
Abstract
Rationale
Major depression has been an area of extensive research during the last decades, for it represents a leading cause of disability and suicide. The stark rise of depression rates influenced by life stressors, economic threats, pandemic era, and resistance to classical treatments, has made the disorder rather challenging. Adult hippocampal neurogenesis and plasticity are particularly sensitive to the dynamic interplay between autophagy and inflammation. In fact, the intricate balance between the two processes contributes to neuronal homeostasis and survival.
Objectives
Having demonstrated promising potentials in AMPK activation, a major metabolic sensor and autophagy regulator, empagliflozin (Empa) was investigated for possible antidepressant properties in the reserpine rat model of depression.
Results
While the reserpine protocol elicited behavioral, biochemical, and histopathological changes relevant to depression, Empa outstandingly hindered these pathological perturbations. Importantly, hippocampal autophagic response markedly declined with reserpine which disrupted the AMPK/mTOR/Beclin1/LC3B machinery and, conversely, neuro-inflammation prevailed under the influence of the NLRP3 inflammasome together with oxidative/nitrative stress. Consequently, AMPK-mediated neurotrophins secretion obviously deteriorated through PKCζ/NF-κB/BDNF/CREB signal restriction. Empa restored hippocampal monoamines and autophagy/inflammation balance, driven by AMPK activation. By promoting the atypical PKCζ phosphorylation (Thr403) which subsequently phosphorylates NF-κB at Ser311, AMPK successfully reinforced BDNF/CREB signal and hippocampal neuroplasticity. The latter finding was supported by hippocampal CA3 toluidine blue staining to reveal intact neurons.
Conclusion
The current study highlights an interesting role for Empa as a regulator of autophagic and inflammatory responses in the pathology of depression. The study also pinpoints an unusual contribution for NF-κB in neurotrophins secretion via AMPK/PKCζ/NF-κB/BDNF/CREB signal transduction. Accordingly, Empa can have special benefits in diabetic patients with depressive symptoms.
Limitations
The influence of p-NF-κB (Ser311) on NLRP3 inflammasome assembly and activation has not been investigated, which can represent an interesting point for further research.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Depression is one of the most common neuropsychiatric disorders and a principal reason of disability worldwide. According to the World Health Organization estimation, depression affects globally more than 264 million people of all ages (Hanifiha et al. 2022; James et al. 2018). The disorder is highly heterogeneous with clinical manifestations characterized by a triad of emotional, neurovegetative, and neurocognitive symptoms which considerably deteriorate patients’ quality of life and social functioning (Park et al. 2018). Suicidal ideation and attempts are further attributes of depression, where up to 15% of severely depressed patients commit suicide. Additionally, depression has long been linked with developing other cardiovascular, metabolic, and central nervous system (CNS) diseases (Cizza et al. 2001; Hoerster et al. 2019).
The most accepted theories of depression include the historic monoamine neurotransmission deficiency, impaired hippocampal neurogenesis, and abnormal synaptic plasticity hypotheses, in addition to a subsidiary role for oxidative stress and neuro-inflammation (Jeon and Kim 2016; Liu et al. 2020). Nonetheless, significant concerns have been raised against the monoamine theory which left behind many unanswered questions. Importantly, this theory does not explain why many patients are considered "partial responders" to traditional antidepressants, and some are rendered with "treatment-resistant depression" (Otte et al. 2016; Pitsillou et al. 2020). Accordingly, it sounds imperative to search for new therapies that delve beyond monoamines and that address the underlying pathophysiology of depression. Given the well-established role of the monoamine theory, different animal models have been replicated to produce a depressive phenotype through depletion of central monoamines. Reserpine (Res), a no longer used antihypertensive alkaloid drug, acts mainly by depleting catecholamine stores in the CNS by acting as an irreversible inhibitor of the vesicular amine pump (Ikram and Haleem 2019). Therefore, single and repeated administration of Res is utilized as an animal model that mimics depression (Antkiewicz-Michaluk et al. 2014).
Although neuro-inflammation has been strongly associated with neurodegenerative disorders (Borikar et al. 2022), its significant contribution in depression and other neuropsychiatric diseases is still under debate (Hassamal 2023; Hurley and Tizabi 2013). In fact, the inflammatory response can start in the periphery and culminate in the brain or break out within the CNS under certain stimuli. Despite being a tightly regulated process that can be rather adaptive, negative aspects of neuro-inflammation lead to neurodegeneration and alterations in synaptic plasticity, resulting in dysfunction of emotion processing (Cai et al. 2019; Llorens-Martín et al. 2014). The nod-like receptor protein 3 (NLRP3) inflammasome is the most protein complex expressed during inflammation; motivating the secretion of interleukin (IL)-18 and IL-1β via caspase-1 activation (Cao et al. 2019; Tian et al. 2021). These released ILs have been strongly associated with stress response and depression by means of inhibiting hippocampal neurogenesis and disrupting neural networks that are involved in reward processing (Hassamal 2023). Accordingly, it is postulated that evidence of neuro-inflammation in depressed patients reflects poor prognosis and response to treatment (Troubat et al. 2020). It has been also believed that antidepressants exert their clinical benefit partly through hindering the NLRP3 inflammasome cascade by stimulating cellular autophagy (Trojan et al. 2019). Autophagy is a vital process for cellular homeostasis and it has been linked to several pathologies, including those which are stress-related (Gassen and Rein 2019). Just like inflammation, autophagy is a strictly controlled endeavor which is mainly responsive to nutrient and growth factors-sensitive pathways, like adenosine monophosphate-activated protein kinase (AMPK) and mechanistic target of rapamycin (mTOR), the master regulator of autophagy. AMPK is a pivotal adjuster of autophagy-related proteins, including microtubule-associated protein light chain 3 (LC3B) and Beclin1. In the same context, both pharmacological and non-pharmacological interventions have been shown to trigger different autophagic pathways to modulate depression-associated pathological mechanisms and behavior (Tang et al. 2021). It is worth noting that the autophagy protein LC3B has been negatively correlated with NLRP3 inflammasome activation in the spinal cord of mice with autoimmune encephalomyelitis (Shao et al. 2014), and in cerebral ischemia/reperfusion injury (Wang et al. 2019). Furthermore, reactive oxygen species (ROS) are known to thrive when autophagy deteriorates (Trocoli and Djavaheri-Mergny 2011). Interestingly, activation of AMPK has been reported to ameliorate depressive-like behavior via activating key neurogenesis pathways (Odaira et al. 2019). Nonetheless, besides to the chief role of AMPK and mTOR in orchestrating autophagy, complex interplay between nuclear factor kappa B (NF-κB) and autophagic assembly has been suggested (Youssef et al. 2021). In fact, due to its well-established role in mediating inflammation, it sounds relevant that NF-κB can repress autophagy (Singh and Singh 2020). Nevertheless, studies have revealed that the two players can control each other through positive and negative feedback mechanisms to achieve homeostasis. In addition, it has been concluded that NF-κB can indirectly activate or inhibit autophagy depending on the driving stimulus and cellular context (Mattson and Camandola 2001; Zhang and Hu 2012). Moreover, in the CNS, NF-κB plays a critical role in the coordinate expression of anti-apoptotic and anti-inflammatory genes, which promotes neuronal survival and, on the contrary, its inhibition causes neuronal death and neurodegenerative milieu (Koulich et al. 2001). In the same context, NF-κB is a potent inducer of Beclin1 expression; where suppression of NF-κB could disturb the expression of Beclin1, resulting in impairment of phagocytic clearance of neuritic plaque (Copetti et al. 2009). Most importantly, if phosphorylated at Ser311, NF-κB mediates the secretion of two critical neurotrophic factors: the nerve growth factor and brain-derived neurotrophic factor (BDNF) under the influence of protein kinase C (PKC)—an outcome that enhances neurogenesis and neuroplasticity (Ji et al. 2010; Lin et al. 2014; Obara et al. 2001).
Empagliflozin (Empa) is a novel oral anti-hyperglycemic agent that selectively inhibits sodium − glucose cotransporter-2 (SGLT2) in proximal renal tubules, thus promoting urinary glucose excretion and overcoming hyperglycemia, which is independent from insulin (Steven et al. 2017). Acknowledging the fact that SGLT2 inhibitors (gliflozins) can access the brain where their molecular target has been identified, research attention has been directed towards the extra cardiovascular and renal benefits of these drugs. However, cumulative evidence has revealed that SGLT2 inhibitors can modify various cellular, metabolic, and bioenergetic mechanisms to amend disease-associated pathological changes in the CNS (Pawlos et al. 2021; Rizzo et al. 2022). Apart from SGLT2 inhibition, gliflozins have been reported to activate AMPK and to replenish autophagy in various organs to achieve cellular protection and homeostasis (Chen et al. 2023; Fukushima et al. 2021; Packer 2022; Safaie et al. 2024). In recent years, growing studies have been unmasking significant neuroprotective properties of SGLT2 inhibitors in different CNS-related disorders, such as Alzheimer’s disease, Huntington's disease, Parkinson's disease, as well as cerebral ischemia, major depression, and epilepsy (Borikar et al. 2024; El-Sahar et al. 2020; Muhammad et al. 2021; Rizzo et al. 2022). In addition, Empa outstandingly refined cognition in a murine mixed model of diabetes and Alzheimer’s disease. The latter effect was linked to its ability to reduce neuronal loss, brain atrophy and hemorrhage, as well as microglial burden and amyloid plaques (Hierro-Bujalance et al. 2020). Moreover, Empa could mitigate cerebral ischemia/reperfusion-induced neuronal degeneration in hyperglycemic rats with consequent neuroprotective effects, which might be attributed not only to glycemic control, but also to its antioxidant, anti-inflammatory, and antiapoptotic properties (Amin et al. 2020). Interestingly enough, in a very recent case–control observational study, the authors have yielded unexpected outcomes, where the use of gliflozins in diabetic patients was associated with higher risks of depression and cognitive decline when compared to the control group (Nodirahon et al. 2024). However, such conflicting studies are rather limited and further investigations are mandatory to support or deny these assumptions.
The current study aims at investigating the effect of Empa in a Res model of depression in rats. It focuses on evaluating the dynamic interplay between autophagy and neuro-inflammation in the pathogenesis of depression. Moreover, the study unmasks the promising role of Empa as a fine-tuning agent that can regulate both processes to achieve neural resilience.
Materials and methods
Animals
This research was implemented according to the principles of The Guide for Care and Use of Laboratory Animals published in (2011) and was accepted by the Ethical Committee for Animal Experimentation of the Faculty of Pharmacy, Cairo University (Permit number: PT 3108). All efforts were exerted to minimize animals' suffering during the investigation duration.
Adult male Wistar rats, weighing 180 − 200 g, were obtained from the animal facility of the Egyptian Drug Authority (Giza, Egypt). Rats were adapted to the animal facility circumstances for 2 weeks before the beginning of the study. Animals were conserved under controlled environmental conditions of temperature (23 ± 2 °C), humidity (60 ± 10%), and light/dark (12/12 h) cycle. Food and water were allowed without hindrance during the experimental period.
Drugs and chemicals
In the current investigation, Res, escitalopram (Esc), and Empa were obtained from Sigma-Aldrich Chemical Co. (MO, USA), Apex Pharmaceutical Co. (Cairo, Egypt), and Boehringer Ingelheim Pharmaceuticals (CT, USA), respectively. The drugs were freshly prepared daily by dissolving in saline just before administration. All other chemicals were of the highest analytical grade.
Experimental design
As shown in Fig. 1, rats were randomly assigned into 4 groups (n = 13/group) as follows: Group I received an appropriate volume of saline intraperitoneally (i.p) and served as the negative (control) group. In group II, Res was given i.p at a dose of 0.2 mg/kg/day for 14 successive days, which counted as the positive control (model) subset. The protocol of Res administration for inducing a depressive phenotype was chosen based on prior studies (Antkiewicz-Michaluk et al. 2014; Khadrawy et al. 2018). Rats in group III received Esc (10 mg/kg/day) orally (Waugh and Goa 2003); one hour before Res injection. Finally, in group IV, rats received oral doses of Empa (10 mg/kg/day); one hour before receiving Res. The Empa dose (10 mg/kg) was selected based on prior studies (Abdel-latif et al. 2020; Amin et al. 2020; Hierro-Bujalance et al. 2020). Empa has been reported to alleviate neuronal apoptosis and enhance neurobehavioral functions in a dose-dependent manner in cerebral/ischemia reperfusion-injured rats, where the higher dose (10 mg/kg) was more protective than the lower one (1 mg/kg) (Abdel-latif et al. 2020). Treatment with Empa (10 mg/kg; i.p) also showed significant amelioration of behavioral/neurological functions and histopathological changes observed in brain tissues of hyperglycemic rats subjected to cerebral ischemi/reperfusion injury via suppressing oxidative/inflammatory/apoptotic pathways (Amin et al. 2020). In addition, it has been reported that Empa (10 mg/kg) attenuated neuronal damage, hemorrhage, and microglial burden, as well as cognitive deficits in a mixed murine model of Alzheimer’s disease and type 2 diabetes (Hierro-Bujalance et al. 2020).

Graphical illustration for the experimental timeline. Empa; empagliflozin, Esc; escitalopram, FST; forced swimming test, H & E; hematoxylin & eosin, IHC; immunohistochemistry, OFT; open field test; qRT-PCR; quantitative (real-time) reverse transcription polymerase chain reaction, Res; reserpine, TST; tail suspension test, WB; western blotting
Drugs administration lasted for 14 days and, on the 15th day, animals were subjected to behavioral assessments to verify the depressive phenotype. Eventually, rats were scarificed by decapitation under anesthesia using phenobarbitone (40 mg/ kg; i.p) (El-Sahar et al. 2020).
For histopathological investigations, whole brain tissues of 3 rats/group were directly immersed in 10% phosphate-buffered formalin solution until being processed. Meanwhile, the hippocampi of 10 rats/group were rapidly dissected and subdivided into two subsets; the first subset (n = 6/group) was used to prepare 10% hippocampal homogenates using phosphate-buffered saline, and the second subset (n = 4/group) was used for subsequent western blotting and PCR assessments. All hippocampal specimens were kept at -80° C and only thawed at the time of the respective analysis.
Behavioral assessments
After 24 h of the last injection day, locomotor function of the animals was evaluated through the open field test (OFT), and they were examined for their depressive behavior using tail suspension and forced swimming tests. All tests were performed in a sound-isolated room starting at 12 p.m. with a two-hour resting period between the tests.
Open Field Test (OFT)
The OFT was accomplished to evaluate spontaneous locomotor function of rats using a square wooden box (80 × 80 × 40 cm) with red walls and a black polished floor divided by white lines into 16 equal squares (4 × 4). Individually, animals were placed gently into the center of the open field apparatus and the behavior of each rat was recorded for 3 min using an overhead camera existing in the room. The floor and walls were cleaned after each testing session using 70% ethyl alcohol to remove possible odors left by prior rats. The ambulation frequency, which is the number of times each rat crosses one of the grid lines with all four paws, was assessed (Khadrawy et al. 2018; Yu et al. 2019).
Tail Suspension Test (TST)
The TST was implemented to assess behavioral despair by measuring the immobility time displayed by rats. Each rat was fixed upside down about 50 cm above the floor using adhesive tape placed approximately 1 cm from the tip of the tail in such a position that rendered rats unable to escape or to hold on to nearby surfaces. Rats were considered immobile when they stopped struggling to overcome this abnormal position or when they became completely motionless after a period of struggling activity. This test was performed for 5 min and the total duration of immobility was recorded (Takahashi et al. 2018).
Forced Swimming Test (FST)
The FST is a widely used assessment tool for the effect of antidepressants in animal models of depression. In this test, rats were placed individually into a glass cylinder (height: 50 cm; diameter: 20 cm) containing tap water (25 ± 2 °C; depth: 30 cm). These criteria should not allow the animals to support themselves by touching the bottom of the cylinder with their paws or tails. The assessment lasted for 5 min and, after cessation of vigorous activity by rats, the total duration of immobility was recorded with the help of a stopwatch. Rats were considered immobile when they stopped swimming; making only the necessary movements for keeping their heads above the water surface. Of note, swimming water was regularly changed between sessions (Han et al. 2009; Takahashi et al. 2018).
Biochemical measurements
Enzyme-Linked Immunosorbent Assays (ELISAs)
MyBiosource (CA, USA) specific ELISA kits were purchased for determination of hippocampal contents of serotonin (5-HT; Cat. No. MBS9362408), dopamine (DA; Cat. No. MBS7214676), reduced glutathione (GSH; Cat. No. MBS265966), malondialdehyde (MDH; Cat. No. MBS727531), inducible nitric oxide synthase (iNOS; Cat. No. MBS263618), caspase-1 (Cat. No. MBS765838), IL-1β (MBS825017), p-PKC zeta (ζ; Thr403; Cat. No. MBS725403), and BDNF (Cat. No. MBS824814), each according to the manufacturer's instructions.
Additionally, hippocampal contents of noradrenaline (NE) and tumor necrosis factor (TNF)-α were measured using Cusabio ELISA kits (Wuhan, China; Cat. No. CSB-E07022r and CSB E11987r, respectively), and that of IL-18 was estimated by the specific ELISA kit obtained from Elabscience (Wuhan, China; Cat. No. E-EL-R0567). Meanwhile, the protein content of each sample was detected in the aliquots according to the method of Lowry et al. (2014).
Western blot analysis
Estimation of the phosphorylated forms of AMPK (Thr172) and cAMP response element-binding protein (CREB; Ser133), as well as NF-κB p65 (Ser311), and finally mTOR hippocampal protein expressions were accomplished using western blot analysis. The right-sided hippocampi of 4 rats per group were homogenized using ReadyPrep™ protein extraction kit (Bio-Rad Inc., CA, USA). Following protein quantification using Bradford protein assay kit (ThermoFisher Scientific Inc., MA, USA), equal protein amounts from each sample were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a nitrocellulose membrane using a semi-dry transfer apparatus (Bio-Rad, CA, USA). The membranes were then blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline containing Tween 20 (TBST) at room temperature for 1 h to prevent non-specific protein binding. Afterwards, the membranes were incubated overnight at 4 °C on a roller shaker with the corresponding primary antibodies: anti-β-actin (Cat. No. MA1-91,399), anti-p-AMPK (Thr172; Cat. No. PA5-37,821), anti-p-CREB (Ser133; Cat. No. MA5-11,192), anti-p-NF-kB p65 (S311; Cat. No. PA5-104,959), and anti-mTOR antibodies (Cat. No. AHO-1232). All the primary antibodies were obtained from ThermoFisher Scientific (MA, USA). Subsequently, the generated blots were rinsed 3 − 5 times for 5 min with TBST and incubated for 2 h at room temperature with horseradish peroxidase-conjugated secondary antibody (Dianova, Hamburg, Germany). Finally, the chemiluminescence substrate reaction was applied to the blot according to the manufacturer's recommendation (Amersham Biosciences, IL, USA). The corresponding intensities of the established protein bands were measured using densitometric analysis by the aid of a scanning laser densitometer (Biomed Instrument Inc., CA, USA). The results were represented as arbitrary units related to β-actin bands’ intensities.
Quantitative real-time PCR analysis of NLRP3
All samples were assayed in duplicates, where total RNA was extracted from left-sided hippocampal tissues (n = 4/group) using SV Total RNA Isolation System (ThermoFisher Scientific, MA, USA). RNA purity was estimated by 260/280 nm absorption ratio using ThermoFisher Scientific NanoDrop™ spectrophotometer. Using 1 μg of the extracted RNA per sample, reverse transcription (RT) of RNA into complementary DNA (cDNA) was developed by the aid of the Reverse Transcription System (Promega, Leiden, Netherlands) in accordance with the manufacturer's guidelines. NLRP3 gene expression was determined via quantitative (q) real-time PCR using Maxima SYBR Green qPCR Master Mix (ThermoFisher Scientific, MA, USA), where 5 μL cDNA was used and exposed to 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 60 s, and extension at 72 °C for 60 s. The specific primers and probes were chosen from Applied Biosystems TaqMan Assays inventories. The sequences of the used primers are listed in Table 1. Data quality check was performed using amplification, primer melting temperature, and cycle threshold values to exclude any outliers before calculation. The 2−ΔΔCT formula was used to obtain the expression of target genes relative to the housekeeping gene, β-actin (Livak and Schmittgen 2001).
Histopathological inspections
The brains of 3 rats per group were removed and fixed in 10% buffered formalin for 72 h. Afterwards, the specimens were washed, dehydrated in serial grades of ethanol, and cleared in xylene. Samples were then infiltrated and embedded into Paraplast tissue embedding media (Leica Biosystems, Wetzlar, Germany). Serial sagittal brain sections of 5 μm thickness were cut by a rotatory microtome and stained with hematoxylin and eosin (H & E) for histopathological examination of the hippocampal regions under light microscope. For detecting surviving neurons, 5 μm-thick paraffinized sections were stained with a toluidine blue stain, then seven random non-overlapping fields/tissue section were examined for quantification of intact neurons in the hippocampal Cornu Ammonis (CA3) region. All micrographs were obtained using a full HD microscopic imaging system operated by Leica application module for histological analysis (Leica Microsystems GmBH, Wetzlar, Germany). All standard procedures and protocols for sample fixation and staining were carried out according to the previously described method (Culling 2013). Histopathological handling and assessment of specimens were accomplished by a professional observer who was unaware of samples identity to avoid any bias.
Immunohistochemical examination of Beclin1 and LC3B
Immunohistochemical staining for Beclin1 and LC3B was done as stated in the manufacturer’s guidelines. Four μm-thick paraffinized tissue sections were first dewaxed with xylene, then rehydrated in graded ethanol, and finally heated for 5 min in citrate buffer. Hydrogen peroxide (0.3%) was used to block antigen-retrieved brain tissue sections that were then incubated with the primary antibodies; anti-Beclin1 (1:100; GenTex, CA, USA; Cat. No. GTX55535) and anti-LC3B antibody (1:200; Bioss Antibodies, MA, USA; Cat. No. bs-11731R). Thereafter, sections were washed with phosphate-buffered saline and incubated for 20 min with the biotinylated link antibody and peroxidase-labelled streptavidin (Dako Carpinteria, CA, USA). The reaction was visualized with diaminobenzidine tetrahydrochloride (DAB) substrate kit (Vector Laboratories Inc., CA, USA). Counter staining with hematoxylin was accomplished, followed by dehydration and clearing in xylene. Tissue sections were then slipcovered for microscopic examination.
For immunohistochemical quantitative analysis, six random non-overlapping fields/immunostained tissue section were analyzed for determining Beclin1 and LC3B levels. All morphological examinations, photographs, as well as quantitative analysis were recorded using Leica's application system modules for histological analysis (Leica Microsystems GmbH, Wetzlar, Germany).
Statistical analysis
Results are illustrated as means ± SD. One − way ANOVA followed by Tukey’s multiple comparison test were used for analyzing all results. Statistical analysis was accomplished using GraphPad Prism software (version 9) with the probability level of significance less than 0.05. For each effect, the F-value (F), degrees of freedom (df), the effect size partial eta squared (ηp2), and statistical significance (p) were reported. Additionally, pairwise comparisons were provided in each relevant figure.
Results
Effect of Empa on Res-induced alterations in OFT, TST, and FST
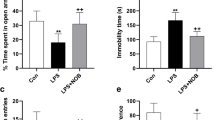
Rats exposed to i.p Res injections exhibited behavioral alterations signifying depression-like symptoms, which was reflected in the considerable prolongation of immobility durations in TST as well as FST, when compared to the control group (for immobility duration in TST: F(3, 48) = 18.85, ηp2 = 0.5409, p < 0.0001 and for immobility duration in FST: F(3, 48) = 32.42, ηp2 = 0.6695, p < 0.0001). Treatment with either Esc or Empa showed equivalent efficacy in terms of decreasing the immobility periods and restoring them to the normal values (Fig. 2B & C).

Effects of Empa on Res-induced alterations in (A) ambulation frequency in the OFT, (B) immobility time during the TST, and (C) immobility time during the FST. Data is expressed as mean ± SD (n = 13), using one-way ANOVA with Tukey − Kramer multiple comparison post-test, (p < 0.05). Empa; empagliflozin, Esc; escitalopram, FST; forced swimming test, OFT; open field test, Res; reserpine, TST; tail suspension test
Of note, rats subjected to Res injection showed lower ambulation frequency in the OFT as compared to the control group (F(3, 48) = 27.59, ηp2 = 0.6330, p < 0.0001). Moreover, no significant difference was elucidated in the number of squares crossed during the 3-min test when treating rats with Esc or Empa, as related to the Res group (Fig. 2A).
Effects of Empa and Res on hippocampal histopathology and histomorphometry
Microscopic examination of H & E brain sections (Fig. 3) was carried out in addition to using toluidine-blue stain (Fig. 4) to determine the mean count of intact neurons in CA3 hippocampal areas. While the control group demonstrated normal organized morphological structures of hippocampal layers (black arrows) and intact intercellular matrix with minimal reactive glial cell infiltrates, on the contrary, the Res group rats revealed severe neuropathic alterations. These modifications included severe neuronal degeneration with abundant records of hyperesenophilic, pyknotic, and structureless pyramidal neurons that lack distinct subcellular details (red arrows) and few dispersed apparent intact cells (black arrows). In addition, severe perineuronal edema accompanied with obviously high reactive glial cell infiltrates (arrowheads) with congested blood vessels (star) were observed. Moreover, the toluidine blue-stained sections of Res group showed decreased count of intact neurons (black arrows) with higher records of degenerated neurons (red arrows), as compared to the control group (F(3, 24) = 472.7, ηp2 = 0.9834, p < 0.0001). Interestingly, administration of either Esc or Empa in Res-injected rats demonstrated noticeable neuroprotective efficacy. Improvement of the microscopic picture was visually signified by minimal sporadic records of degenerated neurons in some tissue sections (red arrows), relatively higher figures of intact neurons (black arrows), and milder persistent records of reactive glial cell infiltrates (arrowheads), along with normal appearance of brain matrix.

Effects of Empa and Res on hippocampal histopathology. Representative H & E-stained photomicrographs (CA3 region, n = 3, 400 ×) for: (A) panel from the control group demonstrating normal organized morphological structures of hippocampal layers (black arrows), intact intercellular matrix, and minimal reactive glial cell infiltrates. (B) The Res group reveals severe neuronal degeneration, abundant records of hyperesenophilic, pyknotic, and structureless pyramidal neurons (red arrows), few dispersed apparent intact cells (black arrows), severe perineuronal edema, high reactive glial cell infiltrates (arrowheads), along with congested blood vessels (star). Sections from (C) Esc and (D) Empa showing marked regression of the neuropathological alterations signified by minimal sporadic records of degenerated neurons (red arrows), relatively higher figures of intact neurons (black arrows), and milder persistent records of reactive glial cell infiltrates (arrowheads) along with normal appearance of brain matrix. Empa; empagliflozin, Esc; escitalopram, Res; reserpine

Effects of Empa and Res on hippocampal histomorphometry. Illustrative toluidine blue-stained photomicrographs (CA3 region, n = 7, 400 ×) from (A) control, (B) Res, (C) Res + Esc, and (D) Res + Empa-treated groups. Black arrows represent intact neurons, whereas red arrows indicate degenerated ones. The number of intact neurons per group is represented in panel (E), where data is presented as mean ± SD using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). Empa; empagliflozin, Esc; escitalopram, Res; reserpine
Effect of Empa on Res-induced alterations in hippocampal neurotransmitters
Dysregulation of neurotransmitters was shown in Res-induced model of depression as evidenced by the significant reductions in the hippocampal contents of 5-HT, NE, and DA, which were almost halved under the effect of Res, when compared to the control group (for 5-HT: F (3, 20) = 32.91, ηp2 = 0.8316, p < 0.0001; for NE: F (3, 20) = 125.8, ηp2 = 0.9497, p < 0.0001; and for DA: F (3, 20) = 46.01, ηp2 = 0.8734, p < 0.0001). Such effects were remarkably reversed by treatment with Empa which produced remarkable upsurges in hippocampal 5-HT, NE, and DA. Of note, Empa produced analogous results to those offered by Esc in terms of its effect on hippocampal neurotransmitters (Fig. 5).

Effects of Empa on Res-induced changes in hippocampal contents of (A) 5-HT, (B) NE, and (C) DA. Data is expressed as mean ± SD (n = 6) using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). 5-HT; serotonin, DA; dopamine, Empa; empagliflozin, Esc; escitalopram, NE; norepinephrine, Res; reserpine
Impact of Empa on Res-induced alterations in hippocampal p-AMPKα1 expression
Hippocampal expression of p-AMPKα1 (Thr172) was significantly reduced in rats, as compared to the control group when treated with Res, for it reached one-third the normal values (F (3, 12) = 44.32, ηp2 = 0.9172, p < 0.0001). On the contrary, compared to the Res group, rats treated with Esc showed more than two-fold elevation of p-AMPKα1. Likewise, p-AMPKα1 levels in the hippocampi of Empa-treated rats were significantly higher than those of the Res group but were not significant from Esc (Fig. 6).

Effects of Res and co-treatments on hippocampal p-AMPKα1 (Thr172) gene expression. Data is expressed as mean ± SD (n = 4) using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). AMPK; adenosine monophosphate-activated protein kinase, Empa; empagliflozin, Esc; escitalopram, Res; reserpine
Effect of Empa on Res-induced alterations in hippocampal autophagic regulators
Herein, the autophagic markers Beclin1 and LC3B were visualized and quantified by the immunohistochemical technique, while hippocampal expression of mTOR was assessed via western blotting analysis. Remarkable dysregulations in hippocampal autophagic machinery were observed through Res, where Beclin1 and LC3B declined and mTOR flourished as compared to the control group (for Beclin1: F(3, 20) = 134.4, ηp2 = 0.9528, p < 0.0001; for LC3B: F(3, 20) = 223.7, ηp2 = 0.9711, p < 0.0001; and for mTOR: F(3, 12) = 31.01, ηp2 = 0.8857, p < 0.0001). These alterations were equally amended by Esc and Empa, except for Beclin1 which was more impacted by Esc than by Empa. In this context, both Esc and Empa significantly replenished the immunohistochemical expressions of Beclin1 and LC3B. Meanwhile, they both reduced the protein expression of mTOR to one-half its value as compared to the Res group (Fig. 7).

Effects of Empa and Res on hippocampal (A) mTOR expression as well as immunoreactivity of (B) Beclin1 and (C) LC3B, where the representative photomicrographs of Beclin1 and LC3B immuno-staining show microsections of (i) control, (ii) Res, (iii) Res + Esc, and (iv) Res + Empa (n = 6, 400 ×), with (v) showing the percentage area staining for each of Beclin1 and LC3B. For mTOR (n = 4), Beclin1 (n = 6), and LC3B (n = 6), data is expressed as mean ± SD using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). Empa; empagliflozin, Esc; escitalopram, LC3B; microtubule-associated protein light chain 3, mTOR; mechanistic target of rapamycin, Res; reserpine
Effect of Empa on Res-induced oxidative burden and neuro-inflammation
Administration of Res instigated a profound state of redox imbalance as exemplified by the significant depletion in hippocampal GSH along with marked elevations in MDA and iNOS levels, which came to 45%, 231%, and 354% their normal magnitudes, respectively (for GSH: F(3, 20) = 73.86, ηp2 = 0.9172, p < 0.0001; for MDA: F(3, 20) = 31.96, ηp2 = 0.8274, p < 0.0001; and for iNOS: F(3, 20) = 98.82, ηp2 = 0.9368, p < 0.0001). Empa exerted outstanding antioxidant potentials, equivalent to Esc, which was reflected in the remarkable increment in hippocampal GSH and the mitigation of MDA and iNOS levels, as compared to the Res group (Fig. 8). In line with the aforementioned results, Res promoted a state of inflammatory perturbation driven by the NLRP3 inflammasome pathway. Hence, as compared to the control group, hippocampal gene expression of NLRP3 showed an eight-fold increase in the Res group, with a consequent upsurge in caspase-1 concentration (for NLRP3: F(3, 12) = 18.08, ηp2 = 0.8189, p < 0.0001 and for caspase-1: F(3, 20) = 17.85, ηp2 = 0.7281, p < 0.0001). The inflammatory disturbance entailed hippocampal contents of IL-1β, IL-18, as well as TNF-α, which reached about 2-, 3-, and 4-folds their normal values, respectively (for IL-1β: F(3, 20) = 143.7 ηp2 = 0.9557, p < 0.0001; for IL-18: F(3, 20) = 110.5, ηp2 = 0.9431, p < 0.0001; and for TNF-α: F(3, 20) = 97.71, ηp2 = 0.9361, p < 0.0001). The NLRP3 inflammasome activation pathway was effectively dampened upon Empa administration resulting in significant decrements in the NLRP3/caspase-1/IL-1β/IL-18/TNF-α cascade versus the Res group. Importantly, both Empa and Esc were equally efficient in terms of their ability to halt the NLRP3 inflammasome overactivation (Fig. 9).

Effects of Empa on Res-induced changes in hippocampal levels of (A) GSH, (B) MDA, and (C) iNOS. Data is expressed as mean ± SD (n = 6) using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). Empa; empagliflozin, Esc; escitalopram, GSH; glutathione, iNOS; inducible nitric oxide synthase, MDA; malondialdehyde, Res; reserpine

Effects of Empa and Res on hippocampal (A) NLRP3 gene expression in addition to hippocampal contents of (B) caspase-1, (C) IL-1β, (D) IL-18, and (E) TNF-α. For NLRP3 (n = 4), caspase-1, IL-1β, IL-18, and TNF-α (n = 6), data is expressed as mean ± SD using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). Empa; empagliflozin, Esc; escitalopram, IL; interleukin, NLRP3; nod-like receptor protein 3, Res; reserpine, TNF; Tumor necrosis factor
Effects of Esc and Empa on Res-induced alterations in hippocampal p-PKCζ/p-NF-κB/BDNF/p-CREB pathway
Injection with Res induced marked decline in hippocampal p-PKCζ (Thr403), p-NF-κB p65 (Ser311), BDNF, and p-CREB (Ser133), as compared to the control group (for p-PKCζ: F(3, 20) = 87.09, ηp2 = 0.9289, p < 0.0001; for p-NF-κB p65: F(3, 12) = 40.30, ηp2 = 0.9097, p < 0.0001; for BDNF: F(3, 20) = 60.65, ηp2 = 0.9010, p < 0.0001; and for p-CREB: F(3, 12) = 33.38, ηp2 = 0.8930, p < 0.0001). Administration of Esc resulted in remarkable increments in the concentrations of p-PKC and BDNF, as well as the protein expressions of p-NF-κB p65 and p-CREB when related to the Res group. Notably, the effects of Empa were comparable to those displayed by Esc, and both drugs outstandingly normalized hippocampal BDNF, as compared to the Res group (Fig. 10).

Effects of Empa and Res on hippocampal (A) p-PKCζ (Thr403) levels, (B) p-NF-kB p65 (S311) expression, (C) BDNF, and (D) p-CREB (Ser133) expression. For p-PKCζ (n = 6), p-NF-kB p65 (n = 4), BDNF (n = 6), and p-CREB (n = 4), data is expressed as mean ± SD using one-way ANOVA followed by Tukey's post-hoc test, (p < 0.05). BDNF; brain derived neurotrophic factor, CREB; cAMP response element-binding protein, Empa; empagliflozin, Esc; escitalopram, NF-kB; nuclear factor kappa B, PKCζ; protein kinase C zeta, Res; reserpine
Discussion
In light of the growing studies that have been investigating the promising health benefits of SGLT2 inhibitors, the current work puts insights into the neuroprotective potentials of Empa as a driving force for neuroplasticity and resilience. Using Res as a pharmacological insult for depression in rats, Empa was explored as a fine regulator of autophagy and inflammation—two major and opposite processes that largely influence adult neurogenesis. Meanwhile, Esc was implemented as a reference antidepressant drug.
Our results imply that mitigating the depressive behavior in rats by Empa was early recognized in the FST and the TST, in which Empa improved rats' mobility and, convincingly, ameliorated their despair—findings that are constantly reported with standard antidepressants (Chen et al. 2001; Odaira et al. 2019). Also, in a previous study, dapagliflozin, a SGLT2 inhibitor, was able to mitigate the despair behavior of rats exposed to stress in FST (Muhammad et al. 2021). Of note, Res demonstrated a negative outcome on the locomotor function of rats in the OFT, while Empa, on the opposite side, successfully overcame this effect and enhanced the ambulation frequency of rats. Although the influence of depression on locomotor activity has not been consistent among studies due to many variables, Res has been repeatedly linked with motor function deterioration mostly due to DA depletion (Khadrawy et al. 2018; Leao et al. 2015; Lima et al. 2021).
At the biochemical level, the current findings insinuate that hippocampal neuro-inflammation outweighs the autophagic process under Res administration. As previously mentioned, autophagy, the programmed cell survival mechanism, plays a protective role against cell damage in the CNS and throughout the body (Wu et al. 2016; Yazdankhah et al. 2014). In the current work, administration of Empa mitigated Res-induced reduction of hippocampal immunoreactivity of the two autophagy-related proteins, Beclin1 and LC3B which are key agents of early and late autophagic assembly, respectively (Chifenti et al. 2013). These outcomes are in convention with previous reports which demonstrate that rats exposed to chronic mild stress (CMS) acquired depressive symptoms through hindering the autophagic machinery (Yang et al. 2017; Zhao et al. 2017). Likewise, the antidiabetic drug rosiglitazone showed significant antidepressant and neuroprotective properties in unpredictable CMS-triggered depression in mice via maintaining autophagy in neurons and mitigating astrocyte-mediated apoptosis (Zhao et al. 2017). Moreover, classical antidepressants such as amitriptyline and citalopram were found to play a crucial role in stimulating autophagy through upregulation of Beclin1 and LC3B (Gulbins et al. 2018; Zschocke et al. 2011). Of note, Beclin1 is fundamental to autophagy-mediated cytoprotection and to opposing apoptotic cell death (Zeng and Zhou 2008). Conversely, our results demonstrate significant decline in mTOR protein expression in response to Empa treatment, which was analogous to the effect generated by Esc. Importantly, inhibition of mTOR pathway has a bona fide role in boosting autophagy (Ravikumar et al. 2004). Additionally, the normal activity of mTOR is pivotal for a sound cognitive function (Uddin et al. 2020). Interestingly, Stanciu and colleagues have established that SGLT2 inhibitors are capable of repressing mTOR, which seems to bring benefits to patients with Alzheimer’s disease (Stanciu et al. 2021). Preclinically, dapagliflozin was found to enhance autophagy through mTOR inhibition in D-galactose-injected/ovariectomized rats (Ibrahim et al. 2022). Moreover, a previous study has revealed that treatment with Empa led to upregulation of AMPK phosphorylation by inhibiting the activation of mTOR (Zheng et al. 2021). In fact, throughout the years, research has been revealing that SGLT2 inhibitors are outstanding AMPK activators (Packer 2020; Yaribeygi et al. 2023; Yu et al. 2022). Of note, phosphorylation of AMPK by upstream kinases at Thr172 can increase its activity by more than 100 folds (Ross et al. 2016).
Besides its chief role in controlling the autophagic reaction, a dynamic interplay exists among AMPK, autophagy, and ROS propagation. In fact, the functional relationship between oxidative stress and autophagy is rather complex. Upon mitochondrial ROS production, redox-sensitive autophagy-related proteins, including AMPK, are rapidly activated to elucidate fast autophagic response. In addition, given that autophagy is a crucial process to eradicate damaged cellular components in response to oxidative stress, it makes sense that ROS are positive effectors for activation of autophagy (Filomeni et al. 2015). Although ROS can initially oxidize, and subsequently, inactivate autophagy-related proteins, ROS ultimately trigger signaling pathways to activate autophagy which in turn suppresses ROS propagation and oxidative damage (Chang et al. 2022). Accordingly, oxidative stress and autophagy regulation can be viewed as a two-way process (Redza-Dutordoir and Averill-Bates 2021). Nonetheless, the story differs with nitric oxide (NO) which can act both as a positive or a negative effector for autophagy. In HeLa cells, NO was found to prevent autophagy by means of halting AMPK and Beclin1 activation, which is in contrast with the well-documented role of NO and nitrative stress in the activation of AMPK in response to DNA damage. This inconsistency is mostly attributed to the fact that NO can range from a pro-survival to a pro-apoptotic molecule depending on its concentration (Filomeni et al. 2015).
Our findings imply that Res administration created a sharp state of oxidative stress in rats' hippocampi, which was revealed by the eminent decline in GSH and the prompt increase in iNOS and MDA. Notably, compared with endothelial and neuronal NOS, iNOS causes nitrative stress and generates more superoxide, which is effective for hindering pathogens but drives inflammation and cell injury as well (Huang et al. 2023). Oxidative stress and neuro-inflammation have been considerably associated with many neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, and Huntington’s diseases (Butterfield 2002; Pizzino et al. 2017; Tripathi et al. 2019). In depression research, the inflammatory cytokines IL-6 and TNF-α have been strongly correlated with the pathology and severity of major depression (Dowlati et al. 2010; Min et al. 2023), and the role of oxidative stress in the progression of depression has long been appreciated (Correia et al. 2023). Noteworthy, the collaborative effects of oxidative/nitrative stress and NLRP3 inflammasome activation result in neuro-inflammation, impaired neurogenesis, and monoamine neurotransmission decline (Correia et al. 2023; Kaufmann et al. 2017). In light of neuro-inflammation, the current results reveal that treatment with Empa plays an important role in dampening the gene expression of the NLRP3 inflammasome, caspase-1 levels, IL-1β/18, and TNF-α. Meanwhile, Empa reversed oxidative stress of the hippocampus, which was observed through the decrements in iNOS and MDA, and alternatively, the potentiation of GSH. In line with our study, Empa was found to prevent cognitive impairment in Alzheimer’s disease as well as in diabetes via attenuating cerebral oxidative stress and inflammation (Wicinski et al. 2020; Yaribeygi et al. 2024). In a recent study by Vercalsteren et al. (2024), the authors confirmed that Empa enhanced post-stoke outcomes in diabetic mice partly through hindering neuro-inflammation. In addition, scientists have highlighted the anti-inflammatory and antioxidant potentials of SGLT2 inhibitors, which largely contributes to their health benefits in aging (Schönberger et al. 2023) and in neurological disorders (Tsai et al. 2021). Moreover, researchers have declared that the clinical benefit of antidepressants is partly mediated through alleviating microglial reactivity, oxidative stress, and the release of inflammatory cytokines (Mariani et al. 2022). Importantly, the histopathological and histomorphometric findings reported herein came to support the observed behavioral and biochemical modifications pertinent to either Res or Esc and Empa treatments.
Growing body of evidence have been showing that depressed patients experience reductions in CREB expression and phosphorylation together with BDNF in the hippocampus (Cunha et al. 2006; Zhang et al. 2017). CREB is a transcription factor that has been extensively studied and attested in the pathogenesis of depression and response to therapy. In fact, chronic antidepressant treatment has been verified to upregulate BDNF and its tyrosine kinase receptor B (TrkB) by augmenting phosphorylation of CREB at Ser133 (Tsuchimine et al. 2018). Additionally, it has been reported that BDNF itself stimulates CREB phosphorylation and activation, suggesting that BDNF is capable of regulating its own expression by forming a positive feedback loop (Esvald et al. 2020; Finkbeiner et al. 1997). Noteworthy, one of the involved enzymes in activating CREB is PKC. In its active form, CREB plays a fundamental role in CNS development, cognitive function, and neural survival (Ren et al. 2011). In the present study, Empa administration restored hippocampal p-CREB (Ser133) expression and BDNF concentration in Res model of depression, and thus, affording comparable effects to those of Esc. Empa has been previously reported to replenish BDNF in diabetic obese mice and to improve their cognitive function (Lin et al. 2014; Piątkowska-Chmiel et al. 2023). In addition, SGLT2 inhibitors have been found to relieve hyperuricemia in diabetic mice (Lu et al. 2020), and obesity in high fat-fed mice via enhancing CREB phosphorylation (Yang et al. 2021). Indeed, the aforementioned effects are not independent from AMPK. Through activating the atypical PKCζ signaling, AMPK plays an indispensable role in boosting hippocampal neurogenesis (Wang et al. 2012). Intriguingly, AMPK has been reported to mediate exercise-induced BDNF augmentation in hippocampi of stressed mice (Kim and Leem 2016). In the same context, PKCζ-mediated BDNF potentiation is thought to arise under the influence of NF-κB (Odaira et al. 2019). In fact, the role of NF-κB in BDNF secretion has not been thoroughly investigated, although it represents an attractive point for research. To be more specific, phosphorylation of NF-κB p65 at Ser311 residue, rather than the famous Ser536 site, has been shown to play a fundamental role in neuronal survival, defending neurons against oxidative stress, and extenuating pyramidal neuronal death (Kim et al. 2013). Importantly, phosphorylation of NF-κB p65 at Ser311 specifically occurs upon PKCζ activation and phosphorylation at Thr403 (Wang et al. 2012). Nonetheless, some authors have documented conflicting results. In this context, Yi et al. (2021) argue against sustained AMPK activation which has negatively affected BDNF/TrkB signaling due to mTOR inhibition in stressed mice. Additionally, NF-κB has been well reported to upregulate NLRP3 expression, which might contradict the current results (Song et al. 2017). Although NF-κB p65 with or without Ser536 phosphorylation has been confirmed to induce NLRP3 expression and to provoke the inflammatory response (Boaru et al. 2015; Perkins and Gilmore 2006) the correspondence between p-NF-κB p65 Ser311 and NLRP3 has not been addressed so far.
Conclusions
The current study emphasizes the role of Empa in neuroprotection and neural resilience potentiation in the Res animal model of depression. Mastered by AMPK activation, the SGLT2 inhibitor is thought to fine regulate the triad of autophagy, inflammation, and oxidative stress in the brain. Deeper evaluation revealed the ability of Empa to rebalance the disturbed autophagy/inflammation dynamics through manipulation of the autophagic machinery components: mTOR/Beclin1/LC3B and attenuation of the NLRP3 inflammasome cascade. Consequently, reinforcement of hippocampal neurogenesis was demonstrated as a valuable outcome of Empa treatment. This latter effect is thought to be mediated partly through AMPK which orchestrates PKCζ phosphorylation and highlights an unusual role for NF-κB in boosting BDNF production—an outcome that necessitates NF-κB phosphorylation at Ser311 upon interaction with PKCζ.
Data availability
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
References
Abdel-latif RG, Rifaai RA, Amin EF (2020) Empagliflozin alleviates neuronal apoptosis induced by cerebral ischemia/reperfusion injury through HIF-1α/VEGF signaling pathway. Arch Pharm Res 43:514–525. https://doi.org/10.1007/s12272-020-01237-y
Amin EF, Rifaai RA, Abdel-latif RG (2020) Empagliflozin attenuates transient cerebral ischemia/reperfusion injury in hyperglycemic rats via repressing oxidative–inflammatory–apoptotic pathway. Fundam Clin Pharmacol 34(5):548–558. https://doi.org/10.1111/fcp.12548
Antkiewicz-Michaluk L, Wąsik A, Możdżeń E, Romańska I, Michaluk J (2014) Antidepressant-like effect of tetrahydroisoquinoline amines in the animal model of depressive disorder induced by repeated administration of a low dose of reserpine: Behavioral and neurochemical studies in the rat. Neurotox Res 26(1):85–98. https://doi.org/10.1007/s12640-013-9454-8
Boaru SG, Borkham-Kamphorst E, Van De Leur E, Lehnen E, Liedtke C, Weiskirchen R (2015) NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem Biophys Res Commun 458(3):700–706. https://doi.org/10.1016/j.bbrc.2015.02.029
Borikar SP, Dongare SI, Danao KR (2022) Reversal of lipopolysaccharide-induced learning and memory deficits by agmatine in mice. Int J Neurosci 132(6):621–632. https://doi.org/10.1080/00207454.2020.1830086
Borikar SP, Sonawane DS, Tapre DN, Jain SP (2024) Exploring the neuropharmacological potential of empagliflozin on nootropic and scopolamine-induced amnesic model of Alzheimer’s like conditions in rats. Int J Neurosci 6:1–13. https://doi.org/10.1080/00207454.2024.2342973
Butterfield DA (2002) Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res 36(12):1307–1313. https://doi.org/10.1080/1071576021000049890
Cai B, Seong K, Bae S, Kook MS, Chun C, Lee JH, Choi W, Jung J, Kim W (2019) Water-soluble arginyl–diosgenin analog attenuates hippocampal neurogenesis impairment through blocking microglial activation underlying NF-κB and JNK MAPK signaling in adult mice challenged by LPS. Mol Neurobiol 56:6218–6238. https://doi.org/10.1007/s12035-019-1496-3
Cao Z, Wang Y, Long Z, He G (2019) Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim Biophys Sin 51(11):1087–1095. https://doi.org/10.1093/abbs/gmz098
Chang KC, Liu PF, Chang CH, Lin YC, Chen YJ, Shu CW (2022) The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci 12(1). https://doi.org/10.1186/s13578-021-00736-9
Chen ACH, Shirayama Y, Shin KH, Neve RL, Duman RS (2001) Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry 49(9):753–762. https://doi.org/10.1016/S0006-3223(00)01114-8
Chen H, Teng D, Xu B, Wang C, Wang H, Jia W, Gong L, Dong H, Zhong L, Yang J (2023) The SGLT2 inhibitor canagliflozin reduces atherosclerosis by enhancing macrophage autophagy. J Cardiovasc Transl Res 16(5):999–1009. https://doi.org/10.1007/s12265-023-10390-w
Chifenti B, Locci MT, Lazzeri G, Guagnozzi M, Dinucci D, Chiellini F, Filice ME, Salerno MG, Battini L (2013) Autophagy-related protein LC3 and Beclin-1 in the first trimester of pregnancy. Clin Exp Reprod Med 40(1):33–37. https://doi.org/10.5653/cerm.2013.40.1.33
Cizza G, Ravn P, Chrousos GP, Gold PW (2001) Depression: a major, unrecognized risk factor for osteoporosis? Trends Endocrinol Metab 12(5):198–203. https://doi.org/10.1016/S1043-2760(01)00407-6
Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C (2009) p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 29(10):2594–2608. https://doi.org/10.1128/mcb.01396-08
Correia AS, Cardoso A, Vale N (2023) Oxidative stress in depression: the link with the stress response, neuroinflammation, serotonin, neurogenesis and synaptic plasticity. Antioxidants 12(2):1–18. https://doi.org/10.3390/antiox12020470
Culling CFA (2013) Handbook of histopathological and histochemical techniques: including museum techniques. Butterworth-Heinemann, vol 94. Elsevier Science & Technology Books, pp 1–94. https://doi.org/10.1016/C2013-0-04011-X
Cunha ABM, Frey BN, Andreazza AC, Goi JD, Rosa AR, Gonçalves CA, Santin A, Kapczinski F (2006) Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett 398(3):215–219. https://doi.org/10.1016/j.neulet.2005.12.085
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctôt KL (2010) A meta-analysis of cytokines in major depression. BPS 67(5):446–457. https://doi.org/10.1016/j.biopsych.2009.09.033
El-Sahar A, Rastanawi AA, El-Yamany MF, Saad MA (2020) Dapagliflozin improves behavioral dysfunction of Huntington’s disease in rats via inhibiting apoptosis-related glycolysis. Life Sci 257:118076. https://doi.org/10.1016/j.lfs.2020.118076
Esvald E, Tuvikene J, Sirp A, Patil X, Bramham X, Timmusk T (2020) CREB family transcription factors are major mediators of BDNF transcriptional autoregulation in cortical neurons. J Neurosci 40(7):1405–1426. https://doi.org/10.1523/JNEUROSCI.0367-19.2019
Filomeni G, Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22(3):377–388. https://doi.org/10.1038/cdd.2014.150
Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19(5):1031–1047. https://doi.org/10.1016/S0896-6273(00)80395-5
Fukushima K, Kitamura S, Tsuji K, Wada J (2021) Sodium-glucose cotransporter 2 inhibitors work as a “Regulator” of autophagic activity in overnutrition diseases. Front Pharmacol 21(12):761842. https://doi.org/10.3389/fphar.2021.761842
Gassen NC, Rein T (2019). Is there a role of autophagy in depression and antidepressant action? Front Psychiatry 1–8. https://doi.org/10.3389/fpsyt.2019.00337
Gulbins A, Schumacher F, Becker KA, Wilker B, Soddemann M, Boldrin F, Müller CP, Edwards MJ, Goodman M, Caldwell CC, Kleuser B, Kornhuber J, Szabo I, Gulbins E (2018) Antidepressants act by inducing autophagy controlled by sphingomyelin–ceramide. Mol Psychiatry 23(12):2324–2346. https://doi.org/10.1038/s41380-018-0090-9
Han F, Nakano T, Yamamoto Y, Shioda N, Lu YM, Fukunaga K (2009) Improvement of depressive behaviors by nefiracetam is associated with activation of CaM kinases in olfactory bulbectomized mice. Brain Res 1265:205–214. https://doi.org/10.1016/j.brainres.2009.02.014
Hanifiha M, Ghanbari A, Keykhaei M, Moghaddam SS, Rezaei N, Zanous MP, Yoosefi M, Ghasemi E, Rezaei N, Shahin S, Rashidi MM, Ghamari A, Haghshenas R, Kompani F, Farzadfar F (2022) Global, regional, and national burden and quality of care index in children and adolescents: A systematic analysis for the global burden of disease study 1990–2017. PLoS One 17:1–14. https://doi.org/10.1371/journal.pone.0267596
Hassamal S (2023) Chronic stress, neuroinflammation, and depression: an overview of pathophysiological mechanisms and emerging anti-inflammatories. Front Psych 11(14):1130989. https://doi.org/10.3389/fpsyt.2023.1130989
Hierro-Bujalance C, Infante-Garcia C, Del Marco A, Herrera M, Carranza-Naval MJ, Suarez J, Alves-Martinez P, Lubian-Lopez S, Garcia-Alloza M (2020). Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res Ther 12(1). https://doi.org/10.1186/s13195-020-00607-4
Hoerster KD, Campbell S, Dolan M, Stappenbeck CA, Yard S, Simpson T, Nelson KM (2019) PTSD is associated with poor health behavior and greater Body Mass Index through depression, increasing cardiovascular disease and diabetes risk among U.S. veterans. Prev Med Rep 15. https://doi.org/10.1016/j.pmedr.2019.100930
Huang K, Luo X, Liao B, Li G, Feng J (2023). Insights into SGLT2 inhibitor treatment of diabetic cardiomyopathy: focus on the mechanisms. Cardiovasc Diabetol 22(1). https://doi.org/10.1186/s12933-023-01816-5
Hurley LL, Tizabi Y (2013) Neuroinflammation, neurodegeneration, and depression. Neurotox Res 23(2):131–144. https://doi.org/10.1007/s12640-012-9348-1
Ibrahim WW, Kamel AS, Wahid A, Abdelkader NF (2022) Dapagliflozin as an autophagic enhancer via LKB1/AMPK/SIRT1 pathway in ovariectomized/D-galactose Alzheimer’s rat model. Inflammopharmacology 30(6):2505–2520. https://doi.org/10.1007/s10787-022-00973-5
Ikram H, Haleem DJ (2019) Repeated treatment with a low dose of reserpine as a progressive model of Parkinson’s dementia. Pak J Pharm Sci 32(2):555–562. https://doi.org/10.1016/j.bbr.2012.03.008
James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J, Abdelalim A, Abdollahpour I, Abdulkader RS, Abebe Z, Abera SF, Abil OZ, Abraha HN, Abu-Raddad LJ, Abu-Rmeileh NME, Accrombessi MMK, Murray CJL (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392(10159):1789–1858. https://doi.org/10.1016/S0140-6736(18)32279-7
Jeon SW, Kim YK (2016) Neuroinflammation and cytokine abnormality in major depression: cause or consequence in that illness? World J Psychiatry 6(3). https://doi.org/10.5498/wjp.v6.i3.283
Ji Y, Lu Y, Yang F, Shen W, Tang T, Feng L, Duan S, Lu B (2010) Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci 13(3):302–309. https://doi.org/10.1038/nn.2505
Kaufmann F, Costa A, Ghisleni G, Diaz A, Rodrigues AL, Peluffo H, Kaster MP (2017) NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun 367–383. https://doi.org/10.1016/j.bbi.2017.03.002
Khadrawy YA, Sawie HG, Hosny EN, Mourad HH (2018) Assessment of the antidepressant effect of caffeine using rat model of depression induced by reserpine. Bull Natl Res Centre 42(1). Springer Science and Business Media LLC. https://doi.org/10.1186/s42269-018-0034-1
Kim DM, Leem YH (2016) Chronic stress-induced memory deficits are reversed by regular exercise via AMPK-mediated BDNF induction. Neuroscience 324:271–285. https://doi.org/10.1016/j.neuroscience.2016.03.019
Kim JE, Kim DS, Jin RH, Il KW, Kim MJ, Won KD, Young CS, Kang TC (2013) The effect of P2X7 receptor activation on nuclear factor-kappa B phosphorylation induced by status epilepticus in the rat hippocampus. Hippocampus 23(6):500–514. https://doi.org/10.1002/hipo.22109
Koulich E, Nguyen T, Johnson K, Giardina CA, D’Mello SR (2001) NF-κB is involved in the survival of cerebellar granule neurons: association of Iκβ phosphorylation with cell survival. J Neurochem 76(4):1188–1198. https://doi.org/10.1046/j.1471-4159.2001.00134.x
Leao AHFF, Sarmento-Silva AJ, Santos JR, Ribeiro AM, Silva RH (2015) Molecular, neurochemical, and behavioral hallmarks of reserpine as a model for Parkinson’s disease: new perspectives to a long-standing model. Brain Pathol 25(4):377–390. https://doi.org/10.1111/bpa.12253
Lima AC, Meurer YSR, Bioni VS, Cunha DMG, Gonçalves N, Lopes-Silva LB, Becegato M, Soares MBL, Marinho GF, Santos JR, Silva RH (2021) Female rats are resistant to cognitive, motor and dopaminergic deficits in the reserpine-induced progressive model of parkinson’s disease. Front Aging Neurosci 13:757714. https://doi.org/10.3389/fnagi.2021.757714
Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K, Ma M, Nakagawa T, Kusaka H, Kim-Mitsuyama S (2014) Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol 13:1–15. https://doi.org/10.1186/s12933-014-0148-1
Liu JJ, Wei Y, Bin SR, Bao Y, Chang S, Shi L, Que J, Gadad BS, Trivedi MH, Kelsoe JR, Lu L (2020) Peripheral cytokine levels and response to antidepressant treatment in depression: a systematic review and meta-analysis. Mol Psychiatry 25(2):339–350. https://doi.org/10.1038/s41380-019-0474-5
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Llorens-Martín M, Jurado-Arjona J, Fuster-Matanzo A, Hernández F, Rábano A, Ávila J (2014) Peripherally triggered and GSK-3β-driven brain inflammation differentially skew adult hippocampal neurogenesis, behavioral pattern separation and microglial activation in response to ibuprofen. Transl Psychiatry 4(10):E463. https://doi.org/10.1038/tp.2014.92
Lowry O, Rosebrough N, Farr AL, Randall R (2014) Protein measurement with the Folin phenol reagent. Encycl Qual Life Well-Being Res 1:921. https://doi.org/10.1007/978-94-007-0753-5_100521
Lu YH, Chang YP, Li T, Han F, Li CJ, Li XY, Xue M, Cheng Y, Meng ZY, Han Z, Sun B, Chen LM (2020) Empagliflozin Attenuates Hyperuricemia by Upregulation of ABCG2 via AMPK/AKT/CREB Signaling Pathway in Type 2 Diabetic Mice. Int J Biol Sci 1;16(3):529−542. https://doi.org/10.7150/ijbs.33007
Mariani N, Everson J, Pariante CM, Borsini A (2022) Modulation of microglial activation by antidepressants. J Psychopharmacol 36(2):131–150. https://doi.org/10.1177/02698811211069110
Mattson MP, Camandola S (2001) NF-κB in neuronal plasticity and neurodegenerative disorders. Clin Investig 107(3):247–254. https://doi.org/10.1172/JCI11916
Min X, Wang G, Cui Y, Meng P, Hu X, Liu S, Wang Y (2023) Association between inflammatory cytokines and symptoms of major depressive disorder in adults. Front Immunol 14:1–8. https://doi.org/10.3389/fimmu.2023.1110775
Muhammad RN, Ahmed LA, Abdul Salam RM, Ahmed KA, Attia AS (2021) Crosstalk among NLRP3 inflammasome, ETBR signaling, and miRNAs in stress-induced depression-like behavior: a modulatory role for SGLT2 inhibitors. Neurotherapeutics 18(4):2664-2681.https://doi.org/10.1007/s13311-021-01140-4
Nodirahon A, Majid H, Waghdhare S, Vohora D, Nidhi. (2024) The effect of sodium glucose Co-transport 2 inhibitors on cognitive impairment and depression in type 2 diabetes mellitus patients. Clin Epidemiol Glob Health. https://doi.org/10.1016/j.cegh.2024.101555
Obara Y, Kobayashi H, Ohta T, Ohizumi Y, Nakahata N (2001) Scabronine G-methylester enhances secretion of neurotrophic factors mediated by an activation of protein kinase C-ζ. Mol Pharmacol 59(5):1287–1297. https://doi.org/10.1124/mol.59.5.1287
Odaira T, Nakagawasai O, Takahashi K, Nemoto W, Sakuma W, Lin JR, Tan K (2019) Mechanisms underpinning AMP-activated protein kinase-related effects on behavior and hippocampal neurogenesis in an animal model of depression. Neuropharmacology 150:121–133. https://doi.org/10.1016/j.neuropharm.2019.03.026
Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, Mohr DC, Schatzberg AF (2016) Major depressive disorder. Nat Rev Dis Primers 2:16065. https://doi.org/10.1038/nrdp.2016.65
Packer M (2020) Interplay of adenosine monophosphate-activated protein kinase/sirtuin-1 activation and sodium influx inhibition mediates the renal benefits of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes: a novel conceptual framework. Diabetes Obes Metab 22(5):734–742. https://doi.org/10.1111/dom.13961
Packer M (2022) Critical reanalysis of the mechanisms underlying the cardiorenal benefits of SGLT2 inhibitors and reaffirmation of the nutrient deprivation signaling/autophagy hypothesis. Circulation 46(18):1383–1405. https://doi.org/10.1161/CIRCULATIONAHA.122.061732
Park BK, Kim YR, Kim YH, Yang C, Seo CS, Jung IC, Jang IS, Kim SH, Lee MY (2018) Antidepressant-like effects of gyejibokryeong-hwan in a mouse model of reserpine-induced depression. Biomed Res Int 13:1–13. https://doi.org/10.1155/2018/5845491
Pawlos A, Broncel M, Woźniak E, Gorzelak-Pabiś P (2021) Neuroprotective effect of SGLT2 inhibitors. Molecules 28;26(23):7213. https://doi.org/10.3390/molecules26237213
Perkins ND, Gilmore TD (2006) Good cop, bad cop: the different faces of NF-κB. Cell Death Differ 13(5):759–772. https://doi.org/10.1038/sj.cdd.4401838
Piątkowska-Chmiel I, Herbet M, Gawrońska-Grzywacz M, Pawłowski K, Ostrowska-Leśko M, Dudka J (2023) Molecular and neural roles of sodium-glucose cotransporter 2 inhibitors in alleviating neurocognitive impairment in diabetic mice. Psychopharmacology 240(4):983–1000. https://doi.org/10.1007/s00213-023-06341-7
Pitsillou E, Bresnehan SM, Kagarakis EA, Wijoyo SJ, Liang J, Hung A, Karagiannis TC (2020) The cellular and molecular basis of major depressive disorder: towards a unifed model for understanding clinical depression. Mol Biol Rep 47:753–770. https://doi.org/10.1007/s11033-019-05129-3
Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A (2017) Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. https://doi.org/10.1155/2017/8416763
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6):585–595. https://doi.org/10.1038/ng1362
Redza-Dutordoir M, Averill-Bates DA (2021) Interactions between reactive oxygen species and autophagy: special issue: death mechanisms in cellular homeostasis. Biochim Biophys Acta Mol Cell Res 1868(8):1–12. https://doi.org/10.1016/j.bbamcr.2021.119041
Ren X, Dwivedi Y, Mondal AC, Pandey GN (2011) Cyclic-AMP response element binding protein (CREB) in the neutrophils of depressed patients. Psychiatry Res 185:108–112. https://doi.org/10.1016/j.psychres.2010.04.013
Rizzo MR, Meoa I, Di Politoa R, Auriemmaa MC, Gambardellab A, Di MG, Capuanoc A, Paolissoa G (2022) Cognitive impairment and type 2 diabetes mellitus: focus of SGLT2 inhibitors treatment. Pharmacol Res 176:106062. https://doi.org/10.1016/j.phrs.2022.106062
Ross FA, MacKintosh C, Hardie DG (2016) AMP-activated protein kinase: a cellular energy sensorthat comes in 12 flavours. FEBS J 283(16):2987–3001. https://doi.org/10.1111/febs.13698
Safaie N, Masoumi S, Alizadeh S, Mirzajanzadeh P, Nejabati HR, Hajiabbasi M, Alivirdiloo V, Basmenji NC, Derakhshi Radvar A, Majidi Z, Faridvand Y (2024) SGLT2 inhibitors and AMPK: the road to cellular housekeeping? Cell Biochem Funct 42(1):e3922. https://doi.org/10.1002/cbf.3922
Schönberger E, Mihaljević V, Steiner K, Šarić S, Kurevija T, Majnarić LT, Bilić Ćurčić I, Canecki-Varžić S (2023) Immunomodulatory effects of SGLT2 inhibitors-targeting inflammation and oxidative stress in aging. Int J Environ Res Public Health 29;20(17):6671. https://doi.org/10.3390/ijerph20176671
Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C (2014) Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther 20(12):1021–1028. https://doi.org/10.1111/cns.12349
Singh S, Singh TG (2020) Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: an mechanistic approach. Curr Neuropharmacol 18(10):918–935. https://doi.org/10.2174/1570159X18666200207120949
Song L, Pei L, Yao S, Wu Y, Shang Y (2017) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci 11. https://doi.org/10.3389/fncel.2017.00063
Stanciu GD, Rusu RN, Bild V, Filipiuc LE, Tamba BI, Ababei DC (2021) Systemic actions of sglt2 inhibition on chronic mtor activation as a shared pathogenic mechanism between alzheimer’s disease and diabetes. Biomedicines 9(5). https://doi.org/10.3390/biomedicines9050576
Steven S, Oelze M, Hanf A, Kröller-Schön S, Kashani F, Roohani S, Welschof P, Kopp M, Gödtel-Armbrust U, Xia N, Li H, Schulz E, Lackner KJ, Wojnowski L, Bottari SP, Wenzel P, Mayoux E, Münzel T, Daiber A (2017) The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol 13:370–385. https://doi.org/10.1016/j.redox.2017.06.009
Takahashi K, Nakagawasai O, Nemoto W, Kadota S, Isono J, Odaira T, Sakuma W, Arai Y, Tadano T, Tan-No K (2018) Memantine ameliorates depressive-like behaviors by regulating hippocampal cell proliferation and neuroprotection in olfactory bulbectomized mice. Neuropharmacology 141–155. https://doi.org/10.1016/j.neuropharm.2018.04.013
Tang M, Liu T, Jiang P, Dang R (2021) The interaction between autophagy and neuroinflammation in major depressive disorder: From pathophysiology to therapeutic implications. Pharmacol Res 168:105586. https://doi.org/10.1016/j.phrs.2021.105586
Tian DD, Wang M, Liu A, Gao MR, Qiu C, Yu W, Wang WJ, Zhang K, Yang L, Jia YY, Yang CB, Wu YM (2021) Antidepressant effect of paeoniflorin is through inhibiting pyroptosis CASP-11/GSDMD pathway. Mol Neurobiol 58(2):761–776. https://doi.org/10.1007/s12035-020-02144-5
Tripathi PN, Srivastava P, Sharma P, Tripathi MK, Seth A, Tripathi A, Rai SN, Singh SP, Shrivastava SK (2019) Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg Chem 85:82–96. https://doi.org/10.1016/j.bioorg.2018.12.017
Trocoli A, Djavaheri-Mergny M (2011) The complex interplay between autophagy and NF-kB signaling pathways in cancer cells. Am J Cancer Res 1(5):629–649
Trojan E, Chamera K, Bryniarska N, Kotarska K, Leśkiewicz M, Regulska M, Basta-Kaim A (2019) Role of chronic administration of antidepressant drugs in the prenatal stress-evoked inflammatory response in the brain of adult offspring rats: involvement of the nlrp3 inflammasome-related pathway. Mol Neurobiol 56(8):5365–5380. https://doi.org/10.1007/s12035-018-1458-1
Troubat R, Barone P, Leman S, Desmidt T, Cressant A, Atanasova B, Brizard B, El Hage W, Surget A, Belzung C, Camus V (2020) Neuroinflammation and depression: a review. Eur J Neurosci 53(1):151–171. https://doi.org/10.1111/ejn.14720
Tsai K-F, Chen Y-L, Chiou TT-Y, Chu T-H, Li L-C, Ng H-Y, Lee W-C, Lee C-T (2021) Emergence of SGLT2 inhibitors as powerful antioxidants in human diseases. Antioxidants 10(8):1166. https://doi.org/10.3390/antiox10081166
Tsuchimine S, Sugawara N, Yasui-Furukori N (2018) Increased levels of CREB in major depressive patients with antidepressant treatment. Psychiatry Res 296−297. https://doi.org/10.1016/j.psychres.2017.12.077
Uddin MS, Rahman MA, Kabir MT, Behl T, Mathew B, Perveen A, Barreto GE, Bin-Jumah MN, Abdel-Daim MM, Ashraf GM (2020) Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 72(9):1843–1855. https://doi.org/10.1002/iub.2324
Vercalsteren E, Karampatsi D, Buizza C, Nyström T, Klein T, Paul G, Patrone C, Darsalia V (2024) The SGLT2 inhibitor Empagliflozin promotes post-stroke functional recovery in diabetic mice. Cardiovasc Diabetol 23:88.https://doi.org/10.1186/s12933-024-02174-6
Wang J, Gallagher D, Devito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD (2012) Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11(1):23–35. https://doi.org/10.1016/j.stem.2012.03.016
Wang Y, Meng C, Zhang J, Wu J, Zhao J (2019) Inhibition of GSK-3β alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int Immunopharmacol 68:234–241. https://doi.org/10.1016/j.intimp.2018.12.042
Waugh J, Goa KL (2003) Escitalopram: a review of its use in the management of major depressive and anxiety disorders. CNS Drugs 17(5):343–362. https://doi.org/10.2165/00023210-200317050-00004
Wicinski M, Wodkiewicz E, Gorski K, Walczak M, Malinowski B (2020) Perspective of SGLT2 inhibition in treatment of conditions connected to neuronal loss: focus on Alzheimer’s disease and ischemia-related brain injury. Pharmaceuticals 13(11):379. https://doi.org/10.3390/ph13110379
Wu X, Fleming A, Ricketts T, Pavel M, Virgin H, Menzies FM, Rubinsztein DC (2016). Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun 7. https://doi.org/10.1038/ncomms10533
Yang X, Liu Q, Li Y, Ding Y, Zhao Y, Tang Q, Wu T, Chen L, Pu S, Cheng S, Zhang J, Zhang Z, Huang Y, Li R, Zhao Y, Zou M, Shi X, Jiang W, Wang R, He J (2021) Inhibition of the sodium-glucose co-transporter SGLT2 by canagliflozin ameliorates diet-induced obesity by increasing intra-adipose sympathetic innervation. Br J Pharmacol 178(8):1756–1771. https://doi.org/10.1111/bph.15381
Yang Y, Hu Z, Du X, Davies H, Huo X, Fang M (2017) miR-16 and fluoxetine both reverse autophagic and apoptotic change in chronic unpredictable mild stress model rats. Front Neurosci 11:1–18. https://doi.org/10.3389/fnins.2017.00428
Yaribeygi H, Maleki M, Atkin SL, Jamialahmadi T, Sahebkar A (2023) SGLT2 inhibitors and autophagy in diabetes. Cell Biochem Funct 10:1–10. https://doi.org/10.1002/cbf.3792
Yaribeygi H, Hemmati MA, Nasimi F, Pakdel R, Jamialahmadi T, Sahebkar A (2024) Empagliflozin alleviates diabetes-induced cognitive impairments by lowering nicotinamide adenine dinucleotide phosphate oxidase-4 expression and potentiating the antioxidant defense system in brain tissue of diabetic rats. Behav Brain Res 5(460):114830. https://doi.org/10.1016/j.bbr.2023
Yazdankhah M, Farioli-Vecchioli S, Tonchev AB, Stoykova A, Cecconi F (2014) The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis 5(9):1–8. https://doi.org/10.1038/cddis.2014.358
Yi LT, Wang SS, Cheng J, Chen M, Zhu JX, Li CF, Dong SQ, Liu Q (2021) Sustained AMP-activated protein kinase activation attenuates the activity of brain-derived neurotrophic factor/tyrosine kinase receptor B signaling in mice exposed to chronic stress. Chin Med J 134(15):1874–1876, 10. https://doi.org/10.1097/CM9.0000000000001323
Youssef ME, Abd El-Fattah EE, Abdelhamid AM, Eissa H, El-Ahwany E, Amin NA, Hetta HF, Mahmoud MH, Batiha GES, Gobba N, Ahmed Gaafar AG, Saber S (2021) Interference with the AMPKα/mTOR/NLRP3 signaling and the IL-23/IL-17 axis effectively protects against the dextran sulfate sodium intoxication in rats: a new paradigm in empagliflozin and metformin reprofiling for the management of ulcerative colitis. Front Pharmacol 12. https://doi.org/10.3389/fphar.2021.719984
Yu H, Zhang F, Guan X (2019) Baicalin reverse depressive-like behaviors through regulation SIRT1-NF-kB signaling pathway in olfactory bulbectomized rats. Phytother Res 33(5):1480–1489. https://doi.org/10.1002/ptr.6340
Yu X, Meng Z, Fang T, Liu X, Cheng Y, Xu L, Liu X, Li X, Xue M, Li T, Sun B, Chen L (2022) Empagliflozin inhibits hepatic gluconeogenesis and increases glycogen synthesis by AMPK/CREB/GSK3β signalling pathway. Front Physiol 13. https://doi.org/10.3389/fphys.2022.817542
Zeng M, Zhou JN (2008) Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal 20(4):659–665. https://doi.org/10.1016/j.cellsig.2007.11.015
Zhang Q, Wang X, Bai X, Xie Y, Zhang T, Bo S, Chen X (2017) Resveratrol reversed chronic restraint stress-induced impaired cognitive function in rats. Mol Med Rep 16(2):2095–2100. https://doi.org/10.3892/mmr.2017.6851
Zhang Y, Hu W (2012) NFκB signaling regulates embryonic and adult neurogenesis. Front Biol 7(4):277–291. https://doi.org/10.1007/s11515-012-1233-z
Zhao Z, Zhang L, Guo XD, Cao LL, Xue TF, Zhao XJ, Yang DD, Yang J, Ji J, Huang JY, Sun XL (2017) Rosiglitazone exerts an anti-depressive effect in unpredictable chronic mild-stress-induced depressive mice by maintaining essential neuron autophagy and inhibiting excessive astrocytic apoptosis. Front Mol Neurosci 10:1–16. https://doi.org/10.3389/fnmol.2017.00293
Zheng J, Hu S, Wang J, Zhang X, Yuan D, Zhang C, Liu C, Wang T, Zhou Z (2021) Icariin improves brain function decline in aging rats by enhancing neuronal autophagy through the AMPK/mTOR/ULK1 pathway. Pharm Biol 59(1):183–191. https://doi.org/10.1080/13880209.2021.1878238
Zschocke J, Zimmermann N, Berning B, Ganal V, Holsboer F, Rein T (2011) Antidepressant drugs diversely affect autophagy pathways in astrocytes and neurons-dissociation from cholesterol homeostasis. Neuropsychopharmacology 36(8):1754–1768. https://doi.org/10.1038/npp.2011.57
Acknowledgements
The authors are thankful for Dr. Mohamed A. Salah Eldin, Department of Pathology, Faculty of Veterinary Medicine, Cairo University for performing the histopathological investigations.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conceptualization: Ibrahim W.W. and Albahairy M.A.; methodology: Albahairy M.A.; data curation and writing-original draft: Muhammad R.N., Ibrahim W.W., and Albahairy M.A.; reviewing and editing: Ibrahim W.W., Albahairy M.A., Muhammad R.N., and Abd El Fattah M.A. Authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muhammad, R.N., Albahairy, M.A., Abd El Fattah, M.A. et al. Empagliflozin-activated AMPK elicits neuroprotective properties in reserpine-induced depression via regulating dynamics of hippocampal autophagy/inflammation and PKCζ-mediated neurogenesis. Psychopharmacology (2024). https://doi.org/10.1007/s00213-024-06663-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00213-024-06663-0