Gründer G, Fellows C, Janouschek H, Veselinovic T, Boy C, Bröcheler A, Kirschbaum KM, Hellmann S, Spreckelmeyer KM, Hiemke C, Rösch F, Schaefer WM, Vernaleken I (2008) Brain and plasma pharmacokinetics of aripiprazole in patients with schizophrenia: an [18F]fallypride PET study. Am J Psychiatry 165:988–995
Abilify® (aripiprazole) tablets (2020) Prescribing information. https://www.otsuka-us.com/media/static/Abilify-PI.pdf. Accessed 27 July 2020
Kane JM, Skuban A, Hobart M, Ouyang J, Weiller E, Weiss C, Correll CU (2016) Overview of short- and long-term tolerability and safety of brexpiprazole in patients with schizophrenia. Schizophr Res 174:93–98
Nelson JC, Zhang P, Skuban A, Hobart M, Weiss C, Weiller E, Thase ME (2016) Overview of short-term and long-term safety of brexpiprazole in patients with major depressive disorder and inadequate response to antidepressant treatment. Curr Psychiatr Rev 12:278–290
Cumming P, Wong DF, Dannals RF, Gillings N, Hilton J, Scheffel U, Gjedde A (2002) The competition between endogenous dopamine and radioligands for specific binding to dopamine receptors. Ann N Y Acad Sci 965:440–450
Cumming P, Wong DF, Gillings N, Hilton J, Scheffel U, Gjedde A (2002) Specific binding of [11C]raclopride and N-[3H]propyl-norapomorphine to dopamine receptors in living mouse striatum: occupancy by endogenous dopamine and guanosine triphosphate-free G protein. J Cereb Blood Flow Metab 22:596–604
Acknowledgments
Special thanks to Robert Dannals, PhD, the radiochemistry team, and the nuclear medicine PET and magnetic resonance imaging research technologists at the Johns Hopkins Hospital.
Writing support was provided by Chris Watling, PhD, assisted by his colleagues at Cambridge Medical Communication Ltd. (Cambridge, UK), and funded by Otsuka Pharmaceutical Development & Commercialization Inc. (Princeton, NJ, USA) and H. Lundbeck A/S (Valby, Denmark).
Funding
This study was funded by Otsuka Pharmaceutical Development & Commercialization Inc. (Princeton, NJ, USA).
Author information
Authors and Affiliations
Contributions
Dean F. Wong, James R. Brašić and Hiroto Kuwabara contributed to the design of the study and were involved in the acquisition, analysis, and interpretation of data. Arash Raoufinia, Patricia Bricmont, Robert D. McQuade and Robert A. Forbes contributed to the design of the study and the interpretation of data. Tetsuro Kikuchi contributed to the interpretation of data. All authors participated in the drafting or the critical review of the article; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
Dean F. Wong received a contract to Johns Hopkins University from Otsuka during this study and has received contracts to Johns Hopkins University from Lilly/Avid, Lundbeck (Denmark/US), Intracellular Technologies NYC, and Roche Neuroscience, and from LB Pharma to Washington University. Arash Raoufinia, Patricia Bricmont, Robert D. McQuade and Robert A. Forbes are full-time employees of Otsuka Pharmaceutical Development & Commercialization Inc. James R. Brašić and Hiroto Kuwabara declare no conflict of interest. Tetsuro Kikuchi is a full-time employee of Otsuka Pharmaceutical Co., Ltd.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable (no individual patient information is included).
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original article was revised: The correct affiliation 1 is presented in this paper.
Electronic supplementary material
ESM 1
(DOCX 880 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wong, D.F., Raoufinia, A., Bricmont, P. et al. An open-label, positron emission tomography study of the striatal D2/D3 receptor occupancy and pharmacokinetics of single-dose oral brexpiprazole in healthy participants. Eur J Clin Pharmacol 77, 717–725 (2021). https://doi.org/10.1007/s00228-020-03021-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-020-03021-9