
Abstract
According to green analytical chemistry principles, the use of agricultural byproducts as sorbent phases is an interesting topic due to their lignocellulosic origin, as they are biodegradable and inexpensive. To the best of our knowledge, this is the first study in which avocado seed and avocado seed activated carbon are proposed as sustainable sorbents for solid-phase microextraction technologies, which were used to assess the proof of concept. Rotating disk sorptive extraction (RDSE) was used as a model technology and ibuprofen (Ibu) and 1-hydroxy-ibuprofen (1-OH-Ibu) as representative analytes. It was found that activated carbon (AC) prepared at 600 °C with an impregnation ratio (raw material/activating agent (ZnCl2), w/w) of 1:1.2 had better extraction efficiency than other ACs obtained at different temperatures, impregnation ratios, and activating agents (K2CO3). Characterization revealed several differences between natural avocado seed, biochar prepared at 600 °C, and selected AC since the typical functional groups of the natural starting material begin to disappear with pyrolysis and increasing the surface area and pore volume, suggesting that the main interactions between analytes and the sorbent material are pore filling and π-π stacking. By using this AC as the sorbent phase, the optimal extraction conditions in RDSE were as follows: the use of 50 mg of sorbent in the disk, 30 mL of sample volume, pH 4, 90 min of extraction time at a rotation velocity of the disk of 2000 rpm, and methanol as the elution solvent. The extracts were analyzed via gas chromatography coupled to mass spectrometry (GC–MS). The method provided limits of detection of 0.23 and 0.07 µg L−1 and recoveries of 81% and 91% for Ibu and 1-OH-Ibu, respectively. When comparing the extraction efficiency of the selected activated carbon with those provided by Oasis® HLB and C18 in RDSE, nonsignificant differences were observed, indicating that avocado seed activated carbon is a suitable alternative to these commercial materials.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Globally, 1.6 million tons of residues are generated worldwide, making its disposal one of the biggest challenges. Valorization of residues is an approach to mitigate this problem. Avocado is a fruit obtained from the Persea americana Mill. tree and is originally from Meso and Central America but found in several countries [1]. In Chile, avocado is widely consumed and cultivated for both domestic consumption and export. In 2019, global exports of this fruit reached 156 thousand tons, and between 2022 and 2023, Chile was responsible for 186 thousand tons of total exports [2, 3]. This fruit, available throughout the year regardless of the season, produces thousands of tons of seeds as a byproduct, with the avocado seed equivalent to almost 15–16% of the total weight of the fruit [4]. This seed is a lignocellulosic material mainly composed of lignin, cellulose, hemicellulose, and other components, such as polyphenols and fatty acids [5]. Due to its composition, avocado seed, which is usually discarded because it has no economic value, has the potential to be used as a sorbent material in various sorption-based techniques [6]. The main advantages of this material as a sorbent are its accessibility, low cost, eco-friendliness, and biodegradability.
Avocado seed activated carbon has been mainly used for water remediation. In 2014, Bhaumik used avocado seed activated carbon in batch experiments to remove Cr(VI) from wastewater through modification with sulfuric acid (H2SO4) at low temperatures (100 °C) [7]. In 2017 and 2018, Leite et al. used activated carbon for the removal of phenolic compounds and emerging organic contaminants, using ZnCl2 as an activation agent at 700 °C in a conventional oven and through microwave heating [8, 9].
In recent years, the concept of green analytical chemistry (GAC) has gained much attention in the scientific community, particularly focused on the sample preparation steps of chemical measurement processes. Different technologies based on solid-phase microextraction (SPME) offer eco-friendly and miniaturized alternatives to traditional sample preparation procedures, as they require a minimal amount of both extractant phase and sample [10].
Despite this, since typical sorbent materials used in microextraction technologies are synthetic, they are not fully environmentally friendly. Therefore, the use of natural sorbent materials that align better with GAC principles is recommended [11]. With respect to the raw material, activated carbon and biochar represent an enhancement of the specific surface area and porosity of the sorbent material, which can improve the extraction efficiency of the analyte(s) of interest [12]. As biochar is the product of the pyrolysis of the material, activated carbon is the product of pyrolysis in the presence of activating reagents. One way to synthesize activated carbon is through the impregnation of the starting material with activation agents such as ZnCl2 and K2CO3 in an appropriate mass ratio, followed by pyrolysis in an inert atmosphere [13, 14]. To date, one study has applied avocado seed as a sorbent phase in SPE for the determination of Cd(II) using an online flow injection system [15].
This work aims to use the rotating disk sorptive extraction (RDSE) technique using avocado seed activated carbon as the sorbent phase to determine ibuprofen and 1-hydroxy ibuprofen from water samples. Avocado seed activated carbon was characterized through N2 adsorption isotherms (to determine the BET area), Fourier transform infrared (FTIR) spectroscopy, scanning electron microscopy (SEM), and the hydrophobicity-hydrophilicity (HI) index. In addition, the synthesis of activated carbon was optimized by varying the temperatures, the activating agents, and their concentrations. Subsequently, the RDSE analytical methodology was developed, optimized, and validated through measurements of the analytes by gas chromatography-mass spectrometry (GC–MS).
Materials and reagents
Avocado was purchased in the local market. Potassium carbonate (K2CO3) (99 + % purity) was obtained from Thermo Scientific (USA). Zinc chloride (ZnCl2) (98 + % purity) was obtained from Sigma-Aldrich (USA). Ibuprofen (Ibu) and 1-OH-ibuprofen (1-OH-Ibu) were obtained from Sigma-Aldrich (Milwaukee, WI, USA). Water from a Millipore Milli-Q Plus water system (Billerica, MA, USA) was used throughout the experiments. Standard stock solutions of the analytes (10 mg L−1) were prepared separately in methanol (MeOH) (Fisher Scientific, Fair Lawn, NJ, USA). Multistandard solutions were prepared in methanol at concentrations of 2, 5, 10, 25, 50, and 100 μg L−1. Nitrogen (99.995% purity) and helium (99.9999% purity) were purchased from Linde (Santiago, Chile) and were used for final extract evaporation and as the chromatographic carrier gas, respectively. Ethyl acetate (EtOAc), acetonitrile (MeCN), chloroform (CHCl3), and n-heptane (HPLC grade) were purchased from Merck (Darmstadt, Germany). N-Methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) was obtained from Sigma-Aldrich (Milwaukee, WI, USA) and used as the derivatizing agent.
Instruments
A Multiposition magnetic stirrer M5-52 M Jeio Tech (Korea) was used for rotating disk extraction. pH values were measured with a Microprocessor pH-Meter 537A (WTW, Germany). A DLAB MX-S vortex (DLAB Instruments, China), a Biotage TurboVap Classic LV concentration evaporation system (Biotage, Sweden), and an analog heat block evaporator (VWR, USA) were used for the derivatization reaction. A Heraeus M110 muffle furnace (Thermo Scientific, USA), a Binder WTC FD-400 oven (Binder, Germany), and a C24 incubator shaker from New Brunswick Scientific (Edison, USA) were used.
For GC–MS determinations, a Thermo Scientific TRACE 1300 gas chromatograph coupled to a Thermo Fisher Scientific ISQ mass-selective detector with an electron impact ionization (70 eV) source was used. The chromatographic column used was a Restek RTX-5MS (30 m × 0.25 mm i.d.; 0.25 μm film thickness). The samples were injected using splitless mode and an injector temperature of 280 °C. The oven program was initiated at 75 °C for 1 min, followed by an increase to 150 °C at 20 °C min−1 and finally an increase to 300 °C at 10 °C min−1. The carrier gas used was helium at 1.0 mL min−1. The solvent delay was 7 min. The MS transfer line and ion source were kept at 300 °C and 250 °C, respectively. A dwell time of 0.2 s was used for each m/z value. Compound quantification was based on matrix-matched calibration using the mass spectrometer in selective ion monitoring (SIM) mode (Table 1). The qualifier ion of Ibu corresponds to the molecular ion of the derivatized analyte, while the target ion corresponds to the original derivatized analyte in which the isopropyl radical is lost. Similarly, in the case of 1-OH-Ibu, the target ion corresponds to the original derivatized analyte with the loss of the isopropyl radical.
For the derivatization reaction, 50 µL of MSTFA and 50 µL of ethyl acetate were added to the vial, vigorously mixed in a vortex for homogenization, and subsequently heated at 60 °C for 30 min [16]. Two microliters of the derivatized extract was injected into GC–MS according to the previously mentioned procedure.
Statistical and graphical software GraphPad Prism 10 (Boston, USA) was used for statistical analysis.
Preparation of avocado seed sorbent phases
As shown in Fig. 1, natural avocado seed (NAS) was prepared by removing the avocado residue with water, and then the seed was soaked in water for 24 h to facilitate grinding. Subsequently, the material was subjected to a solvent treatment ultrasonic process using sequential washes with water and methanol to leach components that could interfere with the instrumental measurement, with methanol being reused in subsequent procedures. Finally, the material was dried and sieved, keeping the fraction within the 35–78 µm range.

General avocado seed treatment to produce natural avocado seed (NAS) and avocado seed activated carbon (AC)
Avocado seed activated carbon (AC) was prepared from particulate NAS impregnated with aqueous solutions of ZnCl2 at 20, 30, and 40% (w/v), ranging from 1 g NAS to 0.8, 1.2, and 1.6 g of activating agent. Impregnation was carried out in an orbital shaker followed by drying at 80 °C for 24 h. Then, the impregnated material underwent a controlled heat treatment process in a muffle furnace within a nitrogen atmosphere, with a temperature program of 25–400 °C, 25–600 °C, and 25–800 °C with a heating rate of 10 °C/min and maintaining the maximum temperature for 30 min. Finally, the material formed was ground in an agate mortar, followed by washing in a shaker with 5 M HCl to remove the ash, and then successive washings with distilled water until a neutral pH was reached. Subsequently, the material was filtered and sieved, maintaining the same fraction as previously employed in the NAS treatment. In addition, K2CO3 was also studied as an activating agent under the same experimental conditions.
Characterization
Thermal gravimetric analysis (TGA) of natural avocado seeds was carried out using a TG209 F1 NIETZSCH (Germany) instrument. The analysis was conducted with a heating rate of 10 °C min−1, reaching a maximum temperature of 800 °C within a nitrogen atmosphere. The data were analyzed by using first derivative (DTG) plots, presenting weight loss percentage related to temperature (% °C−1) against temperature (°C). ICP-MS analysis was performed using a single quadrupole inductively coupled plasma-mass spectrometry (ICP-MS) equipment, iCAP RQ (Thermo Scientific, USA). The methodology is described in Supplementary Information (SI).
Material surface chemical groups were analyzed using a Nicolet iS5 FTIR spectrometer (Thermo Fisher Scientific, USA) equipped with a KBr/Ge beam splitter and a high-performance deuterated triglycine sulfate detector. This instrument was coupled to a smart iTX-iD7 attenuated total reflectance (ATR) sampling accessory with a diamond crystal. The data were acquired using OMNIC 8.0 software (Thermo Fisher Scientific). Spectra were collected in the wavenumber range between 4000 and 400 cm−1 with a resolution of 4 cm−1 and 16 scans per sample. The surface morphology of the materials was analyzed with a high-resolution scanning electron microscope (SEM) INSPECT-F50 model (Thermo Fisher Scientific, FEI, Netherlands) at magnifications ranging from 400 to 4000 × .
The hydrophobicity index (HI) was studied according to the methodology described by Leite et al. (2018) [16]. A 200 mg portion of each material was dried in 10-mL beakers at 70 °C for 24 h and then cooled in a desiccator to reach constant weight. Each 10-mL beaker was placed within a 250-mL beaker with 60 mL of solvent (water or n-heptane) and then placed into an incubator at 25 °C. After 24 h, the sample container beaker was retrieved, carefully dried externally, and weighed again. The amount of vapor adsorbed was obtained by the difference in mg g−1. HI was calculated as the ratio between adsorbed n-heptane (mg g−1) and adsorbed water (mg g−1).
Contact angle (θ) between material and water was studied using a goniometer Model 200 F4 series (ramé-hart instrument co., USA). A 2-µL droplet was placed on a surface covered with the material and adhered with double-contact film. Images were recorded for 2 s. Contact angle values are classified as hydrophobic when θ > 90° and hydrophilic when θ < 90° [17, 18].
The pore distribution and surface characteristics were determined through N2 adsorption isotherms at 77 K using an automated adsorption equipment (Micromeritics, USA). Before the N2 adsorption measurements, the samples underwent a 3-h degassing process under vacuum at 300 °C. The Brunauer–Emmett–Teller (BET) equation was used to estimate the surface area (SBET). The total pore volume (VT) was calculated by assessing the liquid volumes of N2 corresponding to the adsorbed amount at a relatively high pressure of P/P0 ~ 0.99. The micropore volume (Vµ) was determined using the Dubinin-Radushkevich (DR) equation, while the mesopore volume (Vm) was recognized as the difference between VT and Vµ. The equation Dp = 4VT/SBET was used to estimate the average pore diameter (Dp), and the Barrett-Joyner-Halenda (BJH) method was used to assess the pore distribution [19].
General RDSE procedure
The cavity of the disk was filled with 50 mg of AC, covered with a fiberglass filter, and then sealed with a Teflon cap. Preceding extraction, the sorbent was conditioned with ethyl acetate, methanol, and deionized water for 5 min at 2000 rpm each. Extraction was performed with 30 mL of sample adjusted to pH 4, and then the extraction proceeded for 90 min at 2000 rpm in a multiposition magnetic stirrer. Once the extraction was complete, the disk was removed from the extraction vial, given a slight rinse, and then dried before the desorption step. For desorption, the disk was transferred to a dry beaker containing 10 mL of methanol and then stirred for 20 min at 2000 rpm. Then, the solvent was evaporated through N2 flow and derivatized prior to GC–MS injection [20, 21].
Figures of merit of the methodology and application
A matrix-matched calibration was performed with enriched river water samples. The calibration curve was performed in triplicate between 0.5 and 25 µg L−1. Using this curve regression error (S(y/x)) and slope (m), limits of detection (LODs) and quantification (LOQs) were calculated as LOD = 3.3 S(y/x)/m and LOQ = 10 S(y/x)/m. Precision was expressed as %RSD (n = 6). The matrix effect (ME) and recovery (%Re) were obtained by the following equations [22]:
Real samples
Samples were obtained from the Mapocho River, located in Santiago, Chile (Fig. S1). Specifically, samples were collected from the influent and effluent of the “El Trebal” wastewater treatment plant (WWTP, Aguas Andinas S.A. Company). In addition, samples were taken manually from “Canoa,” an upstream sampling site from the WWTP, and “Esperanza,” a downstream sampling site from the WWTP. Remarkably, the “Canoa” upstream site is a downstream site of another WWTP, “La Farfana,” from the same company.
Results and discussion
Selection of the sorbent material
Preliminary comparisons between materials were performed by RDSE [23]. A comparison between the extraction efficiency of the natural phase and AC obtained using two different activation agents (ZnCl2 and K2CO3), both prepared at 400 °C with 1:0.8 NAS/activating agent ratio (denoted by AC400(Z0.8) and AC400(K0.8) in Fig. 2). The result shows that AC is much more efficient for extraction of the analytes. This may be due to the characteristics of AC since the surface area is considerably larger than that of NAS, allowing for a greater contact area with the analytes.

RDSE extraction recovery for the analytes using NAS, AC400(Z0.8), and AC400(K0.8) as the sorbent phase. *Significant difference (ρ-value < 0.05)
ZnCl2 and K2CO3 as activating agents were studied using different impregnation proportions (1:0.8, 1:1.2, and 1:1.6 raw material/activating agent) and activation temperatures (400, 600, and 800 °C) by a univariate optimization in which the recoveries for Ibu and 1-OH-Ibu were compared (Fig. 3).

RDSE extraction recovery for the analytes using different temperatures (vertical), activating reagents (horizontal), and impregnation ratios of 1:0.8, 1:1, and 1:1.2 w/w (NAS to reagent). *Significant difference (ρ-value < 0.05)
Higher recoveries were obtained with ZnCl2 than with K2CO3. This might be due to a greater surface area [9].
To choose between ZnCl2 conditions, two-way ANOVA with Tukey’s multiple comparisons test was carried out for each analyte at each temperature. The results indicated that in general, the proportions of activating reagents were significantly different. These data were analyzed in three steps. First, the proportion 0.8 provided lower recoveries than the other tested proportions, so this condition was discarded. Second, there were no significant differences between the three temperatures for proportions of 1.2 and 1.6; hence, proportion 1.2 was chosen because it used less reagent. Third, to determine the optimal temperature for condition 1.2, no significant difference was found for Ibu, while 1-OH-Ibu showed significantly lower recoveries between 400 °C and the other temperatures (ρ-values of 0.0403 and 0.0264 for 400–600 °C and 400–800 °C, respectively). Even the 600 and 800 °C condition 1.2 did not show any significant differences. A temperature of 600 °C was chosen because it consumes less energy and nitrogen. Finally, the material that was activated with ZnCl2 (1 g of material/1.2 g of activator) at 600 °C was selected for further characterization and microextraction experiments.
Characterization
First, thermogravimetric analysis was performed to study the material’s composition and proportion of biopolymers. The result of the TGA/DGT analysis is shown in Fig. S2: TGA shows three clear weight loss stages: (i) a loss near 10% in the range of 25–150 °C, (ii) a loss near 58.14% in the range of 200–400 °C, and (iii) a loss near 7% above 400 °C. Additionally, DGT shows a first peak in the 50–150 °C range, which is linked to the loss of moisture and degradation of volatile matter related to stage (i) in TGA. The second peak in the range of 150 to 400 °C is due to the decomposition of hemicellulose and cellulose associated with the TGA stage (ii). The third barely visible peak is between 400 and 600 °C and is related to the decomposition of lignin. These results are similar to those reported in previous studies [24]. While the presence of biopolymers can be discerned, it is difficult to accurately determine their proportion in the material since their decomposition is not an isolated process. It has been reported that hemicellulose degrades in a range of 190–320 °C, cellulose degrades between 280 and 400 °C, and lignin degrades from 320 to 450 °C [25]. Therefore, due to the overlapping thermal degradation ranges, attributing the DTG peaks to a single biopolymer is not a precise interpretation [26]. The content of elements was analyzed through microwave digestion and quantified using ICP-MS. The concentration range for Mg, V, Mn, Fe, Cu, Ga, As, Rb, and Sr was found to be between 0.01 and 277 µg g−1 (for more detailed information, refer to Table S1 in the Supplementary Information (SI)).
FTIR was performed to characterize and compare surface chemical groups for NAS and AC600(Z1.2) (Fig. S3). NAS contains a broad and intense band at 3300 cm−1 attributed to a hydroxyl group (-OH), two low-resolution bands near 2900 cm−1 are associated with CH stretching associated with aliphatic carbons, the signal at 1630 cm−1 is attributed to the stretching of the aromatic C = C groups, and the signal at 1000 cm−1 is associated with C-O stretching. In the case of AC, the absence of several signals is evident; however, the signal at 1600 cm−1 remains. In all instances (all temperatures and activating reagents), a similar spectrum was observed. This was due to the decomposition of biopolymers when carbonizing to 600 °C. This behavior was previously observed since activation improves surface physical properties, but it did not develop new chemical groups on the surface [14]. According to this information, it can be inferred that one important interaction between the analytes and the AC occurs via π stacking.
SEM images for NAS and AC600(Z1.2) are shown in Fig. 4. Natural avocado seeds show oval-spherical compact structures with smooth and homogeneous surfaces without visible pores. As a result of the carbonization process, AC600(Z1.2) shows a porous cracked less homogeneous structure with rough and smooth faces. This is likely due to the collapse of the material walls because of the energic activation with ZnCl2, as suggested in previous works [21, 27].

SEM micrograph (4000 ×) of NAS and AC600(Z1.2)
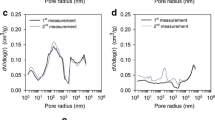
Sorption N2 isotherms were performed to complete morphology analyses. This study was not possible for NAS, confirming the absence of pores mentioned in the SEM interpretation. Table 2 lists the BET surface area, average pore diameter, total pore volume (VT), and micropore volume (Vµ) values.
Table 2 shows that AC600(Z1.2) is a microporous material, with an average pore diameter of 1.93 nm and 80.71% of the total pore volume attributed to this kind of pore. The generation of micropores in AC is due to pyrolysis and chemical activation with ZnCl2, as explained in previous works [27].
The sorption/desorption N2 behavior of AC600(Z1.2) complements this information (Fig. S4): (i) The sorption isotherm fits a type 1(b) isotherm, usually given by microporous materials where the limiting uptake is due to the accessible micropore volume by preference over the internal surface area. In this case, type 1(b) is a subclassification for pores with diameters of 2.5 nm or less, mainly micropores and narrow mesopores, and (ii) hysteresis is a type H4 usually found in micromesoporous carbons [28]. The presence of a high volume of micropores suggest that interactions with analytes occurs also by a pore filling mechanism.
The hydrophobicity of the material was determined by obtaining its HI index (Table 3). The HI index was calculated by dividing the amount of adsorbed n-heptane by the amount of adsorbed water, with higher HI values indicating hydrophobicity. It was observed that the material polarity changed during carbonization, which could be due to certain chemical groups decomposing, as detected previously by FTIR analysis. However, both HI values are less than 1, which means that both materials have hydrophilic characteristics.
Contact angle (θ) results indicate that for NAS and AC-600(Z1.2), the contact angles within the first 2 s were 34° ± 2.3° and 24° ± 4.6° respectively, as shown in Fig. S5. These values suggest that both materials exhibit hydrophilic characteristics, as angles are less than 90° [29]. Additionally, it was observed that the droplet was completely adsorbed after the specified period. These findings are complementary to the HI index analysis.
RDSE variable optimization
The variables of the RDSE methodology were studied and optimized, including the amount of material (sorbent phase), extraction time, sample volume, pH, salting-out effect, and desorption solvent. The amount of material was compared in a univariate study (Fig. 5a) and then analyzed with two-way ANOVA. Both analytes provided better responses with the maximum amount of material, although Ibu recovery was not significantly different. A higher extraction efficiency was expected with an increasing amount of material because it allowed for a larger contact surface.

Effect of different factors on the recovery of the analytes: a the amount of sorbent, b pH, c NaCl concentration, d desorption solvent. *Significant difference (ρ-value < 0.05)
Hydrodynamic variables such as extraction time and sample volume were optimized by a Doehlert assay. Table S2 shows the experimental matrix with a total of 9 experiments, including 3 centers. A total of 5 and 3 levels were assigned for extraction time (X1) and sample volume (X2), respectively. ANOVA was performed to assess the model’s quality, confirming the applicability to the system. The highest level of desirability (D = 0.918435) was achieved in Experiment 3, with an optimum condition of 90 min extraction time and 30 mL of sample (Fig. S6).
The pH optimization was conducted between pH 2 and 9, as shown in Fig. 5b. The findings revealed that pH 4 resulted in significantly higher recoveries, which is explained by the acidic properties of both analytes under study and the dependence of the phase charge on pH. This indicates that at pH 4, the charge equilibrium between the analytes and the phase reaches an optimum value.
Subsequently, salting-out effect experiments were studied. As shown in Fig. 5c, extraction efficiency decreases when high NaCl concentrations are used. This was expected, as both compounds exhibit hydrophobic characteristics (log P for Ibu 3.97 and 1-OH-Ibu 2.4). However, since there was no significant difference between concentrations of 0 and 10%, the absence of NaCl was selected to reduce reagents and simplify the extraction process.
Finally, the desorption solvent was carried out using MeOH, MeCN, CHCl3, and a mix of MeOH and EtOAc (3:7) (Fig. 5d), and methanol was selected due to its nonsignificant but higher recovery values.
Method validation
Table 4 presents the analytical parameters obtained for the proposed methodology. Linearities up to 0.99 were found for both analytes in the range of 0.5–25 µg L−1. The LODs were 0.07 and 0.23 µg L−1, while the LOQs were 0.25 and 0.77 µg L−1 for 1-OH-Ibu and Ibu, respectively. These values were confirmed experimentally via the injection of calibrators spiked at the LOD/LOQ levels, obtaining in both instances differences of less than 10% concerning the expected values. These LOD values are acceptable since they are lower than the concentrations typically found in environmental waters. RSD values (< 9%) were considered appropriate for a microextraction technique based on a laboratory-made sorbent phase. The recoveries and matrix effect values were 81% and − 58% for Ibu and 91% and − 55% for 1-OH-Ibu, respectively. High recoveries indicate an efficient method for the determination of both analytes. Finally, the negative matrix effect reflects a significant suppression of the response for both analytes, since the river sample used for validation is directly impacted by the effluents from WWTPs in Santiago, as shown in Fig. S1.
Application in real samples and comparative study
Ibu and its metabolite, 1-OH-Ibu, have been quantified in both river and WWTP waters (Table 5). These findings are consistent with those of previous studies that suggest that the treatment process used in WWTPs is not effective for this kind of emerging contaminant [16, 18]. The presence of both analytes is concerning, as ibuprofen is one of the most widely used nonprescription drugs, and its detection and quantification indicate a need to enhance regulations in Chile.
Additionally, both contaminants were found in the upstream sampling site (Canoa upstream site). This is likely due to the presence of a previous WWTP (La Farfana) that discharges its effluent into the same river as El Trebal (Mapocho River).
Comparative studies using AC600(Z1.2), HLB, C18, and S-DVB were performed (Fig. 6), confirming the differences by a two-way ANOVA, finding the following results: (i) S-DVB results in significantly lower recoveries for both analytes when compared with each sorbent material, (ii) no significant difference was found between AC600(Z1.2), HLB, and C18. This last result shows the potential of this material, as it is easy to obtain and process, in contrast with high-cost synthetic materials such as Oasis® HLB, C18, and S-DVB.

Recovery (%) for Ibu and 1-OH-Ibu by RDSE methodology using AC600(Z1.2) and synthetic sorbent phases as HLB, C18, and S-DVB. ns, no significant difference (ρ-value > 0.05)
This kind of sorbent phase offers some advantages, as it is easy to acquire and prepare. Further research might be conducted to assess the possibility of using this kind of material in other microextraction techniques, such as thin-film microextraction (TF-ME), pipette tip solid-phase extraction (PT-SPE), and disposable pipette extraction (DPX). Alternatively, GC–MS could be replaced by LC–MS to improve the greenness of the whole methodology.
Conclusion
Avocado seed is a convenient raw material for the development of carbonaceous sorbent phases. Carbonization and chemical activation are suitable modifications for sorption-based techniques because they improve surface properties. Through extensive testing of various activation reagents, proportions, and temperatures, we found that activated carbon prepared with ZnCl2 (1:1.2) at 600 °C provides the most promising results.
Characterization of NAS and AC600(Z1.2) shows chemical and physical differences that explain why AC600(Z1.2) outperforms NAS. Activated carbon has been proven to have a more developed surface structure, as the activating agent causes new pore development, resulting in higher surface areas.
The analytes under study, Ibu and 1-OH-Ibu, were detected in all sample sites, ranging from 0.38 to 1.1 µg/L. AC600(Z1.2) has been demonstrated to be an efficient sorption material, comparable to synthetic sorbent phases such as HLB and C18 in the RDSE technique.
References
Barbosa-Martín E, Chel-Guerrero L, González-Mondragón E, Betancur-Ancona D. Chemical and technological properties of avocado (Persea americana Mill.) seed fibrous residues. Food Bioprod Process. 2016;100:457–63. https://doi.org/10.1016/j.fbp.2016.09.006.
Cabana Villca R, Ortega Marín MI, Gutiérrez Rojas AA, Aguilera Zambra MI, Chiang Guzmán AF. Factors that influence French consumer satisfaction in the preference for Chilean avocados (Persea americana Mill.). Int J Agric Nat Resour. 2021;48(2):45–56. https://doi.org/10.7764/ijanr.v48i2.2285.
González S. Chile: Avocado Annual. USDA Foreign Agricultural Service, Global Agricultural Information Network. CI2022–0027. 2022. https://apps.fas.usda.gov/newgainapi/api/Report/DownloadReportByFileName?fileName=Avocado%20Annual_Santiago_Chile_CI2022-0027.pdf
Ahmad T, Mohammed D. A review of avocado waste-derived adsorbents: characterizations, adsorption characteristics, and surface mechanism. Chemosphere. 2022;296:134036. https://doi.org/10.1016/j.chemosphere.2022.134036.
Mora-Sandí A, Ramírez-González A, Castillo-Henríquez L, Lopretti-Correa M, Vega-Baudrit JR. Persea americana agro-industrial waste biorefinery for sustainable high-value-added products. Polymers. 2021;13(11):1727. https://doi.org/10.3390/polym13111727.
Vasić V, Kukić D, Šćiban M, Đurišić-Mladenović N, Velić N, Pajin B, Crespo J, Farre M, Šereš Z. Lignocellulose-based biosorbents for the removal of contaminants of emerging concern (CECs) from water: a review. Water. 2023;15(10):1853. https://doi.org/10.3390/w15101853.
Bhaumik M, Choi HJ, Seopela MP, McCrindle RI, Maity A. Highly effective removal of toxic Cr (VI) from wastewater using sulfuric acid-modified avocado seed. Ind Eng Chem Res. 2014;53:1214–24. https://doi.org/10.1021/ie402627d.
Leite AJB, Carmalin SA, Thue PS, dos Reis GS, Dias SLP, Lima EC, Vaguetti JCP, Pavan FA, Soares de Alencar W. Activated carbon from avocado seeds for the removal of phenolic compounds from aqueous solutions. Desalination Water Treat. 2017;71:168–81. https://doi.org/10.5004/dwt.2017.20540.
Leite AJB, Saucier C, Lima EC, dos Reis GS, Umpierres CS, Mello BL, Sampaio CH. Activated carbons from avocado seed: optimisation and application for removal of several emerging organic compounds. Environ Sci Pollut Res. 2018;25(8):7647–61. https://doi.org/10.1007/s11356-017-1105-9.
Aly AA, Górecki T. Green approaches to sample preparation based on extraction techniques. Molecules. 2020;25(7):1719. https://doi.org/10.3390/molecules25071719.
Gałuszka A, Migaszewski Z, Namieśnik J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TrAC Trends Anal Chem. 2013;50:78–84. https://doi.org/10.1016/j.trac.2013.04.010.
Sun R, Lu F, Yu C, Yang Y, Qiao L, Liu A. Peanut shells-derived biochars as adsorbents for the pipette-tip solid-phase extraction of endocrine-disrupting phenols in water, milk, and beverage. J Chromatogr A. 2022;1673:463101. https://doi.org/10.1016/j.chroma.2022.463101.
González-García P. Activated carbon from lignocellulosics precursors: a review of the synthesis methods, characterization techniques and applications. Renew Sustain Energy Rev. 2018;82:1393–414. https://doi.org/10.1016/j.rser.2017.04.117.
Gao Y, Yue Q, Gao B, Li A. Insight into activated carbon from different kinds of chemical activating agents: a review. Sci Total Environ. 2020;746:141094. https://doi.org/10.1016/j.scitotenv.2020.141094.
Kudo M, de Oliveira L, Suquila F, de Almeida F, Segatelli M, Lima É, Tarley C. Performance of avocado seed activated carbon as adsorbent for highly sensitive determination of Cd using a flow injection system online coupled to TS-FF-AAS. J Braz Chem Soc. 2020. https://doi.org/10.21577/0103-5053.20190132.
Rojas-Candia V, Arismendi D, Richter P. Determination of ibuprofen and 1-hydroxyibuprofen in aqueous samples using cork as a natural phase in rotating-disk sorptive extraction. J Chil Chem Soc. 2022;66(4):5722–6. https://doi.org/10.4067/S0717-97072022000405722.
Khan MZ, Militky J, Petru M, Tomková B, Ali A, Tören E, Perveen S. Recent advances in superhydrophobic surfaces for practical applications: a review. Eur Polym J. 2022: 111481. https://doi.org/10.1016/j.eurpolymj.2022.111481.
Cui B, Wang H. Molecular interaction of asphalt-aggregate interface modified by silane coupling agents at dry and wet conditions. Appl Surf Sci. 2022;572:151365. https://doi.org/10.1016/j.apsusc.2021.151365.
Mistar EM, Alfatah T, Supardan MD. Synthesis and characterization of activated carbon from Bambusa vulgaris striata using two-step KOH activation. J Mater Res Technol. 2020;9(3):6278–86. https://doi.org/10.1016/j.jmrt.2020.03.041.
Arismendi D, Becerra-Herrera M, Cerrato I, Richter P. Simultaneous determination of multiresidue and multiclass emerging contaminants in waters by rotating-disk sorptive extraction–derivatization-gas chromatography/mass spectrometry. Talanta. 2019;201:480–9. https://doi.org/10.1016/j.talanta.2019.03.120.
Torres-Lara N, Molina-Balmaceda A, Arismendi D, Richter P. Peanut shell-derived activated biochar as a convenient, low-cost, ecofriendly and efficient sorbent in rotating disk sorptive extraction of emerging contaminants from environmental water samples. Green Anal Chem. 2023;6:100073. https://doi.org/10.1016/j.greeac.2023.100073.
Pizzutti IR, Dias JV, Kok AD, Cardoso CD, Vela GME. Pesticide residues method validation by UPLC-MS/MS for accreditation purposes. J Braz Chem Soc. 2016;27(7). https://doi.org/10.5935/0103-5053.20160012.
Richter P, Arismendi D, Becerra-Herrera M. The fundamentals, chemistries and applications of rotating-disk sorptive extraction. TrAC Trends Anal Chem. 2021;137:116209. https://doi.org/10.1016/j.trac.2021.116209.
Bazzo A, Adebayo MA, Dias SL, Lima EC, Vaghetti JC, de Oliveira ER, Leite AJB, Pavan F. Avocado seed powder: characterization and its application for crystal violet dye removal from aqueous solutions. Desalination Water Treat. 2016;57(34):15873–88. https://doi.org/10.1080/19443994.2015.1074621.
Magdziarz A, Wilk M, Straka R. Combustion process of torrefied wood biomass: a kinetic study. J Therm Anal Calorim. 2017;127(2):1339–49. https://doi.org/10.1007/s10973-016-5731-0.
Paniagua S, Reyes S, Lima F, Pilipenko N, Calvo LF. Combustion of avocado crop residues: effect of crop variety and nature of nutrients. Fuel. 2021;291:119660. https://doi.org/10.1016/j.fuel.2020.119660.
Ma Y. Comparison of activated carbons prepared from wheat straw via ZnCl2 and KOH activation. Waste Biomass Valorization. 2016;8(3):549–59. https://doi.org/10.1007/s12649-016-9640-z.
Thommes M, Kaneko K, Neimark AV, Olivier JP, Rodriguez-Reinoso F, Rouquerol J, Sing KS. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl Chem. 2015;87(9–10):1051–69. https://doi.org/10.1515/pac-2014-1117.
Nowak E, Combes G, Stitt EH, Pacek AW. A comparison of contact angle measurement techniques applied to highly porous catalyst supports. Powder Technol. 2013;233:52–64. https://doi.org/10.1016/j.powtec.2012.08.032.
Funding
The authors would like to thank FONDECYT, Chile (Regular Project 1220225, Postdoctoral Project 3210084), FONDEQUIP (Project EQM170141), and ANID, Chile (National PhD scholarship 21200317 and National PhD scholarship 21230953) for financial support.
Author information
Authors and Affiliations
Contributions
Alejandra Molina-Balmaceda: conceptualization, methodology, formal analysis and investigation, writing—original draft preparation, software, visualization. Valentina Rojas Candia: conceptualization, methodology, formal analysis and investigation, writing—original draft preparation, software. Daniel Arismendi: conceptualization, writing—review and editing, supervision, visualization. Pablo Richter: conceptualization, funding acquisition, resources, supervision, writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ABC Highlights: authored by Rising Stars and Top Experts.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Molina-Balmaceda, A., Rojas-Candia, V., Arismendi, D. et al. Activated carbon from avocado seed as sorbent phase for microextraction technologies: activation, characterization, and analytical performance. Anal Bioanal Chem 416, 2399–2409 (2024). https://doi.org/10.1007/s00216-024-05203-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-024-05203-1