
Abstract
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHNNC) is a rare autosomal recessive renal tubulopathy disorder characterized by excessive urinary loss of calcium and magnesium, polyuria, polydipsia, bilateral nephrocalcinosis, progressive chronic kidney disease, and renal failure. Also, sometimes amelogenesis imperfecta and severe ocular abnormalities are involved. The CLDN-16 and CLDN-19 genes encode the tight junction proteins claudin-16 and claudin-19, respectively, in the thick ascending loop of Henle in the kidney, epithelial cells of the retina, dental enamel, etc. Loss of function of the CLDN-16 and/or CLDN-19 genes leads to FHHNC. We present a case of FHHNC type 1, which was first confused with autosomal dominant hypocalcaemia (ADH) due to the presence of a very low serum parathyroid hormone (PTH) concentration and other similar clinical features before the genetic investigations. After the exome sequencing, FHHNC type 1 was confirmed by uncovering a novel homozygous missense mutation in the CLDN-16 gene (Exon 2, c.374 T > C) which causes, altered protein structure with F55S. Associated clinical, biochemical, and imaging findings also corroborate final diagnosis. Our findings expand the spectrum of the CLDN-16 mutation, which will further help in the genetic diagnosis and management of FHNNC.





Similar content being viewed by others

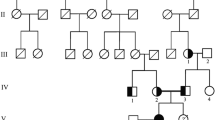
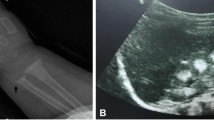
Data Availability
The genetic analysis data file is available on request.
References
Pearce SH, Williamson C, Kifor O et al (1996) A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 335(15):1115–1122. https://doi.org/10.1056/NEJM199610103351505
Nesbit MA, Hannan FM, Howles SA et al (2013) Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N Engl J Med 368(26):2476–2486. https://doi.org/10.1056/NEJMoa1300253
Mittelman SD, Hendy GN, Fefferman RA et al (2006) A hypocalcemic child with a novel activating mutation of the calcium-sensing receptor gene: successful treatment with recombinant human parathyroid hormone. J Clin Endocrinol Metab 91(7):2474–2479. https://doi.org/10.1210/jc.2005-2605
Raue F, Pichl J, Dörr HG et al (2011) Activating mutations in the calcium-sensing receptor: genetic and clinical spectrum in 25 patients with autosomal dominant hypocalcaemia - a German survey. Clin Endocrinol (Oxf) 75(6):760–765. https://doi.org/10.1111/j.1365-2265.2011.04142.x
Sastre A, Valentino K, Hannan FM et al (2021) PTH infusion for seizures in autosomal dominant hypocalcemia type 1. N Engl J Med 385(2):189–191. https://doi.org/10.1056/NEJMc2034981
Gomes V, Silvestre C, Ferreira F, Bugalho MJGM (2020). BMJ Case Rep. https://doi.org/10.1136/bcr-2020-234391
Elston MS, Elajnaf T, Hannan FM, Thakker RV (2022) Autosomal Dominant Hypocalcemia Type 1 (ADH1) associated with myoclonus and intracerebral calcifications. J Endocr Soc. https://doi.org/10.1210/jendso/bvac042
Praga M, Vara J, González-Parra E et al (1995) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int 47(5):1419–1425. https://doi.org/10.1038/ki.1995.199
Claverie-Martin F (2015) Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J 8(6):656–664. https://doi.org/10.1093/ckj/sfv081
Godron A, Harambat J, Boccio V et al (2012) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol 7(5):801–809. https://doi.org/10.2215/CJN.12841211
Weber S, Schneider L, Peters M et al (2001) Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 12(9):1872–1881. https://doi.org/10.1681/ASN.V1291872
Konrad M, Schaller A, Seelow D et al (2006) Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79(5):949–957. https://doi.org/10.1086/508617
Claverie-Martín F, García-Nieto V, Loris C et al (2013) Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS ONE 8(1):e53151. https://doi.org/10.1371/journal.pone.0053151
Vall-Palomar M, Madariaga L, Ariceta G (2021) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Pediatr Nephrol 36(10):3045–3055. https://doi.org/10.1007/s00467-021-04968-2
Prabahar MR, Manorajan R, Fernando ME, Venkatraman R, Balaraman V, Jayakumar M (2006) Nephrocalcinosis in siblings–familial hypomagnesemia, hypercalciuria with nephrocalcinosis (FHHNC syndrome). J Assoc Physicians India 54:497–500
Geethalakshmi S, Bhavani N, Vinayan KP, Nair V (2021) Rare Inherited hypomagnesemias—an endocrine case series. Indian Pediatr 58(5):489–490
Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49(7):433–436. https://doi.org/10.1136/jmedgenet-2012-100918
Geourjon C, Deléage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6):681–684. https://doi.org/10.1093/bioinformatics/11.6.681
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362. https://doi.org/10.1038/nmeth.2890
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. https://doi.org/10.1093/nar/gki375
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
TOPO2 (http://www.sacs.ucsf.edu/TOPO2/).
Zheng W, Zhang C, Li Y, Pearce R, Bell EW, Zhang Y (2021) Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep Methods 1(3):100014. https://doi.org/10.1016/j.crmeth.2021.100014
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12(1):7–8. https://doi.org/10.1038/nmeth.3213
Yang J, Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43(W1):W174–W181. https://doi.org/10.1093/nar/gkv342
Simon DB, Lu Y, Choate KA et al (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285(5424):103–106. https://doi.org/10.1126/science.285.5424.103
Yamaguti PM, Neves FA, Hotton D et al (2017) Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations. J Med Genet 54(11):786
Bardet C, Courson F, Wu Y et al (2016) Claudin-16 deficiency impairs tight junction function in ameloblasts, leading to abnormal enamel formation. J Bone Miner Res 31(3):498–513. https://doi.org/10.1002/jbmr.2726
de Baaij JH, Hoenderop JG, Bindels RJ (2015) Magnesium in man: implications for health and disease. Physiol Rev 95(1):1–46. https://doi.org/10.1152/physrev.00012.2014
de Baaij JH, Dorresteijn EM, Hennekam EA et al (2015) Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant 30(6):952–957. https://doi.org/10.1093/ndt/gfv014
Hou J, Rajagopal M, Yu AS (2013) Claudins and the kidney. Annu Rev Physiol 75:479–501. https://doi.org/10.1146/annurev-physiol-030212-183705
Hou J, Renigunta A, Konrad M et al (2008) Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 118(2):619–628. https://doi.org/10.1172/JCI33970
Dimke H, Schnermann J (2018) Axial and cellular heterogeneity in electrolyte transport pathways along the thick ascending limb. Acta Physiol (Oxf) 223(1):e13057. https://doi.org/10.1111/apha.13057
Muto S (2017) Physiological roles of claudins in kidney tubule paracellular transport. Am J Physiol Renal Physiol 312(1):F9–F24. https://doi.org/10.1152/ajprenal.00204.2016
Tsukita S, Tanaka H, Tamura A (2019) The claudins: from tight junctions to biological systems. Trends Biochem Sci 44(2):141–152. https://doi.org/10.1016/j.tibs.2018.09.008
Mineta K, Yamamoto Y, Yamazaki Y et al (2011) Predicted expansion of the claudin multigene family. FEBS Lett 585(4):606–612. https://doi.org/10.1016/j.febslet.2011.01.028
Meoli L, Günzel D (2020) Channel functions of claudins in the organization of biological systems. Biochim Biophys Acta Biomembr 1862(9):183344. https://doi.org/10.1016/j.bbamem.2020.183344
Suzuki H, Nishizawa T, Tani K et al (2014) Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 344(6181):304–307. https://doi.org/10.1126/science.1248571
Hou J, Paul DL, Goodenough DA (2005) Paracellin-1 and the modulation of ion selectivity of tight junctions. J Cell Sci 118(Pt 21):5109–5118. https://doi.org/10.1242/jcs.02631
García-Castaño A, Perdomo-Ramirez A, Vall-Palomar M et al (2020) Novel compound heterozygous mutations of CLDN16 in a patient with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Mol Genet Genomic Med 8(11):e1475. https://doi.org/10.1002/mgg3.1475
Konrad M, Hou J, Weber S et al (2008) CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 19(1):171–181. https://doi.org/10.1681/ASN.2007060709
Müller D, Kausalya PJ, Claverie-Martin F et al (2003) A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet 73(6):1293–1301. https://doi.org/10.1086/380418
Kausalya PJ, Amasheh S, Günzel D et al (2006) Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J Clin Invest 116(4):878–891. https://doi.org/10.1172/JCI26323
Acknowledgements
The authors acknowledge the necessary support of the Director, NIBMG, authorities of Burdwan Medical College and Hospital, Burdwan, and authorities of Burdwan University, and are thankful to the parents of the patient for agreeing to the present study. RT expresses thanks and acknowledgment to UGC for the NET fellowship.
Funding
The present study was partially supported by the UGC NET fellowship (RT) and intramural grant support of AB from NIBMG.
Author information
Authors and Affiliations
Contributions
RT did the genetic work and wrote the manuscript; AR did the clinical workup; and KN supervised the clinical workup. AB supervises the genetic work and checks the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Rupesh Thapa, Amaresh Roy, Kaustav Nayek and Anupam Basu deny any kind of conflict of interest in the present study.
Human and Animal Rights and Informed Consent
Blood samples was sent to the laboratory for the genetic study as per approval of ethics committee and informed consent was taken from the parents.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Thapa, R., Roy, A., Nayek, K. et al. Identification of a Novel Homozygous Missense Mutation in the CLDN16 Gene to Decipher the Ambiguous Clinical Presentation Associated with Autosomal Dominant Hypocalcaemia and Familial Hypomagnesemia with Hypercalciuria and Nephrocalcinosis in an Indian Family. Calcif Tissue Int 114, 110–118 (2024). https://doi.org/10.1007/s00223-023-01142-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-023-01142-8