
Abstract
Lysergic acid diethylamide (LSD) is an artificial hallucinogenic drug. Thus, we hypothesized that LSD might act 5-HT4 serotonin receptors and/or H2 histamine receptors. We studied isolated electrically stimulated left atrial preparations, spontaneously beating right atrial preparations, and spontaneously beating Langendorff-perfused hearts from transgenic mice with cardiomyocyte-specific overexpression of the human 5-HT4 receptor (5-HT4-TG) or of the H2-histamine receptor (H2-TG). For comparison, we used wild type littermate mice (WT). Finally, we measured isometric force of contraction in isolated electrically stimulated muscle strips from the human right atrium obtained from patients during bypass surgery. LSD (up to 10 µM) concentration dependently increased force of contraction and beating rate in left or right atrial preparations from 5-HT4-TG (n = 6, p < 0.05) in 5-HT4-TG atrial preparations. The inotropic and chronotropic effects of LSD were antagonized by 10 µM tropisetron in 5-HT4-TG. In contrast, LSD (10 µM) increased force of contraction and beating rate in left or right atrial preparations, from H2-TG. After pre-stimulation with cilostamide (1 µM), LSD (10 µM) increased force of contraction in human atrial preparations (n = 6, p < 0.05). The contractile effects of LSD in human atrial preparations could be antagonized by 10 µM cimetidine and 1 µM GR 125487. LSD leads to H2-histamine receptor and 5-HT4-receptor mediated cardiac effects in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lysergic acid diethylamide (LSD, Fig. 1) was studied for use in psychiatry in the 1960s but was largely used in illicit ways and therefore removed from the market worldwide (review: Schlag et al. 2022). Currently, LSD is predominantly used for recreational purposes, and intoxications are recorded (review: Schlag et al. 2022).

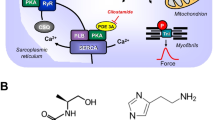
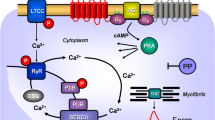
Top: Hypothetical action of lysergic acid diethylamide (LSD). LSD might activate 5-HT4-serotonin receptors (5-HT4-R, stimulated by serotonin and inhibited by tropisetron) or H2-histamine receptors (H2-R, stimulated by histamine and blocked by cimetidine) in the sarcolemma of cardiomyocytes. These receptor stimulations will converge into an activation of adenylyl cyclase activity (AC) by means of stimulatory guanosine triphosphate-binding proteins (G-proteins). AC produces 3´,5´-cyclic adenosine monophosphate (cAMP). This cAMP can then activate a cAMP-dependent protein kinase (PKA). This leads to phosphorylation of many target proteins (in red). For instance, the L-type Ca2+ channel (LTCC) is phosphorylated. This leads to enhanced entrance of trigger Ca2+ into the cell. Ca2+ can release Ca2+ from the sarcoplasmic reticulum. This Ca2+ can bind to myofilaments to generate force (red curve). In diastole, Ca2+ is removed from the cytosol. This leads to relaxation. Ca2+ is pumped by the enzyme SERCA into the sarcoplasmic reticulum where it binds to calsequestrin (CSQ). Ca2+ leaves the sarcoplasmic reticulum via the ryanodine receptor (RYR). Dephosphorylated phospholamban (PLB) inhibits SERCA. Phosphorylated PLB ceases to inhibit SERCA and thus Ca2+ is removed faster from the cytosol. In this way phosphorylation of PLB leads to faster relaxation. Relaxation is further augmented when PKA phosphorylates the troponin inhibitor (TnI) in the myofilaments containing troponin c (TnC). Bottom: Structural formulae of relevant molecules in the present study
Histamine acts via histamine H1, H2, H3 and H4 receptors (Panula et al. 2015; Neumann et al. 2021a, b, c). In the heart of mammals, all four histamine receptor subtypes have been described at RNA and/or protein level.
However, species differences exist in the cardiac effects of histamine (Neumann et al. 2021a, b, c). In the mouse, rat, dog and cat heart, a direct histamine receptor mediated inotropic or chronotropic effect is missing: inotropic effects of histamine were found to be indirect via release of endogenous catecholamines (Flacke et al. 1967; Dai et al. 1976; Laher and McNeill 1980a, 1980b, Gergs et al. 2019).
Moreover, even regional differences in the actions of histamine in the mammalian heart are known: in the rabbit atrium, H1 receptors are more prevalent and a positive inotropic effect mediated by H1 and phospholipase C activation has been described (Hattori et al. 1988). In contrast, in the rabbit ventricle a positive inotropic effect by H2 receptor activation via activation of adenylyl cyclase, a subsequent increase in cAMP and elevation of the activity of a cAMP dependent protein kinase (PKA) was noted by the same group (Hattori et al. 1990, 1991). In humans, H2 receptors were measurable in both the atrium and ventricle (radioligand binding: Baumann et al. 1982, 1983, 1984, antibody and RNA expression: Matsuda et al. 2004). In humans, the cardiac H2 receptors mediate a positive inotropic effect in isolated human atrial cardiac preparations (Levi et al., 1981, Genovese et al. 1988; Zerkowski et al. 1993; Thoren et al. 2011; Sanders et al. 1996). Infusion of histamine in patients led to an increase in heart beat and an increase in the first derivative of pressure development in the left ventricle (Vigorito et al. 1983). These effects of histamine in the human heart are not due to a release of noradrenaline: the human H2 receptor mediates the cardiac actions of histamine in isolated human cardiomyocytes in vitro where a release of noradrenaline from nerve cells was excluded (Sanders et al. 1996). An H2R- agonist called impromidine, has been shown to increase force of contraction in human cardiac preparations (Baumann et al. 1981, 1982, 1983).
LSD was classified by others as a partial agonist at rabbit and guinea-pig cardiac H2 receptors: at low concentrations LSD increases beating rate, and at high concentrations decreases beating rate, in isolated right atrial preparations from rabbits in a cimetidine sensitive fashion (Angus and Black 1980). Moreover, LSD antagonized the positive inotropic effect of histamine in isolated guinea pig papillary muscles (Angus and Black 1980). To the best of our knowledge, the effects of LSD on human cardiac H2 receptors have not been reported.
LSD can also act as an agonist at serotonin receptors. 5-HT2A receptors mediate the hallucinogenic effects of LSD (Schlag et al. 2022). In the human heart, all inotropic and chronotropic effects of serotonin (5-HT) are 5-HT4 receptor mediated (reviews: Neumann et al 2017, 2023). We generated and characterized a mouse model with cardiomyocyte-specific overexpression of the human 5-HT4 receptor (5-HT4-TG) to investigate the actions of the receptor better (Gergs et al. 2010). In a similar fashion as described above for histamine, 5-HT in the heart of wild type littermate mice (WT) has no receptor mediated inotropic effects. However, in 5-HT4-TG, 5-HT exerted positive inotropic effects both in vivo and in vitro (Gergs et al. 2010, 2013). Based on this work from our group, we decided to use 5-HT4-TG to study a putative cardiac role of LSD via 5-HT4-receptors (Fig. 1). To test the clinical relevance of our findings, we set out to measure the effects of LSD under isometric conditions on the force of contraction in the human heart. To this end, we used electrically stimulated right atrial strips obtained rapidly from the surgical theatre.
In summary, we studied the following hypotheses: firstly, LSD stimulates contractility in H2-TG. Secondly, LSD stimulates contractility in 5-HT4-TG. Thirdly, LSD increases the force of contraction in the isolated human atrium via H2–histamine and/or 5-HT4–serotonin receptors.
Methods
Transgenic mice
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Research Council (2011). Animals were maintained and handled according to approved protocols of the animal welfare committees of the University of Halle-Wittenberg, Germany. The generation and initial characterization of the transgenic mice has been described before (Gergs et al. 2010, 2019). In brief, for generation of transgenic mice by pronuclear DNA injection, human H2 -receptor cDNA or human 5-HT4 receptor cDNA were inserted into a mouse cardiac α-myosin heavy chain promoter expression cassette. For all experiments, adult transgenic mice and WT littermates of both sexes were used.
Contractile studies in mice
As described before, the right or left atrial preparations from the mice were isolated and mounted in organ baths (Gergs et al. 2013; Neumann et al. 1998). The bathing solution of the organ baths contained 119.8 mM NaCI, 5.4 mM KCI, 1.8 mM CaCl2, 1.05 mM MgCl2, 0.42 mM NaH2PO4, 22.6 mM NaHCO3, 0.05 mM Na2EDTA, 0.28 mM ascorbic acid and 5.05 mM glucose. The solution was continuously gassed with 95% O2 and 5% CO2 and maintained at 37 °C and pH 7.4 (Neumann et al. 1998, Kim et al. 2004). Spontaneously beating right atrial preparations from mice were used to study any chronotropic effects. After equilibration was reached, ergometrine was cumulatively added to left atrial or right atrial preparations to establish concentration–response curves. Then, where indicated, either serotonin or histamine was additionally applied to the preparations. In separate experiments, concentration–response curves to ergotamine in mouse left atrial preparations were obtained and, after the effect of 10 µM ergotamine had reached a plateau, the atrial strips were rapidly brought to the temperature of liquid nitrogen for further study.
Contractile studies on human preparations
The contractile studies on human preparations used the same setup and buffer as in the mouse studies. The samples were obtained from 3 male patients and 4 female patients, 78–82 years old. Drug therapy included β1-adrenoceptor antagonist metoprolol, the loop diuretic furosemide, the anticoagulant apixaban and the antithrombotic drug acetyl salicylic acid. Our methods used for atrial contraction studies in human samples have been previously published and were not altered in this study (Gergs et al. 2009, 2021b). Patients gave written informed consent.
Langendorff-perfused hearts
Heart preparations were utilized as described previously (Kirchhefer et al. 2014; Gergs et al., 2020). Mice were anesthetized by intraperitoneally administered pentobarbital sodium (50 mg kg-1 body weight) and treated with 1.5 units of heparin. The hearts were removed from the opened chest, immediately attached by the aorta to a 20-gauge cannula, and perfused retrogradely under constant flow of 2 ml min−1 with oxygenized buffer solution (37 °C) containing (in mM): NaCI 119.8, KCI 5.4, CaCl2 1.8, MgCl2 1.05, NaH2P04 0.42, NaHCO3 22.6, Na2EDTA 0.05, ascorbic acid 0.28 and glucose 5.0 in an in-house built isolated heart system equipped with a PowerLab system (ADInstruments, Oxford, United Kingdom). The heart preparations were allowed to equilibrate for 30 min before measurements. Hearts contracted spontaneously in sinus rhythm, and heart rate and force of contraction were measured and monitored continuously. The first derivative of left ventricular contraction (+ dF/dt and − dF/dt) was calculated (LabChart, ADInstruments, Oxford, United Kingdom). This was done to assess the rate of tension development and the rate of relaxation in order to measure the positive inotropic and lusitropic (relaxant) effects of the drugs we studied.
Western blot analysis
Homogenates from ventricular tissue samples were prepared in 300 µl of 10 mM NaHCO3 and 100 µl 20% SDS. Crude extracts were incubated at 25 °C for 30 min before centrifugation to remove debris and thereafter, the supernatants (= homogenates) were separated and stored at -80 °C until further use. Western blot analysis was performed as previously described (Abella et al. 2023). Briefly, aliquots of 20 µg of protein were loaded per lane and finally, bands were detected using enhanced chemiluminescence (ECL, Amersham (Cytiva), Freiburg, Germany) together with an Amersham ImageQuant 800 imager (Cytiva, Freiburg, Germany). The following primary antibodies were used in this study: polyclonal rabbit anti phospho-PLB (antibody was raised against PLB-peptide phosphorylated at serine-16, Badrilla, Leeds, UK) and polyclonal rabbit anti calsequestrin (CSQ, abcam, Cambridge, UK). The characteristics and use of these antibodies has been reported repeatedly by our group (Kirchhefer et al., 2002). The antibody against calsequestrin was used as loading control.
Radioligand competition binding
Radioligand competition binding experiments were performed as previously described by using the HEK293-SP-FLAG-hH2R cell line and [3H]DE257 (Kd = 66.9 nM, c = 40 nM) (Baumeister et al. 2015; Pockes et al. 2018; Rosier et al. 2021). Ligand dilutions were prepared tenfold concentrated in L-15 with 1% BSA, and 10 μL/ well was transferred to a flat-bottom polypropylene 96-well microtiter plate (Greiner Bio-One, Frickenhausen, Germany), as well as 10 μL/ well of the respective radioligand. The cells were adjusted to a density of 1.25 × 106 cells/mL, and 80 μL of the cell suspension was added to each well (total volume of 100 μL). All data were analyzed using GraphPad Prism 9 software (San Diego, CA, USA). The normalized competition binding curves were then fitted with a four-parameter logistic fit yielding pIC50 values. These were transformed into pKi values using the Cheng − Prusoff equation (Cheng and Prusoff 1973).
Data analysis
Data shown are means ± SEM. Statistical significance was estimated by analysis of variance followed by Bonferroni´s t-test. A p-value < 0.05 was considered as significant.
Drugs and materials
LSD was supplied as a stock solution from Merck, Germany. All other chemicals were of analytical grade. Demineralized water was used throughout the experiments. Stock solutions were freshly prepared daily.
Results
-
1.
Left atrium from H2-TG
We first cumulatively applied LSD (0.1 µM to 10 µM). Subsequently, histamine was additionally and cumulatively applied. In left atrial preparations from H2-TG (Fig. 2A) but not from WT (Fig. 2B), LSD exhibited a time- and concentration-dependent positive inotropic effect that was not stimulated further by histamine (Fig. 2A). In WT, neither histamine nor LSD had a positive inotropic effect (Fig. 2B). These data are summarized with regard to the force of contraction for H2-TG (Fig. 2C). Moreover, in the same samples, LSD shortened the time to peak tension (Fig. 2D) and the time of relaxation Fig. 2E). Furthermore, LSD also augmented the absolute values of the rate of tension development (Fig. 2F) and the rate of relaxation (Fig. 2G). Subsequently applied histamine (1 nM to 10 µM) did not add to the effect of previously applied LSD.
-
2.
Right atrium from H2-TG

Original recording depicting the effect of LSD and additionally applied histamine on force of contraction in isolated electrically stimulated left atrial preparations from mice with cardiac overexpression of the human H2-histamine receptor (A) or littermate wild-type mice (WT, B). Arrows indicate drug application. Concentrations are given in negative decadic logarithms of lysergic acid diethyl amide (LSD) or histamine. Horizonal bars: ten minutes (10 min). Vertical bars force of contraction in milli Newton (mN). Force generated by LSD alone or additionally applied histamine (C) in milli Newton (mN) or time to peak tension in milliseconds (ms) (D), or time of relaxation (E), or rate of tension development (F in mN/ms), or rate of tension relaxation (G in mN/ms) in left atrial preparations from H2-TG. Ctr indicates pre-drug values. * indicates the first significant (p < 0.05) difference versus Ctr. Number in brackets indicates the number of experiments. The pD2 values for the effect of LSD are given
Next, we were interested in right atrial function, under the same experimental conditions used in the left atrium. LSD time- and concentration-dependently increased the beating rate of right atrial preparations from H2-TG but not from WT. This is displayed in an original muscle strip in Fig. 3A (H2-TG). Summarized data for the beating rates can be found in Fig. 3B (H2-TG).
-
3.
Left atrium from 5-HT4-TG

Original recording depicting the effect of LSD and additionally applied histamine on beating rate (in beats pro minute, bpm) in spontaneously beating isolated right atrial preparations from mice with cardiac overexpression of the human H2-histamine receptor (A). Concentration-dependent effects of LSD alone or in the additional presence of histamine in beats per minutes (bpm) (B). Ctr indicates pre-drug values. * indicates the first significant (p < 0.05) difference versus Ctr. Number in brackets indicates the number of experiments
In separate studies, we first cumulatively applied LSD (0.1 µM to 10 µM). Subsequently, serotonin was cumulatively applied. In left atrial preparations from 5-HT4-TG, LSD exhibited a concentration- and time-dependent positive inotropic effect that was stimulated further by serotonin (Fig. 4A). These data are summarized in Fig. 4B with regard to force of contraction. Moreover, in the same samples, LSD shortened the time to peak tension (Fig. 4C) and the time of relaxation (Fig. 4D) in 5-HT4-TG. Furthermore, LSD also augmented the absolute values of the rate of tension development (Fig. 4E) and the rate of relaxation (Fig. 4F). Subsequently applied serotonin (1 nM to 10 µM) added to the effect of previously applied LSD. Regarding the effect on the increase in force of contraction, we calculated the effect of LSD relative to that of LSD plus additionally applied serotonin, which was 29.37% ± 8.89%.
-
4.
Right atrium from 5-HT4-TG

Original recording depicting the effect of LSD and additionally applied serotonin on force of contraction in isolated electrically stimulated left atrial preparations from mice with cardiac overexpression of the human 5-HT4-serotonin receptor (A). Arrows indicate drug application. Concentrations in negative decadic logarithms of lysergic acid diethyl amide (LSD) or serotonin. Horizonal bars: ten minutes (10 min). Vertical bars force of contraction in milli Newton (mN). Concentration-dependent effects of LSD alone or additionally applied serotonin on isometric force of contraction (B) in milli Newton (mN) or time to peak tension in milliseconds ms (C), or time of relaxation (D), or rate of tension development (E in mN/s), or rate of relaxation (F in mN/s) in left atrial preparations from 5-HT4-TG. Ctr indicates pre-drug values. * indicates the first significant (p < 0.05) difference versus Ctr. # indicates the first significant (p < 0.05) difference versus the highest concentration of LSD. Number in brackets indicates the number of experiments. Furthermore, the pD2 values are given for the effect of LSD and serotonin respectively
Next, we were interested in right atrial function under the same experimental conditions as used in the left atrium. LSD time- and concentration-dependently increased the beating rate of right atrial preparations from 5-HT4-TG (original muscle strip in Fig. 5A). While LSD increased the beating rate in 5-HT4-TG, additionally applied serotonin did not stimulate the beating rate any further (Fig. 5A). Regarding the effect on the beating rate, we calculated the effect of LSD relative to the effect of LSD plus additionally applied serotonin, which was 76.69% ± 6.65%.
-
5.
Antagonists

Original recording depicting the effect of LSD and subsequently applied serotonin on beating rate (in beats pro minute, bpm) in spontaneously beating isolated right atrial preparations from mice with cardiac overexpression of the human 5-HT4-serotonin receptor (A). Concentration-dependent effects of LSD alone or additionally applied serotonin (B) in beats per minutes (bpm) Ctr indicates pre-drug values. * indicates the first significant (p < 0.05) difference versus Ctr. Number in brackets indicates the number of experiments
To confirm the role of the H2 receptor and the 5-HT4 serotonin receptor in transgenic mice, we used antagonists: cimetidine antagonized the positive inotropic effect or the positive chronotropic effect of LSD in H2-TG (original recording: Fig. 6A). Likewise, tropisetron antagonized the positive inotropic effect or the positive chronotropic effect of LSD in 5-HT4-TG (Fig. 6B, C). We chose to use tropisetron because it was the first antagonist used to define 5-HT4 receptors in the human atrium and had a pKb value of 6.7 in human atrium contraction studies (Kaumann et al. 1990).
-
6.
Langendorff perfused hearts

Original recordings: cimetidine antagonizes the positive inotropic effects of LSD (A, top) in left atrial preparations and the positive chronotropic effects of LSD in right atrial preparations from H2-TG (A, bottom). Tropisetron antagonizes the positive inotropic effects of LSD (B) in left atrial preparations and the positive chronotropic effect of LSD in right atrial preparations from 5-HT4-TG. Ordinates give force of contraction in milli Newton (mN) or indicate beats per minute (bpm). Horizontal bars indicate time scales in minutes (min). The arrows indicate the addition of drugs with the concentrations of drugs given in negative decadic logarithms
Next, it was of interest to investigate ventricular effects of LSD. To this end, we used isolated retrogradely perfused hearts (Langendorff preparations), allowed to beat spontaneously. We recorded force of contraction from the apex and measured ventricular function under these conditions. We noted that 10 µM LSD increased force of contraction in hearts from H2-TG and, to a lesser extent, 5-HT4-TG, but not in hearts from WT animals(Table 1).
-
7.
Protein phosphorylation in mouse samples
In freeze-clamped isolated atrial preparations, LSD increased the phosphorylation state of phospholamban at amino acid serine 16 in preparations from H2-TG and, to a lesser extent, 5-HT4-TG. Figure 7A displays a typical Western blot. Data are statistically analyzed in Fig. 7B.
-
8.
Effects in human atrium

Photograph A of a Western blot. Left atrial contracting preparations from mice with cardiac overexpression of human H2-histamine receptors (H2-TG or human 5-HT4 receptors (5-HT4-TG) or wild type (WT) were stimulated with LSD or isoprenaline (positive control), freeze- clamped and homogenized. One sample of an isoprenaline-treated atrial preparation was boiled immediately before running the polyacrylamide gel. Samples were (molecular weight with arrows on the left) transferred to nitrocellulose. The upper part detects calsequestrin (CSQ; see Fig. 1) the lower part serine 16 phosphorylated phospholamban (PLB). Note the molecular weight reduction of the boiled sample which is characteristic of PLB. The gel was quantified B and the ratio of the signal for serine-16 phosphorlyated PLB and CSQ is plotted on the ordinate. Numbers in bars indicate the number of experiments. The ratio in WT samples was arbitrarily set as 100 percent
In isolated human right atrial preparations, we first investigated the effect of LSD only. Usually LSD failed to show any positive inotropic effect (original recording in Fig. 8A, summarized data in Fig. 8B). Even a negative inotropic effect was apparent. Only in one preparation, LSD showed a positive inotropic effect without pre-incubation (Fig. 8C, patient 1). We had expected from the findings in in left atrial preparations from H2-TG and 5-HT4-TG that LSD alone would increase force of contraction. As that was usually not the case, in isolated human atrial preparations, we routinely measured the effect of LSD in the presence of 1 µM cilostamide. Cilostamide inhibits phosphodiesterase III which is the main phosphodiesterase in the human heart (e.g. von der Leyen et al. 1991). We hypothesized therefore that pre-stimulation with cilostamide would sensitize the human atrial preparations for positive inotropic effects of LSD. Initially, cilostamide on its own increased force of contraction, as expected from a PDE III-inhibitor in the human heart. Thereafter, additionally applied LSD raised force of contraction further. This positive inotropic effect of LSD was completely reversed by additionally applied cimetidine (Fig. 8 C, Patient 2) and partially reversible by additionally applied 10 µM tropisetron (Fig. 8D) or 1 µM GR125487 (Fig. 8E). We used here also GR125487 because it has a higher affinity to the 5-HT4-receptor than tropisetron (pKi-values of 10.1 and 6.8, respectively, Brattelid et al. 2004). Similarly, cilostamide and additionally applied LSD raised the absolute maximum rates of tension development (Fig. 8 F) and of relaxation development (Fig. 8 G), while additionally applied tropisetron or GR125487 and cimetidine reduced force of contraction elevated by LSD. One might thus be tempted to conclude that the effect of LSD in human atrial tissue might be mediated via both H2—and 5-HT4-receptors. Moreover, we also studied as a control the effects of tropisetron (10 µM) alone or GR124587 (1 µM) alone on force of contraction in human atrial preparations, which was not decreased significantly (to 97 ± 8.5% and 95 ± 9.3%, each n = 3, respectively). As a further control, we conducted experiments to determine whether the positive inotropic effect of cilostamide 1 µM itself might be reversed by subsequently applied 10 µM cimetidine, which it was not (original recording in Fig. 8H, summarized data in Fig. 8I).
-
9.
Radioligand binding studies

Original recording (A) of the effect of LSD without preincubation with Cilostamide on the force of contraction in isolated electrically stimulated right atrial preparations from patients undergoing bypass surgery. (B) Summarized relative effect of LSD without preincubation with Cilostamide on the force of contraction in human right atrial preparations(absolute mean force at Ctr 4.97 mN, after 10 µM LSD 2.89 mN). (C) Original recording depicting the effect of LSD alone (patient 1) or after pre-stimulation with 1 µM cilostamide and subsequently additionally applied cimetidine (patient 2) on force of contraction in human atrial preparations. (D) Summarized effects of subsequently applied cilostamide (1 µM), LSD (10 µM), tropisetron (10 µM) and cimetidine (10 µM) on the force of contraction of human atrial preparations. (E) Summarized effects of cilostamide (1 µM), LSD (10 µM), GR124587 (10 µM) and cimetidine (10 µM) on the force of contraction of human atrial preparations. (F, G) Summarized effects of cilostamide (1 µM), LSD (10 µM), tropisetron (10 µM) and cimetidine (10 µM) on the maximum rate of tension (F) and relaxation (G) development of contraction in human atrial preparations. (H, I) Original recording (H) and summarized relative data (I) showing the effect of 1 µM cilostamide and additionally applied 10 µM of cimetidine on the force of contraction in human atrial preparations(absolute mean force at Ctr 0.90 mN, after 1 µM cilostamide 3.05 mN, after 10 µM cimetidine 3.04 mN). Effects on force of contraction are given in mN or as % of pre-drug values respectively. Effects on maximum rates of tension or relaxation development are given in mN/s. Arrows indicate drug application. Horizonal bars: ten minutes (10 min). Ctr indicates pre-drug values. *p < 0.05 versus Ctr. Number in brackets indicates the number of experiments
In order to investigate the binding behavior of LSD we tested its affinity to the H2R in a radioligand binding assay using HEK cells in a recombinant expression system. Therefore, we used the selective H2R radioligand [3H]UR-DE257 (N-[6-(3,4-dioxo-2-{3-[3-(piperidin-1-yl-methyl)phenoxy]propylamino}cyclobut-1-enylamino)hexyl]-(2,3-3H2)propionic amide, Kd = 66.9 nM, c = 40 nM, Bmax = 11,122 dpm, corresponding to 990,000 receptor sites/cell), that is useful for the identification and pharmacological characterization of H2R ligands (Baumeister et al. 2015). LSD showed binding at the human H2 receptor (pKi = 4.49 ± 0.09, slope = 1.31 ± 0.11, n = 3, Fig. 9), which is depicted in Fig. 9 in direct comparison to reference compound famotidine (pKi = 7.63). Unspecific binding was detected using famotidine (10 µM).

Displacement curve from a representative radioligand competition binding experiment performed with LSD, famotidine and [3H]DE257 (Kd = 66.9 nM, c = 40 nM, Bmax = 11,122 dpm, corresponding to 990,000 receptor sites/cell) using HEK293-SP-FLAG-hH2R cells
Discussion
The first evidence for LSD action at H2 receptors was found in membranes from guinea pig brains, where LSD increased the activity of adenylate cyclase in a cimetidine sensitive manner, and thus proved to be H2 receptor mediated (Green et al. 1978). Indeed, our data in transgenic animals indicate that LSD is an agonist at human H2 receptors as the effects of a high concentration of LSD reached a plateau, indicating full receptor saturation. Under these conditions, exogenous histamine was ineffective. The evidence that LSD exerted these actions via H2 receptors is several fold. Firstly, LSD was only active in H2-TG samples and not in WT samples. Secondly, the effect of LSD on force and beating rate in H2-TG samples could be antagonized by cimetidine, known as a selective antagonist at H2 receptors. Thirdly, LSD led to an increase in phosphorylation of PLB (Fig. 1), a pathway expected for H2 receptor agonists (Gergs et al. 2019). Fourthly, in transfected cells, LSD could bind to H2 receptors. Moreover, our findings are in line with previous animal studies where LSD could increase force or/and beating rate in rabbit or guinea-pig cardiac muscle strips (Angus and Black 1980). These effects were blocked by cimetidine (Angus and Black 1980).
We proved that stimulation of H2 receptors is clinically relevant in humans when the inotropic effect of LSD in isolated human heart samples was blocked by cimetidine, thus demonstrating a H2 receptor mediated response. Moreover, we would argue the fact that we usually required phosphodiesterase III inhibition by cilostamide argues that also in the human heart H2 receptors couple LSD to force generation. There are data that H2 stimulation in human heart samples leads to cAMP increases and activation of cAMP dependent protein kinase (Fig. 1, review: Neumann et al. 2021a, b, c). H2 activation is expected to activate PKA, which only phosphorylates PLB on serine 16 (Simmerman et al. 1986). This augmented phosphorylation of PLB explains at least in part the relaxant effects and inotropic effects of LSD in the human atrium.
LSD has a role as a recreational drug or drug of abuse (De Gregorio et al. 2016). LSD is used for these purposes probably because LSD is very potent agonist a 5-HT2A receptors, where binding is thought to lead to hallucinations that some users crave for. In conjunction, it is almost certain that more than one receptor is involved in the central nervous system actions of LSD because it has long known that LSD has a high affinity for many G-protein coupled receptors (Roth et al. 2002). For instance, LSD binds with high affinity to all five dopamine receptors and most serotonin receptors (Roth et al. 2002). Hence, the signal transduction of LSD is complex (review: Olson 2022). However, LSD binding to 5-HT4 receptors appears to be unreported.
PLB is only expressed in cardiomyocytes and not in non-cardiomyocytes. The fact that we measured an increase in PLB phosphorylation in atrial samples is strongly indicative that the H2 receptors are present and functional in cardiomyocytes. We also used the alpha myosin promotor, which drives expression in the heart only in cardiomyocytes (Subramaniam et al. 1993).
Clinical relevance
Our data have clinical applications. Firstly, intoxications with LSD have been well reported. Intoxication with LSD were accompanied with tachycardia by 40% of recreational users, demonstrating that cardiac side effects of LSD are clinically relevant (Leonard et al. 2018, Passie et al. 2008).
Should these intoxications be accompanied by cardiac arrhythmias, our data suggest that it might be worthwhile to test cimetidine as an antidote. Cimetidine is an approved drug and, when given intravenously, should block any effects of LSD on the cardiac H2-R that may have directly caused an arrhythmia. Alternatively, one could also use the hH2-R antagonist ranitidine. Ranitidine is more potent than cimetidine and is available in an injectable drug formulation. Likewise, it is conceivable to try to terminate cardiac arrhythmias in LSD overdosing by additionally applying tropisetron intravenously.
As well as intoxication treatment options, our data might supply evidence for LSD to be used as a therapeutic drug. Recently, efforts have been renewed to treat psychiatric patients with LSD (Gasser et al. 2015). Indeed, there are currently 148 studies on clinicaltrials.gov for LSD. Our data will allow calculations for safe plasma concentrations of LSD, to act on high affinity brain receptors but not on cardiac H2 receptors. We also show that in addition, 5-HT4 receptors might similarly affect the human heart. Reported therapeutic plasma levels after taking 100 µg perorally LSD is 1.3 ng/ml (about 4 nM, Dolder et al. 2018), and for recreational purposes about fivefold higher doses were reported, leading to 20 nM plasma concentrations of LSD (Dolder et al. 2018). However, these plasma concentrations might be higher if the metabolism of LSD is impaired either by additionally applied drugs or when (for genetic reasons) patients exhibit low metabolism of LSD (Luethi et al. 2019, Table 2).
Limitations
We have not had the opportunity to study human ventricular samples in our contraction study due to lack of access to that tissue. Hence, we can only extrapolate from our Langendorff data in H2-TG that LSD will also have effects on the human ventricle. Moreover, there is a negative inotropic effect of LSD in WT in Fig. 2B. This cannot result from stimulation of endogenous mouse H2-receptors because histamine itself does not reduce force of contraction in isolated left atrial preparations from wild-type mice (Gergs et al. 2019). In line with negative inotropic effect in left atrium from WT, we report in Fig. 8A a similar cardioinhibitory effect of LSD alone (in the absence of cilostamide) in human atrial preparations. It is generally accepted that histamine only increase and does not decrease force of contraction in the isolated human atrium (e.g. Baumann et al. 1981). We would speculate, the another, presently unknown receptors, to which LSD binds, probably underlies this effect. The actual mechanism(s) need(s) of the negative inotropic effect of LSD remain(s) to be elucidated.
Moreover, we cannot explain, why in the whole set of patients we studied, only in one patient we measured a pronounced positive inotropic effect of LSD alone. We could speculate that this patient has genetically a higher expression of the 5-HT4-receptor in the heart than other patients which would explain this finding.
In conclusion, LSD can increase force of contraction via stimulation of human H2 histamine receptors and human 5-HT4 receptors, in isolated atria from H2-TG and 5-HT4-TG, but also in the isolated human atrium.
Data availability
The data of this study are available from the corresponding author upon reasonable request.
References
Abella LMR, Hoffmann R, Neumann J, Hofmann B, Gergs U (2023) Levosimendan increases the phosphorylation state of phospholamban in the isolated human atrium. Naunyn Schmiedebergs Arch Pharmacol 396(4):669–682. https://doi.org/10.1007/s00210-022-02348-7
Angus JA, Black JW (1980) Pharmacological assay of cardiac H2-receptor blockade by amitriptyline and lysergic acid diethylamide. Circ Res 46(6 Pt 2):I64–I69
Baumann G, Felix SB, Schrader J, Heidecke CD, Riess G, Erhardt WD, Ludwig L, Loher U, Sebening F, Blömer H. (1981) Cardiac contractile and metabolic effects mediated via the myocardial H2-receptor adenylate cyclase system. Characterization of two new specific H2-receptor agonists, impromidine and dimaprit, in the guinea pig and human myocardium. Res Exp Med (Berl). 179,(1):81–98. https://doi.org/10.1007/BF01852128
Baumann G, Felix SB, Riess G, Loher U, Ludwig L, Blömer H (1982) Effective stimulation of cardiac contractility and myocardial metabolism by impromidine and dimaprit–two new H2-agonistic compounds–in the surviving, catecholamine-insensitive myocardium after coronary occlusion. J Cardiovasc Pharmaco 4(4):542–553
Baumann G, Mercader D, Busch U, Felix SB, Loher U, Ludwig L, Sebening H, Heidecke CD, Hagl S, Sebening F, Blömer H (1983) Effects of the H2-receptor agonist impromidine in human myocardium from patients with heart failure due to mitral and aortic valve disease. J Cardiovasc Pharmacol 5(4):618–625
Baumann G, Permanetter B, Wirtzfeld A (1984) Possible value of H2-receptor agonists for treatment of catecholamine-insensitive congestive heart failure. Pharmacol Ther 24(2):165–177
Baumeister P, Erdmann D, Biselli S, Kagermeier N, Elz S, Bernhardt G, Buschauer A (2015) [(3) H]UR-DE257: development of a tritium-labeled squaramide-type selective histamine H2 receptor antagonist. ChemMedChem 10(1):83–93. https://doi.org/10.1002/cmdc.201402344
Brattelid T, Kvingedal AM, Krobert KA, Andressen KW, Bach T, Hystad ME, Kaumann AJ, Levy FO (2004) Cloning, pharmacological characterisation and tissue distribution of a novel 5-HT4 receptor splice variant, 5-HT4(i). Naunyn Schmiedebergs Arch Pharmacol 369(6):616–628. https://doi.org/10.1007/s00210-004-0919-4
Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22(23):3099–3108. https://doi.org/10.1016/0006-2952(73)90196-2
Dai S (1976) A study of the actions of histamine on the isolated rat heart. Clin Exp Pharmacol Physiol. 3(4):359–367. https://doi.org/10.1111/j.1440-1681.1976.tb00612.x
De Gregorio D, Comai S, Posa L, Gobbi G (2016) d-Lysergic Acid Diethylamide (LSD) as a Model of Psychosis: Mechanism of Action and Pharmacology. Int J Mol Sci 17(11):1953. https://doi.org/10.3390/ijms17111953
Dolder PC, Liechti ME, Rentsch KM (2018) Development and validation of an LC-MS/MS method to quantify lysergic acid diethylamide (LSD), iso-LSD, 2-oxo-3-hydroxy-LSD, and nor-LSD and identify novel metabolites in plasma samples in a controlled clinical trial. J Clin Lab Anal 32(2):e22265. https://doi.org/10.1002/jcla.22265
Flacke W, Atanacković D, Gillis RA, Alper MH (1967) The actions of histamine on the mammalian heart. J Pharmacol Exp Ther 155(2):271–278
Gasser P, Kirchner K, Passie T (2015) LSD-assisted psychotherapy for anxiety associated with a life-threatening disease: a qualitative study of acute and sustained subjective effects. J Psychopharmacol 29(1):57–68. https://doi.org/10.1177/0269881114555249
Genovese A, Gross SS, Sakuma I, Levi R (1988) Adenosine promotes histamine H1-mediated negative chronotropic and inotropic effects on human atrial myocardium. J Pharmacol Exp Ther 247(3):844–849
Gergs U, Baumann M, Böckler A, Buchwalow IB, Ebelt H, Fabritz L, Hauptmann S, Keller N, Kirchhof P, Klöckner U, Pönicke K, Rueckschloss U, Schmitz W, Werner F, Neumann J (2010) Cardiac overexpression of the human 5-HT4 receptor in mice. Am J Physiol Heart Circ Physiol 299(3):H788–H798
Gergs U, Bernhardt G, Buchwalow IB, Edler H, Fröba J, Keller M, Kirchhefer U, Köhler F, Mißlinger N, Wache H, Neumann J (2019) Initial characterization of transgenic mice overexpressing human histamine H2 receptors. J Pharmacol Exp Ther 369:129–141
Gergs U, Böckler A, Ebelt H, Hauptmann S, Keller N, Otto V, Pönicke K, Schmitz W, Neumann J (2013) Human 5-HT4-receptor stimulation in atria of transgenic mice. Naunyn Schmiedebergs Arch Pharmacol 386(5):357–367
Gergs U, Büxel ML, Bresinsky M, Kirchhefer U, Fehse C, Höring C, Hofmann B, Marusakova M, Čináková A, Schwarz R, Pockes S, Neumann J (2021) Cardiac effects of novel H2-histamine receptor agonists. J Pharmacol Exp Ther 379(3):223–234. https://doi.org/10.1124/jpet.121.000822
Gergs U, Kirchhefer U, Bergmann F, Künstler B, Mißlinger N, Au B, Mahnkopf M, Wache H, Neumann J (2020) Characterization of Stressed Transgenic Mice Overexpressing H2-Histamine Receptors in the Heart. J Pharmacol Exp Ther 374(3):479–488. https://doi.org/10.1124/jpet.120.000063
Gergs U, Neumann J, Simm A, Silber RE, Remmers FO, Läer S (2009) Phosphorylation of phospholamban and troponin I through 5-HT4-receptors in the isolated human atrium. Naunyn Schmiedebergs Arch Pharmacol 379(4):349–359
Green JP, Weinstein H, Maayani S (1978) Defining the histamine H2-receptor in brain: the interaction with LSD. NIDA Res Monogr 22:38–59
Hattori Y, Sakuma I, Kanno M (1988) Differential effects of histamine mediated by histamine H1- and H2-receptors on contractility, spontaneous rate and cyclic nucleotides in the rabbit heart. Eur J Pharmacol 153(2–3):221–229
Hattori Y, Nakaya H, Endou M, Kanno M (1990) Inotropic, electrophysiological and biochemical responses to histamine in rabbit papillary muscles: evidence for coexistence of H1- and H2-receptors. J Pharmacol Exp Ther 253(1):250–256
Hattori Y, Gando S, Endou M, Kanno M (1991) Characterization of histamine receptors modulating inotropic and biochemical activities in rabbit left atria. Eur J Pharmacol 196(1):29–36
Holze F, Duthaler U, Vizeli P, Müller F, Borgwardt S, Liechti ME (2019) Pharmacokinetics and subjective effects of a novel oral LSD formulation in healthy subjects. Br J Clin Pharmacol 85(7):1474–1483. https://doi.org/10.1111/bcp.13918
Holze F, Vizeli P, Müller F, Ley L, Duerig R, Varghese N, Eckert A, Borgwardt S, Liechti ME (2020) Distinct acute effects of LSD, MDMA, and D-amphetamine in healthy subjects. Neuropsychopharmacology. 45(3):462–471. https://doi.org/10.1038/s41386-019-0569-3
Kaumann AJ, Sanders L, Brown AM, Murray KJ, Brown MJ (1990) A 5-hydroxytryptamine receptor in human atrium. Br J Pharmacol 100(4):879–885. https://doi.org/10.1111/j.1476-5381.1990.tb14108.x.PMID:2169944;PMCID:PMC1917575)
Kim J, Washio T, Yamagishi M, Yasumura Y, Nakatani S, Hashimura K, Hanatani A, Komamura K, Miyatake K, Kitamura S, Tomoike H, Kitakaze M (2004) A novel data mining approach to the identification of effective drugs or combinations for targeted endpoints-application to chronic heart failure as a new form of evidence-based medicine. Cardiovasc Drugs Ther 18(6):483–489
Kirchhefer U, Baba HA, Kobayashi YM, Jones LR, Schmitz W, Neumann J (2002) Altered function in atrium of transgenic mice overexpressing triadin 1. Am J Physiol Heart Circ Physiol 283(4):H1334–H1343
Kirchhefer U, Brekle C, Eskandar J, Isensee G, Kučerová D, Müller FU, Pinet F, Schulte JS, Seidl MD, Boknik P (2014) Cardiac function is regulated by B56α-mediated targeting of protein phosphatase 2A (PP2A) to contractile relevant substrates. J Biol Chem 289(49):33862–33873
Laher I, McNeill JH (1980a) Effects of histamine on rat isolated atria. Can J Physiol Pharmacol 9:1114–1116
Laher I, McNeill JH (1980b) Effects of histamine in the isolated kitten heart. Can J Physiol Pharmacol 58(11):1256–1261
Leonard JB, Anderson B, Klein-Schwartz W (2018) Does getting high hurt? Characterization of cases of LSD and psilocybin-containing mushroom exposures to national poison centers between 2000 and 2016. J Psychopharmacol 32(12):1286–1294. https://doi.org/10.1177/0269881118793086
Luethi D, Hoener MC, Krähenbühl S, Liechti ME, Duthaler U (2019) Cytochrome P450 enzymes contribute to the metabolism of LSD to nor-LSD and 2-oxo-3-hydroxy-LSD: Implications for clinical LSD use. Biochem Pharmacol 164:129–138. https://doi.org/10.1016/j.bcp.2019.04.013
Matthew H (1968) Lysergic acid diethylamide intoxication. Br Med J 1(5588):380. https://doi.org/10.1136/bmj.1.5588.380
Martin R, Schürenkamp J, Gasse A, Pfeiffer H, Köhler H (2013) Determination of psilocin, bufotenine, LSD and its metabolites in serum, plasma and urine by SPE-LC-MS/MS. Int J Legal Med 127(3):593–601. https://doi.org/10.1007/s00414-012-0796-1. (Epub 2012 Nov 27 PMID: 23183899)
Matsuda N, Jesmin S, Takahashi Y, Hatta E, Kobayashi M, Matsuyama K, Kawakami N, Sakuma I, Gando S, Fukui H, Hattori Y, Levi R (2004) Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J Pharmacol Exp Ther 309(2):786–795
McCarron MM, Walberg CB, Baselt RC (1990) Confirmation of LSD intoxication by analysis of serum and urine. J Anal Toxicol. 14(3):165–7. https://doi.org/10.1093/jat/14.3.165
National Research Council (2011) Guide for the Care and Use of Laboratory Animals, 8th edn. The National Academies Press, Washington, DC
Neumann J, Boknik P, DePaoli-Roach A, Field LJ, Rockman HA, Kobayashi Y, Kelley JS, Jones LR (1998) Targeted overexpression of phospholamban to mouse atrium depresses Ca2+ transport and contractility. J Mol Cell Cardiol 30:1991–2002
Neumann J, Hofmann B, Gergs U (2017) Production and function of serotonin in cardiac cells. "Serotonin - A Chemical Messenger Between All Types of Living Cells", Chapter 13; 271–305 ISBN 978–953–51–3361–2 Kaneez Fatima Shad (ed.)
Neumann J, Kirchhefer U, Dhein S, Hofmann B, Gergs U (2021a) Role of cardiovascular H2-histamine-receptors under normal and pathophysiological conditions. Front Pharmacol. https://doi.org/10.3389/fphar.2021.732842
Neumann J, Grobe JM, Weisgut J, Schwelberger HG, Fogel WA, Wache H, Bähre H, Buchwalow IB, Dhein S, Hofmann B, Kirchhefer U, Gergs U (2021) Histamine can be formed and degraded in the human and mouse heart. Front Pharmacol 12:582916. https://doi.org/10.3389/fphar.2021.582916
Neumann J, Binter MBB, Fehse C, Marusakova M, Kirchhefer U, Wache H, Hofmann B, Gergs U (2021) Amitriptyline functionally antagonizes cardiac H2 histamine receptors in transgenic mice and human atria. Naunyn Schmiedeberg’s Arch Pharmacol 394(6):1251–1262. https://doi.org/10.1007/s00210-021-02065-7
Neumann J, Hofmann B, Dhein S (2023) Gergs U (2023) Cardiac roles of serotonin (5-HT) and 5-HT-receptors in health and disease. Int J Mol Sci 24:4765. https://doi.org/10.3390/ijms24054765
Olson DE (2022) Biochemical Mechanisms Underlying Psychedelic-Induced Neuroplasticity. Biochemistry 61(3):127–136. https://doi.org/10.1021/acs.biochem.1c00812
Panula P, Chazot PL, Cowart M, Gutzmer R, Leurs R, Liu WL, Stark H, Thurmond RL, Haas HL (2015) International Union of Basic and Clinical Pharmacology. XCVIII Histamine Receptors Pharmacol Rev 67(3):601–655
Passie T, Halpern JH, Stichtenoth DO, Emrich HM, Hintzen A (2008) The pharmacology of lysergic acid diethylamide: a review. CNS Neurosci Ther 14(4):295–314. https://doi.org/10.1111/j.1755-5949.2008.00059.x
Pockes S, Wifling D, Keller M, Buschauer A, Elz S (2018) Highly Potent, Stable, and Selective Dimeric Hetarylpropylguanidine-Type Histamine H2 Receptor Agonists. ACS Omega 3(3):2865–2882. https://doi.org/10.1021/acsomega.8b00128
Rosier N, Grätz L, Schihada H, Möller J, Işbilir A, Humphrys LJ, Nagl M, Seibel U, Lohse MJ, Pockes S (2021) A Versatile Sub-Nanomolar Fluorescent Ligand Enables NanoBRET Binding Studies and Single-Molecule Microscopy at the Histamine H3 Receptor. J Med Chem 64(15):11695–11708. https://doi.org/10.1021/acs.jmedchem.1c01089
Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB (2002) Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A 99(18):11934–11939. https://doi.org/10.1073/pnas.182234399
Sanders L, Lynham JA, Kaumann AJ (1996) Chronic beta 1-adrenoceptor blockade sensitises the H1 and H2 receptor systems in human atrium: rôle of cyclic nucleotides. Naunyn Schmiedebergs Arch Pharmacol 353(6):661–670
Schlag AK, Aday J, Salam I, Neill JC, Nutt DJ (2022) Adverse effects of psychedelics: From anecdotes and misinformation to systematic science. J Psychopharmacol 36(3):258–272. https://doi.org/10.1177/02698811211069100
Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR (1986) Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J BiolChem 261(28):13333–41
Subramaniam A, Gulick J, Neumann J, Knotts S, Robbins J (1993) Transgenic analysis of the thyroid-responsive elements in the alpha-cardiac myosin heavy chain gene promoter. J Biol Chem 268(6):4331–4336
Thoren FB, Aurelius J, Martner A (2011) Antitumor properties of histamine in vivo. Nat Med 17(5):537
Vigorito C, Russo P, Picotti GB, Chiariello M, Poto S, Marone G (1983) Cardiovascular effects of histamine infusion in man. J Cardiovasc Pharmacol 5(4):531–537
VdLeyen H, Mende U, Meyer W, Neumann J, Nose M, Schmitz W, Scholz H, Starbatty J, Stein B, Wenzlaff H, Döring V, Kalmár P, Haverich A (1991) Mechanism underlying the reduced positive inotropic effects of the phosphodiesterase III inhibitors pimobendan, adibendan and saterinone in failing as compared to nonfailing human cardiac preparations. Naunyn-Schmiedebergs Arch Pharmacol 344(1):90–100
Zerkowski HR, Broede A, Kunde K, Hillemann S, Schäfer E, Vogelsang M, Michel MC, Brodde OE (1993) Comparison of the positive inotropic effects of serotonin, histamine, angiotensin II, endothelin and isoprenaline in the isolated human right atrium. Naunyn Schmiedebergs Arch Pharmacol 347(4):347–352
Acknowledgements
The authors thank Pia Willmy for expert technical assistance. This work contains parts of theses of JH and PB.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Authors´s contributions: JN and UK devised the study, JN wrote the first draft, draft was improved written by LH, SP, UK, UG, BH. Supplied material (UK) and clinical data (BH), performed experiments: HJ, JN, LH, PB. Analyzed data: UK, PB, HJ, SP. Graphed data: UG, SP, HJ. All authors have read and approved the submission of this version. All authors have read and agree with the submission of the present version of the work. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethical approval
Animals: The investigation conformed to the Guide for the Care and Use of Laboratory Animals as published by the National Research Council (2011). The animals were handled and maintained according to the approved protocols of the Animal Welfare Committee of the University of Halle-Wittenberg, Halle, Germany. Humans: This study complies with the Declaration of Helsinki and has been approved by the local ethics committee (hm-bü 04.08.2005).
Consent to participate
Informed consent was obtained from all patients included in the study.
Consent to publish
All authors declare that they have seen and approved the submitted version of this manuscript.
Competing interests
The authors declare no competing interests of financial or personal nature.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gergs, U., Jacob, H., Braekow, P. et al. Lysergic acid diethylamide stimulates cardiac human H2 histamine and cardiac human 5-HT4-serotonin receptors. Naunyn-Schmiedeberg's Arch Pharmacol 397, 221–236 (2024). https://doi.org/10.1007/s00210-023-02591-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02591-6