
Abstract
The objective of the current research was to develop a liquid chromatography-MSn (LC-MSn) methodology for the determination of free cortisol and its 15 endogenous metabolites (6β-hydroxycortisol, 20α-dihydrocortisol, 20α-dihydrocortisone, 20-β-dihydrocortisol, 20β-dihydrocortisone, prednisolone, cortisone, α-cortolone, β-cortolone, allotetrahydrocortisol, 5α-dihydrocortisol, tetrahydrocortisol, allotetrahydrocortisone, 5β-dihydrocortisol, tetrahydrocortisone) in human urine. Due to its optimal performance, a linear ion trap operating in ESI negative ion mode was chosen for the spectrometric analysis, performing MS3 and MS4 experiments. The method was validated for limit of detection (LOD) and limit of quantification (LOQ) (0.01 ng mL−1 and 0.05 ng mL−1, for all compounds, respectively), intra- and inter-day precision (CV = 1.4–9.2% and CV = 3.6–10.4%, respectively), intra- and inter-day accuracy (95–110%), extraction recovery (65–95%), linearity (R2 > 0.995), and matrix effect that was absent for all molecules. Additionally, for each compound, the percentage of glucuronated conjugates was estimated. The method was successfully applied to the urine (2 mL) of 50 healthy subjects (25 males, 25 females). It was also successfully employed on urine samples of two patients with Cushing syndrome and one with Addison’s disease. This analytical approach could be more appropriate than commonly used determination of urinary free cortisol collected in 24-h urine. The possibility of considering the differences and relationship between cortisol and its metabolites allows analytical problems related to quantitative analysis of cortisol alone to be overcome. Furthermore, the developed method has been demonstrated as efficient for antidoping control regarding the potential abuse of corticosteroids, which could interfere with the cortisol metabolism, due to negative feedback on the hypothalamus-hypophysis-adrenal axis. Lastly, this method was found to be suitable for the follow-up of prednisolone that was particularly important considering its pseudo-endogenous origin and correlation with cortisol metabolism.
Graphical abstract

Similar content being viewed by others
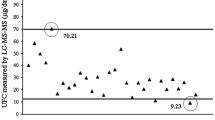

Introduction
Cortisol is a steroid hormone synthesized in the adrenal cortex, involved in glucidic metabolism, lipidic metabolism, and anti-inflammatory and immunosuppressive activity. Its secretion is controlled, through the hypothalamus-hypophysis-adrenal axis, by the corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH), and by cortisol itself that acts with a negative feedback mechanism on its secretion [1].
Cortisol is metabolized by the liver and kidneys. The main enzymes involved in phase 1 metabolic reactions are 11β-hydroxysteroid dehydrogenase isoform 1 (11β-HSD 1) that converts cortisone in cortisol and 11β-hydroxysteroid dehydrogenase isoform 2 (11β-HSD 2) that oxides cortisol in its inactive metabolite cortisone. Other enzymes with reductase and dehydrogenase activity allow the formation of other urinary metabolites. The most important are 5 α/β reductase, 3 α/β hydroxysteroid dehydrogenase, C20 reductase, and CYP3A4 [2, 3]. Interconversion of cortisol/cortisone by 11β-HSDs and formation of their A ring reduced metabolites (tetrahydrocortisol, allo-tetrahydrocortisol, tetrahydrocortisone, and allo-tetrahydrocortisone) as well as corresponding 20β-, 5β- 6β hydroxy metabolites are presented in Fig. 1. Furthermore, the correlation between cortisol/prednisolone must be taken into consideration as one of the important metabolic modifications of cortisol [4]. The metabolism of glucocorticoids includes also phase II conjugation reactions principally with glucuronic acid through UDP-glucuronyltransferase (UDP-GT) enzyme.

Chemical structures of cortisol and metabolites investigated
For their importance in the metabolic processes, many analytical methods have been developed for the detection and measurement of endogenous corticosteroids in human samples [5, 6]. In clinical analysis, cortisol measurement is used in the diagnosis of different endocrine disorders like Cushing’s syndrome, Addison disease, adrenal mineralocorticoid excess syndrome, congenital adrenal hyperplasia, adrenocortical cancer, and adrenal cortical adenoma [6, 7].
In these diagnoses, the three major tests performed are the measurement of urinary free cortisol in 24 h, plasma cortisol, and plasma ACTH [8]. Routine determination of cortisol is commonly performed by immunoassay tests, easy to perform, but lacking specificity due to the cross-reactivity problem of the antibodies used [9, 10]. Indeed, in human matrices, the presence of compounds structurally similar to cortisol is not negligible. Also, the presence of exogenous glucocorticoids, used for therapy, can give interference with the quantification of cortisol itself.
The extreme sensitivity and specificity of mass spectrometry can solve these problems [11]. In particular, for corticosteroid analysis in clinical diagnosis, different gas chromatography-mass spectrometry (GC–MS) methods were developed [12, 13], but, because of low corticosteroid volatilities, a derivatization step was always needed, so the throughput of a routine process can be difficult.
These problems were solved with the introduction of liquid chromatography-mass spectrometry (LC–MS) analysis that provides high analytical sensitivity and specificity, and rapid sample preparation [9, 14,15,16,17,18,19,20,21,22,23,24,25,26]. Thanks to these advantages, different LC–MS methods were proposed in the diagnosis of pathologies in which endogenous corticosteroids can be involved [27,28,29,30,31]. It has largely been demonstrated that LC–MS is the analytical method of choice for endogenous and exogenous corticosteroid measurements in human samples [32].
For LC–MS analysis of corticosteroids, triple quadrupole mass spectrometry operating in multiple reaction monitoring (MRM) in positive ion mode with electrospray source (ESI) is the most frequently used method to analyze these molecules [27,28,29]. On the contrary, there is just a few publications that deal with ion trap mass detector application in cortisol determination [33, 34]. Furthermore, information that regards the usage of LC-MSn methodology in cortisol metabolic profiling is lacking in the available literature. Therefore, this study was designed to develop and validate a LC-MSn method performed in electrospray (ESI) negative ion mode, for the qualitative and quantitative analyses of cortisol and 15 metabolites in human urine. Subsequently, the practical utility of this analytical platform for diagnostic and antidoping purposes will be preliminarily investigated.
Materials and methods
Reagents and chemicals
Cortisol [4-pregnen-11β,17α,21-triol-3,20-dione], prednisolone [1,4-pregnadiene-11β,17α,21-triol-3,20-dione], 6β-hydroxycortisol [4-pregnen-6β,11β,17,21-tetrol-3,20-dione], cortisone [17α,21-dihydroxy-4-pregnene-3,11,20-trione], allotetrahydrocortisol [5α-pregnan-3α,11β,17α,21-tetrol-20-one], tetrahydrocortisol [5β-pregnan-3α,11β,17α,21-tetrol-20-one], allotethrahydrocortisone [5α-pregnan-3α,17α,21-triol-11,20-dione], tetrahydrocortisone [5β-pregnan-3α,17α,21-triol-11,20-dione], 20α-dihydrocortisol [4-pregnen-11β,17α,20α,21-tetrol-3-one], 20-β dihydrocortisol [4-pregnen-11β,17α,20β,21-tetrol-3-one], 5α-dihydrocortisol [5α pregnan-11β,17α,21-triol-3,20-dione], 5β-dihydrocortisol [5β-pregnan-11β,17α,21-triol-3,20-dione], α-cortolone [5β-pregnan-3α,17α,20α,21-tetrol-11-one], β-cortolone [5β-pregnan-3α,17α,20β,21-tetrol-11-one], 20α-dihydrocortisone [4-pregnen-17α, 20α,21-triol-3,11-dione], 20β-dihydrocortisone [4-pregnen-17α,20β,21-triol-3,11-dione], and 6α-methylprednisolone [11β,17α,21-trihydroxy-6α-methyl-1,4-pregnadiene-3,20-dione] as internal standard (IS) were from Steraloids (Newport, RI, USA). β-Glucuronidase (E. coli K12 243 U mg−1) was from Roche (Mannheim, Germany).
All solvents were of HPLC grade. Tert-butyl methyl ether, acetonitrile, formic acid, and ammonium acetate, were from Sigma-Aldrich (St. Louis, MO, USA). Water was freshly prepared with a Milli-Q Advantage A10 Ultrapure Water Purification System (Merck-Millipore, Darmstadt, Germany).
Standard stock solutions (1 mg mL−1) were prepared by dissolving the dry powder of each analyte in methanol; solutions were stored at − 20 °C. Working solutions were prepared daily by diluting the stock solutions with methanol.
Sample collection
The concentrations of free cortisol and 15 free metabolites were calculated in the urine of 50 healthy volunteers (25 males and 25 females, 20 to 70 years old) divided into 5 groups of 10 people each (5 men and 5 women). Each group was subjected to a single urine collection at a given time of the day, i.e., 7 a.m.–11 a.m., 11 a.m.–15 p.m., 15 p.m.–19 p.m., 19 p.m.–23 p.m., and 23 p.m.–7 a.m. The 24-h urine was also collected from two volunteers (one male and one female).
Sample preparation
Two milliliters of human urine was spiked with 50 μL of the internal standard methylprednisolone to a final concentration of 2.5 ng mL−1. Tert-butyl methyl ether (2 mL) was added. After shaking in a vertical rotary shaker for 20 min, the sample was centrifuged at 2000 g for 20 min.
The upper layer was collected with a Pasteur pipette, transferred into a 10-mL glass tube, and dried under nitrogen stream at room temperature. The residue was dissolved in 100 µL of the mobile phase (23% acetonitrile, 77% aqueous solution 0.1% formic acid) and injected into the LC–MS system. The injection volume was 5 µL.
For all the compounds, to have a percentage value of the glucuronic acid derivatives of each compound, a hydrolysis step with β-glucuronidase was performed on 20 urine samples (10 males, 10 females) in which the free-form concentrations of every compound had been previously detected without hydrolysis.
For the hydrolysis procedure, 500 µL of ammonium acetate buffer 1 M (pH = 6.1) and 40 μL of β-glucuronidase were added. Samples were incubated for 60 min at 55 °C ± 5 °C. The extraction steps were the same as described for samples not undergoing deconjugation.
LC–MS parameters
The chromatographic separation was performed at room temperature, in isocratic condition, on a reversed-phase C18 Sunfire® column (150 × 2.1 mm i.d., 3.5 µm particle size; Waters, Milford, MA, USA) equipped with a Sunfire C18 Guard Column® (2.1 × 10 mm i.d., 3.5 µm particle size; Waters, Milford, MA, USA). The mobile phase consisted of a mixture of aqueous solution 0.1% formic acid and acetonitrile (77:23) at a flow rate of 0.3 mL min−1.
An LTQ® linear ion trap mass spectrometer equipped with an Electrospray Source (Thermo Fisher, San José, CA, USA), connected to a Surveyor Autosampler and a Surveyor MS Pump (Thermo Fisher, San José, CA, USA), was used.
The linear ion trap was operated in negative electrospray ionization mode [ESI ( −)] under the following conditions: sheath and auxiliary gas (nitrogen) flow rates of 40 and 20 arbitrary units, respectively; sweep gas was off; spray voltage of 4 kV; ion transfer capillary temperature 275 °C; capillary voltage − 5 V; and tube lens − 30 V. Helium was used as collision gas. Collision energy ranged between 18 and 25%.
Due to the fact that all compounds considered in the study can form a very abundant and stable adduct with formic acid in ESI ( −) mode ([M + HCOO]−), the MS parameters were optimized using the cortisol adduct with formic acid (MW = 407 Da) as reference compound, by direct injection of a standard solution of cortisol (1 µg mL−1) at a flow rate of 20 µL min−1. Data acquisition and analysis were accomplished using Xcalibur® software version 2.1 (Thermo Fisher, San José, CA, USA).
The compounds, their retention times, their molecular weights, the molecular weights of their adducts with formic acid (used as precursor ions), and the product ions with the related molecular weight are shown in Table 1, together with the collision energies (CE%). The product ions obtained after the MS2 experiment were used as precursor ions for the subsequent MS3 and eventual MS4 steps of analysis.
Method validation
Due to the endogenous nature of the corticosteroids involved in the study, an analyte-free matrix (blank) was obtained from a volunteer treated with a synthetic glucorticosteroid by oral administration (betamethasone, 1 mg day−1 for 7 days). On the fourth day after first administration, cortisol and consequently all its metabolites were lower than the LOD, for the effect of the negative feedback of the synthetic glucorticosteroid on the hypothalamus-hypophysis-adrenal axis. Urine collected after this time was used as blank urine to calculate all the validation parameters of the method. Validation was performed following the WADA guideline with more details provided in Eurachem guide [35,36,37]. For each analyte, the method performance was assessed through (1) qualitative parameters achieved by specificity and by the identification of compounds in urine with respect to the retention time and fragmentation pattern of their analytical standards; (2) quantitative parameters, such as the linearity, accuracy in terms of trueness (valued as bias), and precision expressed as the intra- and inter-day repeatability; (3) analytical sensitivity estimated as limit of detection (LOD) and limit of quantification (LOQ). Additionally, the robustness of the method as well as matrix effect was assessed, too.
For the linearity, two calibration curves were prepared on blank urine depending on the concentrations expected for each compound in healthy human urine [17], preliminarily calculated with a semiquantitative approach. The first calibration curve was prepared with six “low” concentrations of the free forms (0.05, 0.10, 0.25, 0.5, 1, 5 ng mL−1) while six “high” concentrations were used for the second calibration curve (5, 10, 50, 100, 250, 500 ng mL−1). Two curves were prepared with the same concentrations also in water.
The comparison between the curves in matrix and in water showed the absence of cortisol and its metabolites, and of matrix effect in urine. The matrix effect absence was also calculated comparing the peak areas of a standard solution at 100 ng mL−1 with the peak areas of the blank urine spiked after extraction with the same standard solution [38].
The limit of detection (LOD), defined as the lowest level at which a compound could be identified with a signal-to-noise (S/N) ratio greater than 3, and the limit of quantification (LOQ), defined as the lowest level at which a compound could be identified and quantified with a signal-to-noise ratio greater than 10, were calculated for each compound. For each corticosteroid, precision was determined by analyzing 3 QC samples at two different concentration levels (5 and 25 ng mL−1). Intra- and inter-assay precisions were expressed as CV%. Recovery (%) was calculated for all the compounds at two different urinary concentrations (5.0 and 25 ng mL−1). Robustness was observed in four different trials, fortifying blank urine samples at a concentration of 5.0 and 25 ng mL−1, changing slightly (± 10%) factors that may influence the outcome of the analysis. The factors were the volume of extraction solvent volume, centrifugation time, the volume of the mobile phase used for resuspension of dry extract and two different persons that performed analysis.
Statistical analysis
Descriptive statistics, Kolmogorov Smirnov test for normality, and Mann Whitney test were performed using GraphPad InStat version 3.10 for Windows (GraphPad Software, San Diego, CA, USA).
Results and discussion
LC-MSn analysis
In biological matrices, like human urine, LC–MS methods for corticosteroid imaging detection are usually performed in ESI ( +) using a triple quadrupole mass spectrometer operating in multiple reaction monitoring (MRM) acquisition mode [4, 39]. In the ESI positive ion mode, the precursor ions for fragmentation are either a pseudomolecular ion [M + H]+ or adducts with ammonium [M + NH4]+, methanol [M H + CH3OH]+, water [M + H − H2O]+, sodium [M + Na]+, etc. Generally, for structures with the carbonyl group on C3 position of steroid moiety the species [M + H]+ is used as precursor ion [15]).
In the ESI negative ion mode, we obtained a clear evidence that corticosteroids do not promptly generate the pseudomolecular ion[M − H]− but a very stable adduct with formic acid. In the present work, 0.1% aqueous solution of formic acid was used as polar phase to form [M + HCOO]−, used as precursor ion for the MS2 fragmentation. The adduct fragmentation tends to generate a base peak as predominant while the abundance of the other products ions is less than 20% of the base peak. The most abundant product ion obtained in the MS2 analysis was used as precursor ion for MS3 analysis. For some molecules, it was also possible to perform MS4 experiments, as shown in Table 1. This possibility of performing consecutive MSn acquisition steps presents a great advantage of the linear ion trap mass spectrometer with respect to common triple quadrupole mass spectrometer.
In Fig. 2, 5β-dihydrocortisol (to represent C 20 keto compounds) and α-cortolone (to represent C 20 hydroxy compounds) full-scan spectra of the different MSn steps of analysis are reported as an example. In the MS2 full-scan spectra that comprise all parent ions, it is possible to observe two different types of fragmentations: all the compounds with a hydroxyl group in C20 (C20 hydroxy compounds), like α and β-cortolone, 20α and 20β-dihydrocortisol, and 20α and 20β-dihydrocortisone, give a very abundant and stable product ion corresponding to a loss of 46 Da (formic acid). All the compounds with a keto group on C20 (C20 keto series), such as 5α and 5β-dihydrocortisol, allotetrahydrocortisol and tetrahydrocortisol, allotetrahydrocortisone and tetrahydrocortisone, prednisolone, cortisol, and cortisone, give a very abundant and stable product ion corresponding to a loss of 76 Da (formic acid and formaldehyde CH2O). For this reason, also the internal standard methylprednisolone for the presence of the keto group on C20 (C20 keto compound), in the MS2 spectrum, gives a very abundant ion (m/z 343) corresponding to a loss of 76 Da from its adduct with formic acid in negative ion mode (m/z 419). For all the compounds, these abundant ions obtained after the first fragmentation (MS2) were used as precursor ions for MS3 analysis.

Full-scan spectra of the different MSn steps for 5β-dihydrocortisol (represent C 20 keto compounds) and α-cortolone (to represent C 20 hydroxy compounds)
Due to the scarcity of product ions in MS2 spectra, MS3 analysis was chosen as the best compromise for specificity and sensitivity of the proposed method. Nevertheless, extreme specificity of MS3 with a very high S/N ratio allowed the achievement of an outstanding sensitivity. The differences in sensitivity operating in positive or negative mode were also checked working in MRM with a triple quadrupole, confirming the greater sensitivity of the negative mode for detection and quantification of glucocorticoids.
Figure 3 shows an LC-MS3 analysis of a blank human urine spiked at a concentration of 25 ng mL−.1 with all the 16 compounds and IS. The chromatographic conditions were carefully optimized until they permitted the complete separation of the isomers [40]

Full-scan LC-MS3 analysis of a blank human urine spiked at a concentration of 25 ng mL−1 with all the 16 compounds and IS. In order for retention time (Rt): 6β-hydroxycortisol (1), 20α-dihydrocortisol (2), 20α-dihydrocortisone (3), 20-β dihydrocortisol (4), 20β-dihydrocortisone (5), prednisolone (6), cortisol (7), cortisone (8), α-cortolone (9), β-cortolone (10), allotetrahydrocortisol (11), 5α-dihydrocortisol (12), tetrahydrocortisol (13), 6α-methylprednisolone as I.S. (14), allotetrahydrocortisone (15), 5β-dihydrocortisol (16), and tetrahydrocortisone (17)
Method validation
Validation was performed according to WADA criteria [41] and is presented in Table 2. A correlation coefficient higher than 0.995 was obtained in both calibration curves (urine and water) for all the compounds except 6β-hydroxycortisol. This compound was discharged from validation because of its polarity that did not permit obtaining in liquid–liquid extraction a stable repartition between polar and organic phases. For each compound, the LOD was 0.01 ng mL−1 and the LOQ was 0.05 ng mL−1. Regarding cortisol these values are lower than those recently obtained with LC–MS/MS tandem mass spectrometry [12]. Intra- and inter-day precision (expressed as % CV) was in the range 1.4–9.2 and 3.6–10.4, respectively. Intra- and inter-day accuracies (expressed as % bias) ranged from 95 to 110 for all the analytes. Regarding the robustness of the method, none of the factors (volume of extraction solvent, centrifugation time, volume of resuspension solvent, persons that executed the sample preparation procedure) showed a significant variation in the concentration measurements.
Recoveries ranged from 65 to 95% at two different urinary concentrations (5.0 and 25 ng mL−1) for all corticosteroids were in line with data reported by others [39].The lowest recoveries were achieved for those compounds with the 20α or 20β-hydroxyl group (C 20 hydroxyl series). This polar chemical group could explain the lowest distribution in the organic phase during liquid–liquid extraction and consequently the lower recovery. Matrix effect was completely absent for all compounds working in ESI negative ion mode respect of the positive ion mode where the matrix effect influenced notably. Our validation data are comparable with those obtained by Wang et al. [42]. The authors reported that cortisol and metabolites were detected by means of high-resolution mass spectrometry with Orbitrap technology, which is recognized for its high specificity. Nevertheless, liner ion trap analysis preceded by almost complete chromatographical separation has exhibited a high specificity and sensitivity to candidate this methodology as a reliable alternative to high-resolution mass spectrometry (HRMS).
Method application and results on healthy subjects
With the collection protocol adopted, all possible urine concentrations during the 24 h were obtained and the highest and the lowest levels for each molecule were determined. For all the free analytes in 50 healthy volunteers, the concentration range, expressed in nanograms per milliliter, is reported in Table 3. For the main compounds (cortisol and cortisone), the values found are in the line with recently reported concentration range [12, 15, 31]. The greatest concentration was detected for cortisone followed by cortisol. The 24-h urine from the two volunteers completely demonstrated the correlation of the circadian excretion profile of all the metabolites with the circadian rhythm of excretion of cortisol.
The normal distribution for steroid concentration in male and female urine was checked through the Kolmogorov Smirnov test, and, due to the lack of it in most of the groups, the Mann–Whitney test for unpaired samples was performed to check for differences between male and female urine. No difference (P > 0.05) was observed in urinary concentrations for all the steroids, between male and female. The total ion current (TIC) of an LC-MS3 analysis of a human urine after liquid–liquid extraction is shown in Fig. 4.

Full-scan LC-MS3 analysis of a human urine after liquid–liquid extraction
The percentage estimation of the conjugated form with glucuronic acid of all the compounds was performed on 20 urine samples (10 males, 10 females).
The concentrations of some conjugated metabolites, such as tetrahydrocortisol and tetrahydrocortisone, α-cortolone, and allotetrahydrocortisol, were higher than the highest calibration curve level (500 ng mL−1) used to calculate the free form. Therefore, a semiquantitative approach was applied, because the study aimed at the quantitation of the free forms, just to bypass the hydrolysis step, in a future routine procedure. After the hydrolysis step, tetrahydrocortisone and tetrahydrocortisol showed the highest urinary concentrations. The total ion current (TIC) of an LC-MS3 analysis of a human urine after the hydrolysis step is shown in Fig. 5.

Full-scan LC-MS3 analysis of a human urine after hydrolysis step and liquid–liquid extraction
About conjugation percentage, we experimentally observed 30–70% range of the glucuronated forms of cortisol, cortisone, 20α-dihydrocortisol, 20-β dihydrocortisol, 20α-dihydrocortisone, and 20β-dihydrocortisone. These molecules, on the A ring of the cyclopentanoperhydrophenanthrene nucleus, are characterized by a C4–C5 double bond and by the presence of a C3-keto group. On the other hand, a higher glucuronidation percentage, near to 100%, was calculated for allotetrahydrocortisol, tetrahydrocortisol, allotethrahydrocortisone, tetrahydrocortisone, α-cortolone, and β-cortolone. In these molecules, a α-hydroxyl group is bound to C3 of the A ring. This group chemically is favored for glucuronic acid conjugation by UDP glucuronyl transferase enzyme.
It is important to note that, considering a urine average volume of 1.5 L produced in 24 h, our computed concentrations agreed with the values expressed in micrograms per day reported by other authors [17].
Clinical application
To evaluate the possible application of the method in clinical diagnosis, two urine samples from Cushing patients and one sample from an Addison patient were analyzed.
Cushing’s syndrome is characterized by chronic glucocorticoid excess. It can be adrenocorticotropic hormone (ACTH) dependent (Cushing’s disease or ectopic ACTH production) or ACTH independent (adrenal adenoma or carcinoma). In patients with this syndrome, cortisol concentration is higher than in healthy people [7]. Consequently, also, the cortisol metabolite concentration is usually expected to be high [10]. Without any kind of clinical treatment, Cushing effect syndrome may include osteoporosis, hypertension, type 2 diabetes, frequent or unusual infections, and muscle mass loss and strength [6]. The diagnosis is really complex, and a lot of techniques are used. The recommended screening test is an overnight dexamethasone suppression test or a 24-h urinary free cortisol collection quantification [43].
Addison’s disease, also known as primary adrenal insufficiency and hypocortisolism, is a long-term disease characterized by reduced production of steroid hormones. Symptoms may include abdominal pain, weakness, and weight loss. Under certain circumstances, symptoms such as low blood pressure, vomiting, lower back pain, and loss of consciousness occur. An adrenal crisis can be triggered by stress, such as from an injury, surgery, or infection. Death may occur without treatment. The diagnosis made by biochemical laboratory tests in which blood and/or urine cortisol levels are measured before and after a synthetic form of ACTH is given by injection [44]. In urine the cortisol concentration is expected to be extremely low and consequently also all its metabolite concentration.
The urinary corticosteroid concentrations of tree patients (two with Cushing’s syndrome and one with Addison’s disease) are presented in the Table 3. The revealed concentrations clearly showed the macroscopic differences between healthy and pathological conditions. In the two Cushing urine samples, it was undoubtedly evident the overproduction of cortisol and of the metabolites with respect the healthy subjects. In the urine from the Addison patient, we obtained the opposite situation with an evident suppression of cortisol and metabolites with respect to the concentrations calculated in healthy people.
It is clearly evident that it is possible to select other potential biomarkers, in addition to cortisol, effective in recognizing a non-physiological situation. As defined by Kushnir et al. [16]: “biochemicals markers are endogenous compounds that are either not present in a normal physiological state or present within certain range of concentrations (e.g. intermediate and product of metabolic pathways).” Among these, “prednisolone” was also included. It was demonstrated that this molecule, very commonly used in therapy as a synthetic corticosteroid, has furthermore an endogenous production related to cortisol metabolism. In a normal subject, in urine, its concentration is usually lower than 0.5 ng mL−1. It was moreover possible to observe that, in Cushing patients, the calculated values were higher than the highest value (1 ng mL−1) calculated on 50 healthy subjects’ urine. This evidence can be considered as an additional confirmation of our study [34], about its endogenous origin and correlation with cortisol metabolism. Its high level in a pathological condition makes also prednisolone a candidate biomarker.
Antidoping application
In the World Anti-Doping Agency (WADA’s) list, glucocorticoids (e.g., prednisolone, dexamethasone, etc.) are considered doping substances and they are prohibited for the in-competition sport activities. Nevertheless, they are frequently abused due to their common utilization as anti-inflammatory drugs. The consumption of these drugs prompts a negative feedback inhibition of hypothalamus-hypophysis-adrenal axis, causing a lower production of cortisol and consequently of all its metabolites. When used in doping purposes, usually, they are given in small doses over a relatively long time period [45]. Because of their low concentrations and the fact that there is only limited understanding of their metabolism, the determination of exogenously administered corticosteroids remains difficult and rather problematic. The question that arises is whether the absence of these substances actually means that the subject had not abused them? An answer to this question cannot be given with certainty; studies on the impact of synthetic analogs on the metabolism of endogenous glucocorticosteroids would perhaps provide some explanations. Therefore, the accurate and precise measurement of endogenous corticosteroids can be considered a powerful tool in the identification of illegal glucocorticoid treatments. For this reason, the here-described technique for screening of cortisol and its metabolites can be used in antidoping analysis [42].
To test the feasibility of our method, two different human urine samples aimed for the antidoping control were analyzed. Those two samples were chosen appropriately, as the presence of betamethasone and dexamethasone was confirmed previously. In Table 3, the concentrations of each compound calculated in those two urine samples are reported. Doping case 1 shows a clear suppression of all steroids while in case 2, 20α-dihydrocortisol, 20α-dihydrocortisone, 20β-dihydrocortisol, and 20β-dihydrocortisone underwent a suppression, with other steroids in the low range value. It is therefore evident that biomarkers, different from cortisol and revealed by our method, could be useful to identify a pharmacologic treatment.
Conclusion
In this study, an LC-MSn method for simultaneous quantification of urinary cortisol and its 15 metabolites in human urine was developed and validated. The excellent sensitivity, accompanied by extreme specificity due to complete chromatographical separation of all species with subsequent detection in ESI negative ion mode with a linear ion trap like analyzer, undoubtedly showed the suitability of this method for different purposes. The method was used to calculate the concentration range of all free compounds in human urine of 50 healthy subjects and found its application in the identification of pathological conditions in which the corticosteroid metabolism is involved (ex. Cushing and Addison disease), as well as to identify urine of a sportsmen doped with corticosteroids.
The method evidently shows a high potential for diagnostics as only 2 mL urine from a single sampling was needed instead of the 24-h collection, commonly used for the assay of free cortisol in urine. Therefore, especially for its potential clinical application, it could be considered as a rapid screening tool, followed in a second time by further medical diagnostic investigations.
Moreover, the possibility of using cortisol metabolites as specific biomarkers of cortisol metabolism disbalance allows solving and bypassing some analytical problems that can be related to qualitative and quantitative analyses of cortisol alone [46, 47]. It remains to perform the studies on a huge number of human urines deriving from subjects with various pathophysiological conditions caused by disturbances and lack of regulation in cortisol metabolism.
References
Lemke TL, Williams DA, Foye WO, Roche VF, Zito SW. Foye’s principles of medicinal chemistry. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2013.
Kamrath C, Hartmann MF, Wudy SA. Quantitative targeted GC-MS-based urinary steroid metabolome analysis for treatment monitoring of adolescents and young adults with autoimmune primary adrenal insufficiency. Steroids. 2019;150: 108426. https://doi.org/10.1016/j.steroids.2019.108426.
Araujo-Castro M, Valderrábano P, Escobar-Morreale HF, Hanzu FA, Casals G. Urine steroid profile as a new promising tool for the evaluation of adrenal tumors. Literature review. Endocrine. 2021;72:40–8. https://doi.org/10.1007/s12020-020-02544-6.
Pavlovic R, Cannizzo FT, Panseri S, Biolatti B, Trutic N, Biondi PA, Chiesa L. Tetrahydro-metabolites of cortisol and cortisone in bovine urine evaluated by HPLC–ESI-mass spectrometry. J Steroid Biochem Mol Biol. 2013;135:30–5. https://doi.org/10.1016/j.jsbmb.2012.12.015.
Shackleton C. Clinical steroid mass spectrometry: a 45-year history culminating in HPLC–MS/MS becoming an essential tool for patient diagnosis. J Steroid Biochem Mol Biol. 2010;121:481–90. https://doi.org/10.1016/j.jsbmb.2010.02.017.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–17. https://doi.org/10.1016/S0140-6736(06)68699-6.
Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocr. 2008;93:1526–40. https://doi.org/10.1210/jc.2008-0125.
de Vries Lv, de Jong WHA, Touw DJ, Berger SP, Navis G, Kema IP, Bakker SJL. Twenty-four hour urinary cortisol excretion and the metabolic syndrome in prednisolone-treated renal transplant recipients. Steroids. 2017;127:31–9. https://doi.org/10.1016/j.steroids.2017.09.001.
Wood L, Ducroq DH, Fraser HL, Gillingwater S, Evans C, Pickett AJ, Rees DW, John R, Turkes A. Measurement of urinary free cortisol by tandem mass spectrometry and comparison with results obtained by gas chromatography-mass spectrometry and two commercial immunoassays. Ann Clin Biochem. 2008;45:380–8. https://doi.org/10.1258/acb.2007.007119.
Oßwald A, Wang R, Beuschlein F, Hartmann MF, Wudy SA, Bidlingmaier M, Zopp S, Reincke M, Ritzel K. Performance of LC–MS/MS and immunoassay based 24-h urine free cortisol in the diagnosis of Cushing’s syndrome. J Steroid Biochem Mol Biol. 2019;190:193–7. https://doi.org/10.1016/j.jsbmb.2019.04.004.
Luo A, el Gierari ETM, Nally LM, Sturmer LR, Dodd D, Shi R-Z. Clinical utility of an ultrasensitive urinary free cortisol assay by tandem mass spectrometry. Steroids. 2019;146:65–9. https://doi.org/10.1016/j.steroids.2019.03.014.
McDonald JG, Matthew S, Auchus RJ. Steroid profiling by gas chromatography–mass spectrometry and high performance liquid chromatography–mass spectrometry for adrenal diseases. Horm Cancer. 2011;2:324–32. https://doi.org/10.1007/s12672-011-0099-x.
Christakoudi S, Cowan DA, Taylor NF. Steroids excreted in urine by neonates with 21-hydroxylase deficiency. 2. Characterization, using GC–MS and GC–MS/MS, of pregnanes and pregnenes with an oxo- group on the A- or B-ring. Steroids. 2012;77:382–93. https://doi.org/10.1016/j.steroids.2011.12.018.
Kushnir MM, Rockwood AL, Bergquist J. Liquid chromatography-tandem mass spectrometry applications in endocrinology. Mass Spec Rev. 2010;29:480–502. https://doi.org/10.1002/mas.20264.
Raffaelli A, Saba A, Vignali E, Marcocci C, Salvadori P. Direct determination of the ratio of tetrahydrocortisol+allo-tetrahydrocortisol to tetrahydrocortisone in urine by LC–MS–MS. J Chromatogr. 2006;830:278–85. https://doi.org/10.1016/j.jchromb.2005.11.011.
McCann SJ, Gillingwater S, Keevil BG. Measurement of urinary free cortisol using liquid chromatography-tandem mass spectrometry: comparison with the urine adapted ACS:180 serum cortisol chemiluminescent immunoassay and development of a new reference range. Ann Clin Biochem. 2005;42:112–8. https://doi.org/10.1258/0004563053492775.
Marcos J, Renau N, Casals G, Segura J, Ventura R, Pozo OJ. Investigation of endogenous corticosteroids profiles in human urine based on liquid chromatography tandem mass spectrometry. Anal Chim Acta. 2014;812:92–104. https://doi.org/10.1016/j.aca.2013.12.030.
Deventer K, Delbeke FT. Validation of a screening method for corticosteroids in doping analysis by liquid chromatography/tandem mass spectrometry. RCM. 2003;17:2107–14. https://doi.org/10.1002/rcm.1157.
Cuzzola A, Mazzini F, Petri A. A comprehensive study for the validation of a LC–MS/MS method for the determination of free and total forms of urinary cortisol and its metabolites. J Pharm Biomed Anal. 2014;94:203–9. https://doi.org/10.1016/j.jpba.2014.02.003.
Cuzzola A, Petri A, Mazzini F, Salvadori P. Application of hyphenated mass spectrometry techniques for the analysis of urinary free glucocorticoids. RCM. 2009;23:2975–82. https://doi.org/10.1002/rcm.4214.
Kosicka K, Siemiątkowska A, Pałka D, Szpera-Goździewicz A, Bręborowicz GH, Główka FK. Detailed analysis of cortisol, cortisone and their tetrahydro- and allo-tetrahydrometabolites in human urine by LC–MS/MS. J Pharm Biomed Anal. 2017;140:174–81. https://doi.org/10.1016/j.jpba.2017.03.039.
Zhai X, Chen F, Zhu C, Lu Y. A simple LC–MS/MS method for the determination of cortisol, cortisone and tetrahydro-metabolites in human urine: assay development, validation and application in depression patients. J Pharm Biomed Anal. 2015;107:450–5. https://doi.org/10.1016/j.jpba.2015.01.041.
Turpeinen U, Stenman UH. Determination of urinary free cortisol by liquid chromatography-tandem mass spectrometry. Scand J Clin Lab Invest. 2003;63:143–50.
Kushnir MM, Rockwood AL, Roberts WL, Yue B, Bergquist J, Meikle AW. Liquid chromatography tandem mass spectrometry for analysis of steroids in clinical laboratories. Clin Biochem. 2011;44:77–88. https://doi.org/10.1016/j.clinbiochem.2010.07.008.
Dasenaki M, Papatzani M, Gounari E, Magnisali P, Papadopoulou-Marketou N, Kanaka-Gantenbein C, Moutsatsou P, Thomaidis NS. Simultaneous determination of free cortisol, cortisone and their tetrahydrometabolites in urine by single solvent extraction and liquid chromatography–tandem mass spectrometry. Anal Lett. 2019;52:2764–81. https://doi.org/10.1080/00032719.2019.1602629.
Chiesa L, Nobile M, Panseri S, Vigo D, Pavlovic R, Arioli F. Suitability of bovine bile compared to urine for detection of free, sulfate and glucuronate boldenone, androstadienedione, cortisol, cortisone, prednisolone, prednisone and dexamethasone by LC-MS/MS. Food Chem. 2015;188:473–80. https://doi.org/10.1016/j.foodchem.2015.04.131.
Lin C-L, Wu T-J, Machacek DA, Jiang N-S, Kao PC. Urinary free cortisol and cortisone determined by high performance liquid chromatography in the diagnosis of Cushing’s syndrome1. J Clin Endocrinol Metab. 1997;82:151–5. https://doi.org/10.1210/jcem.82.1.3687.
Ceccato F, Antonelli G, Barbot M, Zilio M, Mazzai L, Gatti R, Zaninotto M, Mantero F, Boscaro M, Plebani M, Scaroni C. The diagnostic performance of urinary free cortisol is better than the cortisol:cortisone ratio in detecting de novo Cushing’s syndrome: the use of a LC–MS/MS method in routine clinical practice. Eur J Endocrinol. 2014;171:1–7. https://doi.org/10.1530/EJE-14-0061.
Fong BM-W, Tam S, Leung KS-Y. Improved liquid chromatography–tandem mass spectrometry method in clinical utility for the diagnosis of Cushing’s syndrome. Anal Bioanal Chem. 2010;396:783–90. https://doi.org/10.1007/s00216-009-3247-1.
Pavlovic R, Chiesa L, Soncin S, Panseri S, Cannizzo FT, Biolatti B, Biondi PA. Determination of cortisol, cortisone, prednisolone and prednisone in bovine urine by liquid chromatography-electrospray ionisation single quadrupole mass spectrometry. J Liq Chromatogr Relat. 2012;35:444–57. https://doi.org/10.1080/10826076.2011.601496.
Chiesa LM, Nobile M, Biolatti B, Pavlovic R, Panseri S, Cannizzo FT, Arioli F. Detection of selected corticosteroids and anabolic steroids in calf milk replacers by liquid chromatography-electrospray ionisation - tandem mass spectrometry. Food Control. 2016;61:196–203. https://doi.org/10.1016/j.foodcont.2015.09.028.
Agrawal K, Voggu RR, Pisek D, Becht S, Chudnovskiy R, Dufour GM, Arfvidsson C, Thomas CE. A validated surrogate analyte LC-MS/MS assay for quantification of endogenous cortisol in human whole blood. J Pharm Biomed Anal. 2021;198: 114028. https://doi.org/10.1016/j.jpba.2021.114028.
Gaudl A, Kratzsch J, Bae YJ, Kiess W, Thiery J, Ceglarek U. Liquid chromatography quadrupole linear ion trap mass spectrometry for quantitative steroid hormone analysis in plasma, urine, saliva and hair. J Chromatogr A. 2016;1464:64–71. https://doi.org/10.1016/j.chroma.2016.07.087.
Fidani M, Gamberini MC, Pompa G, Mungiguerra F, Casati A, Arioli F. Presence of endogenous prednisolone in human urine. Steroids. 2013;78:121–6. https://doi.org/10.1016/j.steroids.2012.10.020.
WADA Technical Document-TD2019DL v. 2.0. Decision limits for the confirmatory quantification of threshold substances https://www.wada-ama.org/sites/default/files/resources/files/td2019dl_v2_finalb.pdf. Accessed 28 June 2022.
WADA Technical Document TDIDCR: minimum criteria for chromatographic-mass spectrometric confirmation of the identity of analytes for doping control purposes. https://www.wada-ama.org/en/what-we-do/science-medical/laboratorieshttps://www.wada-ama.org/en/anti-doping-partners/laboratories. Accessed 28 June 2022.
Eurachem Guide. The fitness for purpose of analytical methods (2014). https://www.eurachem.org/index.php/publications/guides/mv. Accessed 9 Jul 2022.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC−MS/MS. Anal Chem. 2003;75:3019–30. https://doi.org/10.1021/ac020361s.
de Kock N, Acharya SR, Ubhayasekera SJKA, Bergquist J. A novel targeted analysis of peripheral steroids by ultra-performance supercritical fluid chromatography hyphenated to tandem mass spectrometry. Sci Rep. 2018;8:16993. https://doi.org/10.1038/s41598-018-35007-0.
Chiesa L, Panseri S, Pavlovic R, Cannizzo FT, Biolatti B, Divari S, Villa R, Arioli F. HPLC-ESI-MS/MS assessment of the tetrahydro-metabolites of cortisol and cortisone in bovine urine: promising markers of dexamethasone and prednisolone treatment. Food Addit Contam - Chem Anal Control Expo Risk Assess. 2016;33:1175–89. https://doi.org/10.1080/19440049.2016.1202453.
WADA releases four Laboratory Technical Documents for 2022 | World Anti-Doping Agency. https://www.wada-ama.org/en/news/wada-releases-four-laboratory-technical-documents-2022. Accessed 24 May 2022.
Wang Y, Fujioka N, Xing C. Quantitative profiling of cortisol metabolites in human urine by high-resolution accurate-mass bioanalysis. (2018); 10:2015–2026. https://doi.org/10.4155/bio-2018-0182.
Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:2807–31. https://doi.org/10.1210/jc.2015-1818.
Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, Husebye ES, Merke DP, Murad MH, Stratakis CA, Torpy DJ. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016;101:364–89. https://doi.org/10.1210/jc.2015-1710.
Duclos M. Glucocorticoids: a doping agent? Endocrinol Metab Clin North Am. 2010;39:107–26. https://doi.org/10.1016/j.ecl.2009.10.001.
Pussard E, Travers S, Bouvattier C, Xue Q-Y, Cosson C, Viengchareun S, Martinerie L, Lombès M. Urinary steroidomic profiles by LC-MS/MS to monitor classic 21-hydroxylase deficiency. J Steroid Biochem Mol Biol. 2020;198: 105553. https://doi.org/10.1016/j.jsbmb.2019.105553.
Protti M, Mandrioli R, Mercolini L. Microsampling and LC–MS/MS for antidoping testing of glucocorticoids in urine. Bioanalysis. 2020;12:769–82. https://doi.org/10.4155/bio-2020-0044.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All of the authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Marco Fidani, Giulia Bussei, Francesca Mungiguerra, Radmila Pavlovic, Alessio Casati, and Francesco Arioli. The first draft of the manuscript was written by Maria Cristina Gamberini, Radmila Pavlovic, Federica Di Cesare, Giulia Bussei, and Susanna Draghi and all authors commented on previous versions of the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Human urine was collected anonymously for analytical method validation from informed volunteers. The urine sample collection was approved by the medical director of Unirelab srl on October 2019.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arioli, F., Gamberini, M.C., Pavlovic, R. et al. Quantification of cortisol and its metabolites in human urine by LC-MSn: applications in clinical diagnosis and anti-doping control. Anal Bioanal Chem 414, 6841–6853 (2022). https://doi.org/10.1007/s00216-022-04249-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-04249-3