
Abstract
Cantharidin and sodium fluoride inhibit the activity of serine/threonine protein phosphatases 1 (PP1) and 2A (PP2A) and increase the force of contraction in human atrial preparations. R-phenylisopropyl adenosine (R-PIA) acts as an agonist at A1-adenosine receptors. R-PIA exerts a negative inotropic effect on human atria. The effect of R-PIA—and its various manifestations—are currently explained as a function of the inhibition of sarcolemmal adenylyl cyclase activity and/or opening of sarcolemmal potassium channels. We hypothesise that cantharidin and sodium fluoride may attenuate the negative inotropic effect of R-PIA. During open heart surgery, trabeculae carneae from the right atrium were obtained for human atrial preparations (HAPs). These trabeculae were mounted in organ baths and electrically stimulated at 1 Hz. Furthermore, we studied isolated electrically stimulated left atrial (LA) preparations from female wild-type mice (CD1). The force of contraction was recorded under isometric conditions. R-PIA (1 µM) exerted a rapid negative inotropic effect in the HAPs and mice LA preparations. These negative inotropic effects of R-PIA were attenuated by pre-incubation for 30 min with 100-µM cantharidin in HAPs, but not in mice LA preparations. Adenosine signals via A1 receptors in a species-specific pathway in mammalian atria. We postulate that R-PIA, at least in part, exerts a negative inotropic effect via activation of serine/threonine phosphatases in the human atrium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-Adrenoceptors activate adenylyl cyclases via stimulatory guanosine triphosphate (GTP) binding proteins, thereby inducing the formation of 3′,5′-cyclic adenosine monophosphate (cAMP), which then stimulates cAMP-dependent protein kinase (PKA). PKA phosphorylates specific amino acids in regulatory proteins (serine and threonine) and thereby activates several proteins (Fig. 1), which are dephosphorylated by protein phosphatases (PPs) (Herzig and Neumann 2000; Neumann et al. 2021).

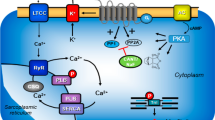
Scheme of a cardiomyocyte The Ca2+ for force generation in the heart is derived in part from trigger Ca2+ via L-type Ca2+ channels and, to a greater extent, via subsequent release of Ca2+ from the sarcoplasmic reticulum (SR) via ryanodine receptors (RYR). Cardiac relaxation is brought about via the phosphorylation of phospholamban (PLB) and the inhibitory subunit of troponin. Phosphorylated PLB de-inhibits the activity of the SR-Ca2+−ATPase (SERCA), and Ca2+ is rapidly moved from the cytosol into the SR, supporting relaxation. Furthermore, phosphorylation of the inhibitory subunit of troponin (TnI) reduces the Ca2+ sensitivity of myofilaments, hastening relaxation. Ca2+ can be released from the SR via ryanodine receptors. The activities of serine/threonine protein phosphatases (PP) PP1 and PP2A are inhibited by cantharidin or by sodium fluoride. R-PIA-stimulating A1-adenosine receptors may inhibit the enzymatic activity of adenylyl cyclases (AC) via a pertussis toxin-sensitive G-protein and/or may open potassium channels (PC), and/or may open the L-type Ca2+ channel (LTCC), and/or may directly or indirectly activate PP. AC can be inhibited by Gi, an inhibitory guanosine triphosphate (GTP) binding protein, and can catalyse cAMP (3′,5′-cyclic adenosine monophosphate) formation. This cAMP activates cAMP-dependent protein kinases (PKA), which phosphorylates several cardiac proteins, such as PLB, RyR, LTCC and TnI, indicated by P in the circle
In human and animal cardiac preparations, both cantharidin and sodium fluoride inhibit PP1 and PP2A (Herzig and Neumann 2000). This inhibition is functionally relevant: cantharidin increases the force of contraction in guinea pig papillary muscle, mice atrial preparations and human atrial muscle and ventricular preparations by increasing the phosphorylation state of regulatory proteins (Neumann et al. 1995b; Schwarz et al. 2023a, b).
Furthermore, our research team, as well as other researchers, has supplied circumstantial evidence that the effect of R-phenylisopropyl adenosine (R-PIA) in reducing the force of contraction—at least concomitantly with β-adrenoceptor stimulation in the mammalian ventricle—involves not only inhibition of cAMP production but also activation of cardiac PPs (Herzig et al. 1995; Gupta et al. 2002).
In this study, we postulate that—in the human atrium—R-PIA acts, in part, by stimulating PP1 and/or PP2A (Fig. 1). Therefore, if we then inhibit PP1 and/or PP2A by means of cantharidin or sodium fluoride, the negative inotropic effect of R-PIA in the human atrium should be attenuated.
Similar studies on carbachol in the human heart (atrium or ventricle) have failed to touch on R-PIA. Nonetheless, R-PIA, like carbachol, can reduce the force of contraction in human atrial preparations in part via the same potassium channel. Interestingly, even in the presence of a phosphodiesterase inhibitor (IBMX), R-PIA has a negative inotropic effect devoid of a reduction in cAMP or cGMP (Böhm et al. 1994), and this prompted all our subsequent studies on cardiac PPs. Alternatively, one can directly increase intracellular cAMP by adding dibutyryl-cAMP. This produces a positive inotropic effect in, for example, Langendorff-perfused guinea pig hearts or isolated rat atrial preparations (Rockoff and Dobson 1980; West et al. 1986). However, when 5 mM dibutyryl-cAMP is added, the applied adenosine—presumably via A1-adenosine receptors—subsequently fails to reduce the force of contraction. This is typically interpreted as evidence against any involvement of PPs in the negative inotropic effect of R-PIA. However, as we show in this study, this may simply result from using a much too high concentration of dibutyryl-cAMP. Indeed, approximately 0.3 mM dibutyryl-cAMP is required to increase the force of contraction half-maximally in isolated human ventricular preparations (Neumann et al. 1999). Therefore, we tested the interaction between R-PIA and such a low concentration of dibutyryl-cAMP in the human atrium. To the best of our knowledge, this has not been studied previously.
Primarily, we test the following hypotheses: (1) cantharidin attenuates the negative inotropic effect of R-PIA in isolated mouse left atria and human atria and (2) sodium fluoride attenuates the negative inotropic effect of R-PIA in isolated mouse left atria and human atria.
Materials and methods
Mice
To avoid gender differences, we used only left atrial preparations from female mice aged 9–17 months. Only wild-type mice with a CD1 background were used in our experiments.
Humans
Human specimens were obtained from male and female patients aged 60–82 years. The cardiac drug therapy of these patients included apixaban, furosemide, metoprolol and acetyl salicylic acid. This study was approved by a local ethical committee, and the patients from whom the human study specimens were obtained gave their written informed consent.
Contractile studies in mice and humans
Our methods for atrial contraction experimentation studies on human samples have been published previously and were not altered in this study (Schwarz et al. 2023a, b). Right atrial preparations from humans and left atrial preparations (auricles) from wild-type mice were isolated and mounted in organ baths as described in a previous study (Neumann et al. 2003). The bathing solution—a modified Tyrode’s solution—in the organ baths contained NaCI (119.8 mM), KCI (5.4 mM), CaCl2 (1.8 mM), MgCl2 (1.05 mM), NaH2PO4 (0.42 mM), NaHCO3 (22.6 mM), Na2EDTA (0.05 mM), ascorbic acid (0.28 mM) and glucose (5.05 mM). The bathing solution was gassed continuously with 95% O2 and 5% CO2 and maintained at 37 ℃ and pH 7.4.
The specimens (human right atrial muscle strips and left atrial preparations from mice) were electrically stimulated at 1 Hz using a Grass Stimulator SD9 (Grass Instruments, Quincy, MA, USA), with 5-ms rectangular impulses of direct current at a 10% higher voltage than required for initiation of the heartbeat. The preparations were stretched carefully in such a way as to ensure that the maximum force (i.e., maximal difference between systolic and diastolic tensions) was generated which was about 4 mN. Force was measured under isometric conditions, and the signals were amplified with a bridge amplifier and fed into a personal computer. The hardware and software utilised were procured from AD Systems, Heidelberg, Australia, through their European outlet.
Drug application was performed as follows: After equilibration was reached, cantharidin (at 100 µM concentration) was added to the mouse left atrial preparations and human right atrial preparations. Then, where indicated, R-PIA was applied cumulatively or as a single concentration to the preparations.
Cantharidin (CANT, 100 mM in dissolved dimethyl sulfoxide (DMSO)) and (R)-N6-(1-methyl-2-phenylethyl)-adenosine (i.e., R-PIA) were purchased from Sigma-Aldrich (now Merck) in Taufkirchen, Germany. DMSO, at the concentrations used, slightly reduced the force of contraction, as reported in a previous study (Schwarz et al. 2023a). All other chemicals were of the highest purity grade commercially available. Deionised water was used throughout the experiments, and stock solutions were prepared fresh daily.
Data analysis
The data are presented as the mean ± standard error of the mean. Statistical significance was estimated using analysis of variance, followed by Dunnett’s or Bonferroni’s t test. A p value of < 0.05 was considered significant.
Results
Cantharidin: mice atria
To electrically stimulate left atrial preparations from mice, 1 µM R-PIA was applied in the absence of CANT and in the presence of CANT. The original tracings are presented in Fig. 2A and B, respectively. The negative inotropic effect of R-PIA was not attenuated in the presence of CANT; the original recordings are presented in Fig. 2A and B, and the data on the force of contraction are summarised in Fig. 2C. We previously reported that CANT, at a 100 µM concentration, by itself increases the force of contraction in left atrial preparations from mice (Schwarz et al. 2023a, b). Under these conditions, R-PIA exerted a negative inotropic effect that was similar in the absence and presence of CANT (Fig. 2C).

Impact of cantharidin on the negative inotropic effect of R-PIA in mouse left atrium. A, B Original recording of the effect of 1 µM R-PIA alone or in the presence of 100-µM cantharidin (CANT) on the force of contraction in milli Newton (mN, ordinate) over time in min (abscissa). C Time course of the negative inotropic effect of 1 µM R-PIA alone or in the presence of CANT (100 µM) in isolated electrically driven left atrial preparations from mice. The ordinate gives the force of contraction in % of pre-drug value before application of R-PIA, * first significant difference versus Ctr. The numbers in brackets indicate the number of experiments. Ctr indicates the value prior to the application of R-PIA
Cantharidin: human atria
For human atrial preparations, we used two different experimental set-ups. First, 100 µM CANT was utilised, and it raised the force of contraction (Ctr1 versus Ctr2 in Fig. 3F), which is consistent with the findings of previous studies (Schwarz et al. 2023a, b). Thereafter, R-PIA was applied noncumulatively or cumulatively. Notably, similar to the effect in mice atria, the effect of R-PIA in human atria is monophasic. This contrasts with carbachol, which initially induces a negative inotropic effect that is subsequently followed by a positive inotropic effect at higher concentrations of carbachol (Schwarz et al. 2023b), indicating that R-PIA stimulates only one type of receptor (i.e., A1-adenosine receptor), while carbachol stimulates two receptor types in the human heart, i.e., the M2- and M3-muscarinic receptors (Du et al. 1995). At a 1 µM concentration, R-PIA elicited a pronounced negative inotropic effect; the original recording is presented in Fig. 3A. However, the negative inotropic effect of 1 µM R-PIA was attenuated in the presence of CANT; the original recording is presented in Fig. 3B. The data on the negative inotropic effect of 1 µM R-PIA are summarised in Fig. 3C. Comparing Ctr1 and Ctr2 in Fig. 3C, the positive inotropic effect of CANT may be determined. Under the given conditions, CANT increased the rate of tension development and the rate of tension relaxation. Next, we cumulatively applied R-PIA to the specimens in the absence and presence of 100 µM CANT (Fig. 3D and E). Again, CANT attenuated the concentration-dependent negative inotropic effect of R-PIA; the data are summarised in Fig. 3F. Reductions in the rate of tension development and the time of relaxation effectuated by R-PIA were also attenuated by CANT (Fig. 3G and H).

Cantharidin attenuates the negative inotropic effect of R-PIA on contractile parameters in human right atria. A, B Original recording of the effect of 1 µM R-PIA alone or with pre-applied cantharidin (100 µM) on the force of contraction in milli Newton (mN, ordinate) over time in min (abscissa). C Time course of the negative inotropic effect of 1 µM R-PIA alone or in the presence of CANT (100 µM) in isolated electrically driven human right atrial preparations. Ordinate gives the force of contraction in % of pre-drug value before application of R-PIA, * and + first significant difference versus Ctr or R-PIA in the presence of CANT, respectively. The numbers in brackets indicate the number of experiments. Ctr indicates the value prior to the application of R-PIA. D, E Original recording of the effect of cumulatively applied R-PIA alone or in the presence of 100-µM cantharidin on the force of contraction in milli Newton (mN, ordinate) over time in min (abscissa). F Concentration-dependent negative inotropic effect of cumulatively applied R-PIA alone or in the presence of CANT (100 µM) in isolated electrically driven human right atrial preparations. Ordinate gives the force of contraction in % of value before R-PIA (Ctr2). * and + first significant difference versus Ctr2 or R-PIA in the presence of CANT, respectively. The numbers in brackets indicate the number of experiments. Ctr1 indicates the pre-drug value. G, H Concentration-dependent effect of cumulatively applied R-PIA alone or in the presence of CANT (100 µM) on rate of tension development and on rate of tension relaxation in isolated electrically driven human right atrial preparations. Ordinate gives the rate of tension development (DF/dtmax) or the rate of tension relaxation (− DF/dtmax) in % of the value before R-PIA (Ctr2). * and + first significant differences versus Ctr2 or R-PIA in the presence of CANT, respectively. The numbers in brackets indicate the number of experiments. Ctr1 indicates the pre-drug value
Sodium fluoride: human atria
For CANT in human atrial preparations, we used two experimental set-ups. Sodium fluoride (3 mM), similar to CANT, exerted a positive inotropic effect (Fig. 4F, Ctr1 versus Ctr2) in human atrial preparations, as has been reported previously (Schwarz et al. 2023b). The original recordings for the application of 1 µM R-PIA in the absence and presence of 3 mM sodium fluoride are presented in Fig. 4A and B. Similar to CANT, sodium fluoride attenuated the negative inotropic effect of R-PIA (Fig. 4C). Cumulatively applied R-PIA, in the absence of sodium fluoride, exerted a concentration-dependent negative inotropic effect over time (Fig. 4D). However, in the presence of sodium fluoride, the negative inotropic effect of R-PIA was attenuated (Fig. 4E). The data on R-PIA are summarised in Fig. 4F. Under these experimental conditions, R-PIA also reduced the rate of tension development (Fig. 4G) and the rate of tension relaxation (Fig. 4H). The effects of R-PIA on the rate of tension relaxation were attenuated by the addition of sodium fluoride, but the effects of R-PIA on the rate of tension development were unaffected (Fig. 4G and H).

Sodium fluoride attenuates the negative inotropic effect of R-PIA on contractile parameters in the human right atrium A, B Original recording of the effect of 1 µM R-PIA alone or with pre-applied sodium fluoride on the force of contraction in milli Newton (mN, ordinate) over time in minutes in human right atrial preparations (min, abscissa). C Time course of the negative inotropic effect of 1 µM R-PIA alone or in the presence of sodium fluoride (3 mM) in isolated electrically driven human right atrial preparations. Ordinate gives the force of contraction in % of pre-drug value before the application of R-PIA, * and # first significant difference versus Ctr or R-PIA in the presence of sodium fluoride. The numbers in brackets indicate the number of experiments. Ctr indicates the value prior to the application of R-PIA. D, E Original recording of the effect of R-PIA (cumulatively applied) alone or in combination with 3 mM sodium fluoride (NaF) on the force of contraction in milli Newtons (mN, ordinate) over time in minutes in human right atrial preparations (min, abscissa). F Concentration-dependent negative inotropic effect of R-PIA alone or in the presence of sodium fluoride (3 mM) in isolated electrically driven human right atrial preparations. Ordinate gives the force of contraction in % of value before R-PIA (Ctr2). * and # first significant difference versus Ctr2 or R-PIA in the presence of sodium fluoride. The numbers in brackets indicate the number of experiments. Ctr1 indicates the pre-drug value. G, H Concentration-dependent effect of cumulatively applied R-PIA alone or with pre-applied sodium fluoride (3 mM) on the rate of tension development and on the rate of tension relaxation in isolated electrically driven human right atrial preparations. Ordinate gives the rate of tension development (DF/dtmax) or the rate of tension relaxation (DF/dtmax) in % of the value before R-PIA (Ctr2). * and # first significant differences versus Ctr2 or R-PIA in the presence of sodium fluoride, respectively. The numbers in brackets indicate the number of experiments. Ctr1 indicates the pre-drug value
Dibutyryl-cAMP (Db-cAMP): human atria
We used dibutyryl-cAMP (Db-cAMP) as a stimulatory drug for PKA experimentation. This was done to intensify the force of contraction without any previous adenylyl cyclase stimulation (Fig. 1). In other words, R-PIA can reduce the force of contraction without any previous stimulation to induce cAMP generation in sarcolemma. Db-cAMP was applied to human atrial preparations at two different concentrations: 100 µM and 300 µM. The original recordings are presented in Fig. 5A and B. Both concentrations—with 300 µM being slightly more efficient—attenuated the negative inotropic effect of R-PIA (Fig. 5C and D).

Dibutyryl-cAMP attenuates the negative inotropic effect of R-PIA on contractile parameters in human right atria. A Original recording of the effect of 1 µM R-PIA on the force of contraction in the presence of 100 µM dibutyryl-cAMP (Db-cAMP) in milli Newton (mN, ordinate) over time in minutes in human right atrial preparations (min, abscissa). B Original recording of the effect of 1 µM R-PIA on the force of contraction in the presence of 300 µM dibutyryl-cAMP (Db-cAMP) in milli Newton (mN, ordinate) over time in minutes in human right atrial preparations (min, abscissa). C, D Time course of the negative inotropic effect of 1 µM R-PIA alone or in the presence of 100 µM or 300 µM dibutyryl-cAMP in isolated electrically driven human right atrial preparations. Ordinate gives the force of contraction in % of pre-drug value before the application of R-PIA, * and § first significant difference versus Ctr or R-PIA in the presence of dibutyryl-cAMP, respectively. The numbers in brackets indicate the number of experiments. Ctr indicates the value prior to the application of R-PIA
Discussion
In our view, the main new finding in this study is the observation that the PP inhibitors CANT and sodium fluoride attenuate the negative inotropic effect of R-PIA in the human atrium.
Specifically, the contractile effects of sodium fluoride are species- and region-dependent. For instance, we have observed that 3 mM sodium fluoride (NaF) has a positive inotropic effect on guinea pig papillary muscle (Neumann et al. 1995a; Neumann and Scholz 1998). In contrast, 3 mM NaF has been observed to decrease the force of contraction in guinea pig and rat atria until the preparations go into asystole (Neumann and Scholz 1998).
In guinea pig ventricular preparations and human ventricular preparations, R-PIA alone does not decrease the force of contraction (Borea et al. 2018). However, when force generation was pre-stimulated by isoprenaline (a β-adrenoceptor agonist) or 3-isobutyl-1-methylxanthine (an unselective phosphodiesterase inhibitor), R-PIA produced a negative inotropic effect in guinea pig papillary muscle that was almost completely neutralised when 3 mM NaF was applied (Neumann et al. 1995a).
In this study, we researched mouse and human atrial preparations. In mice atria and human atria, the direct negative inotropic effect of R-PIA was attenuated by previously applied NaF. However, we cannot be sure that NaF alone inhibits cardiac phosphatases. Indeed, NaF can interfere with mitochondrial function and G-proteins (Neumann and Scholz 1998). However, because CANT acts similarly, we postulate that NaF uses phosphatase inhibition to counteract the contractile effects of R-PIA.
An open question in this field—despite our past efforts and those of other researchers—is the question of how exactly cardiac A1-adenosine receptors couple to phosphatases. Based on our present data, it is uncertain whether genetic manipulation of mice will help answer this question, as mice and humans seem to signal differently via A1-adenosine receptors. We speculate that human cardiomyocytes derived from stem cells may be a more suitable model for further research in this direction. In such human cardiomyocytes, genetic manipulation might be worth the effort.
We have shown previously, at the mRNA and protein levels, that the catalytic subunits of PP1 and PP2A exist in the human atrium and mice atria (Lüss et al. 2000). Hence, the targets of CANT and NaF are expressed in the human atrium and mice atria (Lüss et al. 2000). The activity of PP1 and PP2A (measured together) was higher in the human heart than in the mouse heart (Neumann et al. 1997; Brüchert et al. 2008). This might explain why cantharidin was more potent to raise force of contraction in the mouse preparations than in the human preparations (Schwarz et al. 2023a). One might speculate that 50% of inhibition of PP is required to detect any positive inotropic effect of cantharidin. Hence, conceivably less cantharidin is required (because PP activity in lower) in mouse atrium compared to human atrium and thus less cantharidin is required in mouse than in human atrium. In addition, we cannot rule out that cell permeability differences exist for cantharidin between human heart and mouse hearts which might contribute to the higher potency of cantharidin to elicit a positive inotropic in the mouse atrium compared to the human atrium. Indeed, there is evidence in mouse liver for a membrane transport of cantharidin (Erdödi et al. 1995). Some additional species differences may lie in the accessory proteins that assemble with the catalytic subunits of PP. Our data on dibutyryl-cAMP seem to indicate that there is room for the effect of PPs. When using a small concentration of dibutyryl-cAMP (100 µM or 300 µM) that does not maximally stimulate phosphorylation, R-PIA, presumably via the activation of distally located PPs (Fig. 1), can reduce force generation. This is circumstantial evidence, as the inhibition of adenylyl cyclases is not necessary for the negative inotropic effect of R-PIA because dibutyryl-cAMP is not known to stimulate cardiac adenylyl cyclases.
The cardiac action of R-PIA differs between species and across cardiac regions (i.e., the atrium and the ventricle). In the atrium, the functionally negative inotropic effect of R-PIA is well established in human atria and mice atria (Böhm et al. 1994; Boknik et al. 2009; Du et al. 1994; Neumann et al. 2003). The negative inotropic effect of R-PIA vanishes in A1-adenosine receptor knockout mice and is thus mediated by A1-adenosine receptors (Burnstock 2017). Similar findings have been reported in human atrial samples. In this regard, studies on receptor-specific antagonists have arrived at the same conclusion: the effect of R-PIA is blocked by DPCPX, thus confirming that the effect is mediated by A1-adenosine receptors (Böhm et al. 1994). Thus, on the one hand, the negative inotropic effect of R-PIA in mice and human atria is probably mediated by A1-adenosine receptors. On the other hand, we have repeatedly shown that the negative inotropic effect of R-PIA in the presence of isoprenaline in the guinea pig ventricle involves phosphorylation of phosphatase inhibitor-1 and subsequent inhibition of PP1 (Gupta et al. 1993, 1994, 2002; Neumann et al. 1994). Other studies have presented evidence that in the ventricle, R-PIA can also activate PP2A (Liu and Hofmann 2002). Hence, there is precedence for R-PIA activating PP1 or PP2A in the mammalian heart. In this study, we postulate that the same occurs in the human atrium. R-PIA activates PP1 or PP2A in the human atrium, but this is inhibited by CANT and sodium fluoride. The positive inotropic effect of cantharidin in the human atrial preparations is accompanied by an increase the phosphorylation state of regulatory proteins like phospholamban (Schwarz et al. 2023a). We propose that the remaining negative inotropic effect of R-PIA in the perpetual presence of CANT is due to the activation of potassium ion channels and the subsequent inhibition of calcium ion channels in the atrium in a phosphorylation-independent fashion (Burnstock 2017).
This study has several limitations. We cannot offer an outline of the biochemical chain of events from the receptor to the phosphatase in the human atrium. There are data that acetylcholine can activate PPs in cardiac membranes after perfusion of whole guinea pig hearts (Ahmad et al. 1989). Similar studies in mouse heart with R-PIA are, to the best of our knowledge, lacking and would better define signal transduction pathways. However, it is known that NaF and CANT inhibit phosphatase activity in human and mouse hearts (Neumann et al. 1994, 1995a). Moreover, we have not measured phosphatase activity in extracts from HAPs and extracts from mouse LA. Moreover, CANT and dibutyryl-cAMP in human muscle strips increase the phosphorylation state of phospholamban, for example (Schwarz et al. 2023b; Neumann et al. 1999). At least in guinea pig ventricular cardiomyocytes, acetylcholine reduced the phosphorylation of phospholamban that had been stimulated by dibutyryl-cAMP (Gupta et al. 1998). These animal data suggest that a similar mechanism for dibutyryl-cAMP an R-PIA might exist in the human atrium. Moreover, in atrial cardiac preparations, A1-adenosine receptors can open potassium channels, and this leads to a shortening of the duration of the action potential and thus less time is available for Ca2+ entering the cardiomyocytes leading to decline in force of contraction in the atrium. To what extent any action of R-PIA on potassium channels in human atrium is relevant and is regulated by phosphatases needs to be elucidated. Moreover, the effect of R-PIA was more prominent in single application compared to cumulative application in the presence of cantharidin or NaF. One might speculate that in cumulative application, a receptor desensitisation occurs or coupling to additional G-proteins might take place for which there is some experimental evidence (Boknik et al. 2009). Moreover, basal force was different between patients and this might bias our findings.
In summary, we can now confirm at least in part the hypotheses formulated in the introduction: CANT and NaF attenuate the negative inotropic effect of R-PIA in human right atria but not in mice left atria. We speculate that CANT and NaF, in part, inhibit cardiac phosphatases stimulated by R-PIA—at least in the human right atrium. However, other additional mechanism cannot be ruled out but many contribute to the negative inotropic effects of R-PIA in HAP.
Data availability
No datasets were generated or analysed during the current study.
References
Ahmad Z, Green FJ, Subuhi HS, Watanabe AM (1989) Autonomic regulation of type 1 protein phosphatase in cardiac muscle. J Biol Chem 264(7):3859–3863
Belardinelli L, Isenberg G (1983) Isolated atrial myocytes: adenosine and acetylcholine increase potassium conductance. Am J Physiol 244(5):H734–H737. https://doi.org/10.1152/ajpheart.1983.244.5.H734
Böhm M, Gierschik P, Schwinger RH, Uhlmann R, Erdmann E (1994) Coupling of M-cholinoceptors and A1 adenosine receptors in human myocardium. Am J Physiol 266(5 Pt. 2):H1951-8. https://doi.org/10.1152/ajpheart.1994.266.5.H1951
Boknik P, Grote-Wessels S, Barteska G, Jiang M, Müller FU, Schmitz W, Neumann J, Birnbaumer L (2009) Genetic disruption of Giα2 and Goα does not abolish inotropic and chronotropic effects of M-cholinoceptor stimulation in atria. Br J Pharmacol 158:1557–1564
Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K (2018) Pharmacology of adenosine receptors: the state of the art. Physiol Rev 98(3):1591–1625. https://doi.org/10.1152/physrev.00049.2017
Brüchert N, Mavila N, Boknik P, Baba HA, Fabritz L, Gergs U, Kirchhefer U, Kirchhof P, Matus M, Schmitz W, DePaoli-Roach AA, Neumann J (2008) Inhibitor-2 prevents phosphatase 1 induced cardiac hypertrophy and mortality. Am J Physiol Heart Circ Physio 295:H1539–H1546
Burnstock G (2017) Purinergic signalling in the cardiovascular system. Circ Res 120(1):207–228. https://doi.org/10.1161/CIRCRESAHA.116.309726
Du XY, Schoemaker RG, Bos E, Saxena PR (1994) Different pharmacological responses of atrium and ventricle: studies with human cardiac tissue. Eur J Pharmacol 259(2):173–180. https://doi.org/10.1016/0014-2999(94)90507-x
Du XY, Schoemaker RG, Bos E, Saxena PR (1995) Characterization of the positive and negative inotropic effects of acetylcholine in the human myocardium. Eur J Pharmacol 284(1–2):119–127. https://doi.org/10.1016/0014-2999(95)00384-w
Erdödi F, Tóth B, Hirano K, Hirano M, Hartshorne DJ, Gergely P (1995) Endothall thioanhydride inhibits protein phosphatases-1 and -2A in vivo. Am J Physiol 269(5 Pt 1):C1176–C1184. https://doi.org/10.1152/ajpcell.1995.269.5.C1176
Gupta RC, Neumann J, Watanabe AM (1993) Comparison of adenosine and muscarinic receptor mediated effects on phosphatase inhibitor-1 activity in the heart. J Pharmacol Exp Ther 266:16–22
Gupta RC, Neumann J, Boknik P, Watanabe AM (1994) M2-specific muscarinic cholinergic receptor-mediated inhibition of phosphorylation of cardiac regulatory proteins. Am J Physiol 266:H1138–H1144
Gupta RC, Neumann J, Watanabe AM, Sabbah HN (1998) Muscarinic-cholinoceptor mediated attenuation of phospholamban phosphorylation induced by inhibition of phosphodiesterase in ventricular cardiomyocytes: evidence against a cAMP-dependent effect. Mol Cell Biochem 187(1–2):155–161. https://doi.org/10.1023/a:1006899931151
Gupta RC, Neumann J, Watanabe AM, Sabbah HN (2002) Inhibition of type-1 protein phosphatase activity by activation of β-adrenoceptors in ventricular myocardium. Biochem Pharmacol 63:1069–1076
Herzig S, Neumann J (2000) Effects of serine/threonine phosphatases on ion channels in excitable membranes. Physiol Rev 80:173–210
Herzig S, Meier A, Pfeiffer M, Neumann J (1995) Stimulation of protein phosphatases as a mechanism of the muscarinic receptor-mediated inhibition of cardiac L-type calcium channels. Pflügers Arch (eur J Physiol) 429:531–538
Liu Q, Hofmann PA (2002) Antiadrenergic effects of adenosine A(1) receptor-mediated protein phosphatase 2a activation in the heart. Am J Physiol Heart Circ Physiol 283(4):H1314–H1321. https://doi.org/10.1152/ajpheart.00343.2002
Lüss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, Müller FU, Scheld HH, Schmid C, Schmitz W, Neumann J (2000) Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J Mol Cell Cardiol 32:2349–2359
Neumann J, Scholz H (1998) Deferoxamine blocks interactions of fluoride and carbachol in isolated mammalian cardiac preparations. Eur J Pharmacol 350:189–194
Neumann J, Boknik P, Bodor GS, Jones LR, Schmitz W, Scholz H (1994) Effects of adenosine receptor and muscarinic cholinergic receptor agonists on cardiac protein phosphorylation. Influence of pertussis toxin. J Pharmacol Exp Ther 269:1310–1318
Neumann J, Kaspareit G, Kirchhefer U, Scholz H (1995a) Sodium fluoride attenuates the negative inotropic effects of muscarinic M2 and adenosine receptor agonists. Eur J Pharmacol 294:451–457
Neumann J, Herzig S, Boknik P, Apel M, Kaspareit G, Schmitz W, Scholz H, Zimmermann N (1995b) On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J Pharmacol Exp Ther 274:530–539
Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N (1997) Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol 29:265–272
Neumann J, Bartel S, Eschenhagen T, Haverich H, Hirt S, Kalmár P, Karczewski P, Krause EG, Schmitz W, Scholz H, Stein B, Thoenes M (1999) Dissociation of the effects of forskolin and dibutyryl cAMP on force of contraction and phospholamban phosphorylation in human heart failure. J Cardiovasc Pharmacol 33:157–162
Neumann J, Boknik P, Matherne GP, Lankford A, Schmitz W (2003) Pertussis toxin sensitive and insensitive effects of adenosine and carbachol in murine atria overexpressing A1-adenosine receptors. Br J Pharmacol 138:209–217
Neumann J, Boknik P, Kirchhefer U, Gergs U (2021) The role of PP5 and PP2C in cardiac health and disease. Cell Signal 85:110035
Rockoff JB, Dobson JG Jr (1980) Inhibition by adenosine of catecholamine-induced increase in rat atrial contractility. Am J Physiol 239(3):H365–H370. https://doi.org/10.1152/ajpheart.1980.239.3.H365
Schwarz R, Hofmann B, Gergs U, Neumann J (2023a) Cantharidin increases the force of contraction and protein phosphorylation in the isolated human atrium. Naunyn-Schmiedeberg’s Arch Pharmacol 396:2613–2625. https://doi.org/10.1007/s00210-023-02483-9
Schwarz R, Hofmann B, Gergs U, Neumann J (2023b) Cantharidin and sodium fluoride attenuate the negative inotropic effects of carbachol in the isolated human atrium. Naunyn-Schmiedeberg´s Arch Pharmacol 397:2183–2202. https://doi.org/10.1007/s00210-023-02747-4
West GA, Isenberg G, Belardinelli L (1986) Antagonism of forskolin effects by adenosine in isolated hearts and ventricular myocytes. Am J Physiol 250(5 Pt 2):H769–H777. https://doi.org/10.1152/ajpheart.1986.250.5.H769
Acknowledgements
Designed project: JN and UG. Supplied reagents and clinical data: BH. Performed experiments: RS and JN. Performed statistics and plotted data: RS, UG. Wrote paper: UG, RS and JN. All authors have read and agreed with the submission of the present version of the work. The authors declare that all data were generated in-house and that no research paper mill was used.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Designed project: JN and UG. Supplied reagents and clinical data: BH. Performed experiments: RS and JN. Performed statistics and plotted data: RS, UG. Wrote paper: UG, RS and JN. All authors have read and agreed with the submission of the present version of the work.
The authors declare that all data were generated in-house and that no research paper mill was used.
Corresponding author
Ethics declarations
Ethics approval
This research conformed to the Guide for the Care and Use of Laboratory Animals, as published by the National Research Council (2011). The animals were handled and maintained according to the approved protocols of the Animal Welfare Committee of the University of Halle-Wittenberg, Halle, Germany. This study complies with the Declaration of Helsinki and was approved by a local ethics committee (hm-bü 04.08.2005).
Consent to participate
Informed written consent was obtained from all patients recruited to the study.
Consent for publication
All authors declare that they have seen and approved the version of this manuscript submitted for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwarz, R., Hofmann, B., Gergs, U. et al. Cantharidin and sodium fluoride attenuate the negative inotropic effect of the A1-adenosine receptor agonist N6-(R)-phenylisopropyl adenosine in isolated human atria. Naunyn-Schmiedeberg's Arch Pharmacol (2024). https://doi.org/10.1007/s00210-024-03402-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00210-024-03402-2