
Abstract
Summary
This 78-week (18-month) study conducted in 479 postmenopausal women with osteoporosis evaluated the efficacy, pharmacodynamics, pharmacokinetics, safety, and immunogenicity of candidate biosimilar CT-P41 relative to US reference denosumab. CT-P41 had equivalent efficacy and pharmacodynamics to US-denosumab, with similar pharmacokinetics and comparable safety and immunogenicity profiles.
Purpose
To demonstrate equivalence of candidate biosimilar CT-P41 and US reference denosumab (US-denosumab) in postmenopausal women with osteoporosis.
Methods
This 78-week (18-month), double-blind, randomized, active-controlled Phase 3 study (NCT04757376) comprised two treatment periods (TPs). In TPI, patients (N = 479) were randomized 1:1 to 60 mg subcutaneous CT-P41 or US-denosumab. At Week 52, those who had received CT-P41 in TPI continued to do so. Those who had received US-denosumab were randomized (1:1) to continue treatment or switch to CT-P41 in TPII. The primary efficacy endpoint was percent change from baseline in lumbar spine bone mineral density at Week 52. Efficacy equivalence was concluded if associated 95% confidence intervals (CI) for least squares (LS) mean group differences fell within ± 1.503%. The primary pharmacodynamic (PD) endpoint was area under the effect curve for serum carboxy-terminal cross-linking telopeptide of type I collagen through the first 26 weeks, with an equivalence margin of 80–125% (for 95% CIs associated with geometric LS mean ratios).
Results
Equivalence was demonstrated for CT-P41 and US-denosumab with respect to primary efficacy (LS mean difference [95% CI]: − 0.139 [− 0.826, 0.548] in the full analysis set and − 0.280 [− 0.973, 0.414] in the per-protocol set) and PD (geometric LS mean ratio [95% CI]: 94.94 [90.75, 99.32]) endpoints. Secondary efficacy, PD, pharmacokinetics, and safety results were comparable among all groups up to Week 78, including after transitioning to CT-P41 from US-denosumab.
Conclusions
CT-P41 was equivalent to US-denosumab in women with postmenopausal osteoporosis, with respect to primary efficacy and PD endpoints.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis affects around 20% of the world’s population and is a primary cause of bone fractures in older adults. Under-treatment of osteoporosis, or treatment cessation, increases fracture risk [1,2,3]. In addition to significant effects on patient health, independence, and quality of life, osteoporosis places substantial economic burden on healthcare systems, particularly with regards to fracture treatment. As the global population ages, societal impacts are set to increase [1,2,3,4,5].
Denosumab is a human monoclonal antibody selectively targeting receptor activator of nuclear factor kappa-B ligand (RANKL) [6,7,8]. Binding of denosumab to RANKL prevents RANKL from interacting with its receptor (RANK) on the surface of osteoclasts and osteoclast precursors, thus inhibiting osteoclast formation, function, and survival, and reducing bone resorption [6]. By inhibiting bone resorption, denosumab increases bone mineral density (BMD) and reduces fracture risk [6,7,8,9,10,11]. In the USA and European Union, denosumab is approved for osteoporosis treatment in patients with high risk of fractures [7, 8].
Biologic therapies, such as denosumab, have become increasingly important across a range of therapy areas, but treatment access can be hampered by costs [12]. The availability of biosimilars, developed according to stringent regulations and which must have highly similar properties to the approved reference product, can drive down costs, reducing burden on healthcare systems and enabling more equitable treatment access [13, 14]. The first denosumab biosimilars have recently been approved in the USA [15] and Europe [16]. Several are in clinical development [17,18,19]. CT-P41 is a candidate denosumab biosimilar that, in a Phase 1 study involving male participants (NCT06037395), demonstrated pharmacokinetic (PK) equivalence to US-licensed reference denosumab (US-denosumab) [20].
We report findings of a double-blind, randomized, active-controlled Phase 3 study comparing CT-P41 to US-denosumab in women with postmenopausal osteoporosis. The primary objectives were to demonstrate the equivalence of CT-P41 to US-denosumab, with respect to the primary efficacy endpoint (percent change from baseline in lumbar spine BMD at Week 52) and pharmacodynamics (PD; through the first 26 weeks). Secondary efficacy, PD, PK, and safety endpoints, including immunogenicity, were also evaluated.
Methods
Study design
This double-blind, randomized, active-controlled Phase 3 study (NCT04757376) was conducted at 20 sites in four countries (Supplementary Table 1). The study comprised four periods (Supplementary Fig. 1): an initial 28-day screening period (Day − 28 to Day − 1), followed by Treatment Period I (TPI; Week 0 [Day 1] to Week 52 predose), Treatment Period II (TPII; Week 52 to Week 78), and an end-of-study (EOS) visit at Week 78 (Month 18). On Day 1, eligible patients were randomly assigned (1:1 ratio) to receive CT-P41 (Celltrion Inc., Incheon, Republic of Korea) or US-denosumab (Amgen Inc., Thousand Oaks, CA, USA). Before initiating TPII, all patients underwent a second randomization process to maintain blinding. Patients who received CT-P41 in TPI continued to receive CT‑P41 (“CT-P41 maintenance”); patients who received US-denosumab in TPI were randomized (1:1) to continue US‑denosumab (“US-denosumab maintenance”) or switch to CT-P41 (“switched to CT-P41”).
Study drug was administered in 60 mg doses via a prefilled syringe at Weeks 0 (Day 1), 26, and 52, as a single subcutaneous injection into the upper arm, upper thigh, or abdomen. Administration was carried out by predefined, unblinded clinical staff, since the visual appearance of the two study drugs was not identical. Patients were blinded to treatment assignment using a blindfold, screen, or similar to ensure the prefilled syringe was not visible. Staff conducting study assessments were absent throughout the dosing procedure and remained blinded throughout the study.
Randomization schedules were generated by unblinded biostatisticians using an interactive web response system. Randomizations were balanced using permuted blocks (block size: 2, 4 [mixed]). The first randomization was stratified by age (< 65 years versus ≥ 65 years), baseline BMD T-score at the lumbar spine (≤ − 3.0 versus > − 3.0), and prior bisphosphonate therapy (yes/no). The second randomization was stratified by change from baseline lumbar spine BMD at Week 52 (≥ 3% versus < 3%; centrally read at baseline and locally read at Week 52). Stratification variables were based on international guidelines on the recommended age for osteoporosis screening in women, stringent criteria for osteoporosis definition, and assessment of bone metabolism [21, 22]. Due to the COVID-19 pandemic and conflict in Ukraine, global and country-specific protocol amendments were necessary after the first patient was randomized (Supplementary Methods).
This study was conducted according to Good Clinical Practice, the principles of the Declaration of Helsinki, and national, state, and local laws or regulations. Study materials were approved by independent ethics committees/institutional review boards at each site (Supplementary Table 1). All participants provided written informed consent prior to study initiation.
Participants
Study participants were postmenopausal women aged 50 to 80 years, inclusive. Key inclusion criteria comprised the following: ≥ 3 evaluable vertebrae in the lumbar spine (L1 to L4); lumbar spine (L1 to L4) BMD T‑score ≤ − 2.5 and ≥ − 4.0 at screening, as assessed by dual-energy X-ray absorptiometry (DXA) scan; ≥ 1 evaluable hip by DXA at screening; good general health, determined at the investigator’s discretion by medical history, physical examination, and laboratory tests; and ability to walk without assistance. Exclusion criteria included prior treatment with reference/biosimilar denosumab or other biologics for osteoporosis; receipt of intravenous bisphosphonates, fluoride, or strontium for osteoporosis within the last 5 years prior to first administration of study drug; receipt of oral bisphosphonates ≥ 3 years cumulatively prior to screening or receipt of any dose of oral bisphosphonates within 12 months before screening; and use of oral or parenteral glucocorticoids (> 5 mg/day prednisone or equivalent for > 10 days) within 3 months before first administration of study drug. Other key exclusion criteria included current or historical vertebral fractures (1 severe or > 2 moderate), hip fracture, hyperparathyroidism/hypoparathyroidism, hyperthyroidism/hypothyroidism (unless currently well controlled on stable therapy), bone/metabolic disease (except osteoporosis) that might interfere with interpretation of results, certain oral/dental conditions (including osteomyelitis and osteonecrosis of the jaw [ONJ]), dental conditions requiring oral surgery, and planned invasive dental procedures. Full eligibility criteria are detailed in the Supplementary Methods.
Study endpoints and assessments
Full details of study assessments and time points for evaluation are shown in Supplementary Table 2. In brief, for efficacy, DXA scans and lateral spine X-rays were conducted at screening, Week 26, Week 52, and Week 78 (EOS). Quality-of-life questionnaires were administered at Day 1, Week 26, Week 52, and Week 78. The primary efficacy endpoint was the percent change from baseline in lumbar spine (L1 to L4) BMD by DXA at Week 52. Secondary efficacy endpoints were as follows: percent change from baseline in BMD for lumbar spine, total hip, and femoral neck, as assessed by DXA, at Weeks 26, 52, and 78; incidences of new vertebral, nonvertebral, and hip fractures; and change from baseline in health-related quality of life (HRQoL) at Weeks 26, 52, and 78. Efficacy analysis of new fractures included only vertebral fractures occurring from T4 to L4 and confirmed by the central imaging vendor. A new vertebral fracture was defined as an increase of ≥ 1 grade in any vertebra from T4 to L4 that was normal at screening [23]. The nonvertebral fractures endpoint included fractures other than those of the vertebrae, excluding the skull, facial bones, mandible, metacarpals, and phalanges (fingers or toes) since they are not associated with decreased BMD, and excluded pathologic fractures and those associated with severe trauma acquired from a fall (from a height higher than a stool, chair, or first rung of a ladder) or otherwise [23, 24]. Only nonvertebral fractures confirmed by the central imaging vendor were included in the efficacy analysis. HRQoL was measured using the Osteoporosis Assessment Questionnaire-short version (OPAQ-SV), EQ-5D-5L and EQ visual analog scale (EQ VAS).
Samples were taken for PD analysis at multiple time points through Week 78. Serum carboxy-terminal cross-linking telopeptide of type I collagen (s-CTX) and procollagen type I N-terminal propeptide (PINP) concentrations were measured by Elecsys β-CrossLaps/serum assay and Elecsys total PINP assay, respectively, on a Cobas e602 module (Roche Diagnostics GmbH, Mannheim, Germany). The primary PD endpoint was the area under the effect curve (AUEC) of s-CTX up to Week 26 predose. Secondary PD endpoints included AUEC of PINP up to Week 26 predose, and percent change from baseline in s-CTX and PINP at Weeks 26, 52, and 78.
Blood samples were taken for PK analysis at multiple time points through Week 78. Serum denosumab concentration and serum denosumab trough concentration (Ctrough) were evaluated through Week 78. Other secondary PK endpoints were assessed up to Week 26, including maximum serum denosumab concentration (Cmax) after first study drug administration, truncated area under the concentration–time curve from zero to Week 26 (AUC0–τ), time of observed maximum serum denosumab concentration after first study drug administration (Tmax), volume of distribution (Vd), and terminal elimination half-life (T1/2). PK and PD parameters were estimated using non-compartmental methods in Phoenix WinNonlin version 8.3 (Pharsight, St. Louis, MO, USA).
Safety, assessed throughout the study, included monitoring of adverse events, vital signs, and laboratory parameters. Treatment-emergent adverse events (TEAEs) of special interest were injection-site reaction, drug-related hypersensitivity/allergic reaction, infection, hypocalcemia (defined as albumin-adjusted total serum calcium < 8.5 mg/dL [< 2.125 mmol/L]), ONJ, atypical femur fracture, and dermatologic reaction. Radiography, performed on suspected fractures as required, was analyzed at a central imaging vendor. Pain was assessed by 100 mm VAS immediately after study drug administration at Weeks 0, 26, and 52. All patients received a daily supplement containing calcium (at least 1,000 mg) and vitamin D (at least 400 IU) from randomization to the EOS visit.
Blood samples for immunogenicity analysis were taken at multiple time points. Samples were analyzed for antidrug antibodies (ADAs) using a Meso Scale Discovery – Electrochemiluminescence (MSD–ECL) assay, with a confirmatory assay sensitivity of 4.69 ng/mL in human serum. Confirmed ADA-positive samples were further analyzed for neutralizing antibodies (NAbs) using MSD–ECL with a sensitivity of 79.5 ng/mL in human serum.
Statistical analyses
A sample size of 352 patients (176 per group) was determined to provide 90% statistical power to demonstrate equivalence between CT-P41 and US-denosumab in terms of percent change from baseline in lumbar spine BMD at Week 52, based on a two one-sided 2.5% significance level and equivalence margin of − 1.503 to 1.503%. A common standard deviation (SD) of 3.89% and expected mean difference in percent change from baseline of zero were assumed. Allowing for a hypothesized 20% dropout rate, approximately 440 patients (220 per group) were to be randomized.
For evaluating PD equivalence between CT-P41 and US-denosumab, in terms of the ratio of geometric means of AUEC for s-CTX up to Week 26 predose, a sample size of 396 patients (198 per group) was determined to provide ≥ 90% statistical power, based on a two one-sided 2.5% significance level and equivalence margin of 80 to 125%. The coefficient of variance was assumed to be 50% with an expected ratio of one. Accounting for a 10% dropout rate, 440 patients (220 per group) were to be randomized.
Statistical analysis of the primary efficacy endpoint used analysis of covariance (ANCOVA), with treatment as a fixed effect and age, baseline lumbar spine BMD T-score, and prior bisphosphonate therapy (yes/no) as covariates. Equivalence was concluded if the 95% confidence interval (CI) for the between-group difference fell entirely within an equivalence margin of − 1.503 to 1.503%.
The impact of missing data on primary efficacy results was evaluated under both missing-at-random and missing-not-at-random scenarios. In the missing-at-random scenario, percent changes in BMD from baseline to Week 52 were imputed using a mean imputation method (the imputed value was the mean of the non-missing values). A tipping point analysis was performed to evaluate the impact of missing-not-at-random scenarios. Imputed values for change from baseline in lumbar spine BMD at Week 52 were gradually shifted in each group until the 95% CI for the between-group difference was no longer within the equivalence margin of − 1.503 to 1.503%.
Equivalence for the primary PD endpoint was assessed using ANCOVA, with treatment as a fixed effect and age, baseline lumbar spine BMD T‑score, prior bisphosphonate therapy (yes/no), and baseline s-CTX level as covariates. Equivalence was concluded if the 95% CI for the between-group geometric least squares (LS) mean ratio fell entirely within the 80 to 125% equivalence margin. The secondary endpoint AUEC of PINP was also analyzed by ANCOVA, but with baseline PINP (not s-CTX) level as a covariate.
Analysis sets are described in the Supplementary Methods. All statistical analyses were performed using Statistical Analysis System (SAS®) software version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient disposition, characteristics and demographics
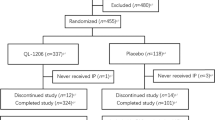
The first patient was randomized on June 17, 2021. The last visit took place on November 16, 2023. A total of 1,238 patients were screened, with 479 randomized in TPI (240 to the CT-P41 group and 239 to the US-denosumab group; Fig. 1). Two patients not meeting eligibility criteria were randomized in error and terminated the study before study drug administration. Thus, 239 and 238 patients initiated treatment in the CT-P41 and US-denosumab groups, respectively. Eighteen (7.5%) and 37 (15.5%) patients in the CT-P41 and US-denosumab groups, respectively, discontinued study drug in TPI, most frequently owing to patient withdrawal. At Week 52, 422 patients continued into TPII, with 221 patients who received CT-P41 in TPI assigned to CT-P41 maintenance, and 201 patients who received US-denosumab in TPI re-randomized 1:1 to either US-denosumab maintenance (n = 100) or switched to CT-P41 (n = 101). One patient (0.5%) in the CT-P41 maintenance group discontinued before dosing at Week 52, due to an ongoing adverse event. A total of 220 (99.5%), 100 (100.0%), and 101 (100.0%) patients completed study treatment in TPII and 215 (97.3%), 97 (97.0%), and 98 (97.0%) patients completed study participation in TPII in the CT-P41 maintenance, US-denosumab maintenance, and switched to CT-P41 groups, respectively.

Patient disposition (ITT set and ITT set – TPII subset). aOne patient in each of the CT-P41 and US-denosumab groups who did not meet eligibility criteria were mistakenly randomized by study center staff. The patients terminated their study participation before first study drug administration. bOne patient in the CT-P41 maintenance group discontinued treatment at Week 52 owing to an ongoing adverse event; however, the patient continued to participate in the study (without study treatment) and completed TPII. ITT, intent-to-treat; TPII, Treatment Period II; US-denosumab, US-licensed reference denosumab
Baseline demographics, disease characteristics, and stratification factors were well balanced between groups at first randomization. Groups were also well balanced at re-randomization (for TPII) with respect to the stratification factor of change from baseline in lumbar spine BMD at Week 52 (Table 1).
Efficacy and HRQoL
The LS mean difference between treatment groups for the percent change from baseline in lumbar spine BMD at Week 52 was − 0.139 (95% CI − 0.826, 0.548) in the full analysis set (FAS) and − 0.280 (95% CI − 0.973, 0.414) in the per-protocol set (PPS) (Table 2). The 95% CIs for these differences were fully contained within the predefined equivalence margin, demonstrating equivalence of CT-P41 to US-denosumab. The number of patients with missing data for change in lumbar spine BMD at Week 52 was 17 (7.1%) and 26 (10.9%) in the CT-P41 and US-denosumab groups, respectively. Tipping point analysis suggested that findings were not impacted by missing data.
For secondary efficacy endpoints, mean ± SD percent changes from baseline in BMD for lumbar spine, total hip, and femoral neck increased during TPI, and were comparable between groups at Weeks 26 and 52 (Fig. 2). In TPII, results were also comparable across groups for lumbar spine, total hip, and femoral neck BMD (Fig. 2).

Mean ± SD percent change from baseline in BMD for lumbar spine (a), total hip (b), and femoral neck (c) from baseline through Week 78 (FAS and FAS – TPII subset). BMD, bone mineral density; FAS, full analysis set; SD, standard deviation; TP, treatment period; US-denosumab, US-licensed reference denosumab
In TPI, 1 (0.4%) patient each in the CT-P41 and US-denosumab groups had new vertebral fractures (L2 and T8, respectively). Nonvertebral fractures were reported in 2 (0.8%) patients in the CT-P41 group (carpus and distal radius in 1 patient and distal radius in 1 patient) and in 4 (1.7%) patients in the US-denosumab group (1 distal fibula, 1 proximal humerus, and 2 distal radius fractures). There were no hip fractures. In the CT-P41 maintenance group in TPII, 1 (0.5%) patient had a new vertebral fracture (L1) and 2 (0.9%) patients had nonvertebral fractures (distal radius in 1 patient, and distal ulna and radius in 1 patient). One (1.0%) patient in the switched to CT-P41 group had a nonvertebral fracture (rib).
Mean ± SD changes from baseline OPAQ-SV scores, EQ-5D-5L index values, and EQ VAS scores were small and generally similar between groups during TPI and across groups during TPII, including after switching from US-denosumab to CT-P41 (Supplementary Table 3).
Pharmacodynamics
Geometric LS mean ratio between treatment groups for the AUEC of s-CTX, up to Week 26 predose, was 94.94 (95% CI 90.75, 99.32; FAS) (Table 3). The 95% CI was fully contained within the predefined equivalence margin, thus demonstrating PD equivalence of CT-P41 to US-denosumab. Geometric LS mean AUEC of PINP up to Week 26 predose (a secondary PD endpoint) was 7663.9 in the CT-P41 group and 9119.8 in the US-denosumab group (Table 3). Median percent changes from baseline in s-CTX and PINP were comparable between groups during TPI and across groups during TPII, including after switching from US-denosumab to CT‑P41 (Fig. 3). Following study drug administration on Day 1, there was a rapid and large decrease in s-CTX levels at Day 3. This was maintained through Week 4, with a subsequent increase until Week 26. After study drug administration at Week 26, the median percent change from baseline in s-CTX decreased up to Week 39, then increased until study drug administration at Week 52, after which the pattern was repeated. For PINP, levels decreased from Week 1 to Week 12, then remained largely stable.

Median (Q1, Q3) percent change from baseline in serum concentrations of s-CTX (a) and PINP (b) during TPI (PD set) and TPII (PD set – TPII subset). Serum concentrations below the LLoQ were set to the LLoQ, and values above the ULoQ were set to the ULoQ. EOS, end of study; LLoQ, lower limit of quantification; PD, pharmacodynamic; PINP, procollagen type I N-terminal propeptide; Q, quartile; s-CTX, serum carboxy-terminal cross-linking telopeptide of type I collagen; TP, treatment period; ULoQ, upper limit of quantification; US-denosumab, US-licensed reference denosumab
Pharmacokinetics
Mean ± SD serum denosumab concentrations and Ctrough were similar between groups during TPI and TPII, including after switching to CT-P41 (Supplementary Fig. 2). PK parameters, including mean ± SD Cmax, AUC0–τ, Tmax, Vd, and T1/2, were similar between groups over the first 26 weeks (Supplementary Table 4).
Safety
During TPI (Table 4), similar proportions of patients in each group experienced TEAEs (75.7% in the CT-P41 group and 70.2% in the US-denosumab group). Treatment-emergent serious adverse events (TESAEs) were experienced by 7 (2.9%) and 10 (4.2%) patients in the CT-P41 and US-denosumab groups, respectively. There was one death owing to a TEAE (coronary artery disease) in the CT‑P41 group, not considered study drug-related by the investigator, as the patient had concomitant coronary artery disease. In TPII (Table 4), generally similar proportions of patients across groups experienced TEAEs (50.9%, 42.0%, and 56.4% in the CT-P41 maintenance, US-denosumab maintenance, and switched to CT-P41 groups, respectively). TESAEs were experienced by 8 (3.6%) and 3 (3.0%) patients in the CT-P41 maintenance and US-denosumab maintenance groups, respectively, and by no patients in the switched to CT-P41 group. There was one death owing to a TEAE of “genital neoplasm malignant female” in the CT-P41 maintenance group; this was considered unrelated to the study drug by the investigator.
The majority of TEAEs throughout the study were Grade 1 or 2 in intensity (Table 4). In TPI, the most frequent TEAE reported in both groups was COVID-19 (28 [11.7%] patients in the CT-P41 group and 26 [10.9%] patients in the US-denosumab group). In TPII, the most frequently reported TEAE across groups was upper respiratory tract infection (13 [5.9%] patients in the CT-P41 maintenance group, 4 [4.0%] patients in the US-denosumab maintenance group, and 11 [10.9%] patients in the switched to CT-P41 group; Supplementary Table 5).
Immunogenicity
Proportions of patients with positive ADA results at each visit were similar among the groups in both TPI and TPII. In TPI, after first study drug administration, 233 (97.5%) and 234 (98.3%) patients in the CT‑P41 and US-denosumab groups, respectively, had ≥ 1 positive ADA result (Supplementary Table 6). In TPII, the proportion of patients with ≥ 1 positive ADA result after first study drug administration was 208 (94.5%), 92 (92.0%), and 93 (92.1%) in the CT-P41 maintenance, US-denosumab maintenance, and switched to CT-P41 groups, respectively (Supplementary Table 6). Most ADA titer values were low, with no positive NAb results in either treatment period (Supplementary Table 7).
Discussion
This double-blind, randomized, active-controlled Phase 3 study confirmed that CT-P41 has equivalent efficacy and PD, as assessed by changes in BMD and markers of bone turnover, to US-denosumab in women with postmenopausal osteoporosis. The LS mean treatment difference for the primary efficacy endpoint, percent change from baseline in lumbar spine BMD at Week 52, was within the predefined 95% CI equivalence margin (LS mean difference [95% CI]: − 0.139 [− 0.826, 0.548] in the FAS and − 0.280 [− 0.973, 0.414] in the PPS. This finding was supported by sensitivity analysis. PD equivalence was demonstrated, per the primary PD endpoint of AUEC for s-CTX over the initial 26 weeks (geometric LS mean ratio [95% CI]: 94.94 [90.75, 99.32]). During TPI, a smaller proportion of patients in the CT-P41 than US-denosumab group discontinued treatment (7.5% and 15.5%, respectively).
Primary efficacy findings were reinforced by those for secondary efficacy endpoints. Percentage increases in lumbar spine BMD from baseline were comparable for CT-P41 and US-denosumab at further time points (Weeks 26 and 78) and in other skeletal regions (total hip and femoral neck), including when comparing patients who switched from US-denosumab to CT-P41 with those who maintained initial CT-P41 or US-denosumab treatment. The largest gains in BMD occurred in the lumbar spine, and the magnitude of changes observed at Week 52 (i.e. an approximately 5% increase in lumbar spine BMD) were similar to those at 1 year in the pivotal Phase 3 FREEDOM trial of reference denosumab [23]. Likewise, fracture rates were low in all groups throughout the current study, in line with low levels (≤ 2%) reported in the FREEDOM trial [23]. Small, comparable changes in HRQoL measures were observed in all groups and both treatment periods. This was true for measures of physical function, emotional status, and pain.
Comparable results were obtained between treatment groups throughout both treatment periods for PD markers of bone turnover and osteoclast activity. Early and large changes observed in s-CTX levels were in line with those previously reported for reference denosumab [25]. This effect was also observed in the switched to CT-P41 group. Similarly, the observed decline in PINP levels is expected with denosumab [26] and was consistently observed across the CT-P41 and US-denosumab groups. Levels of s-CTX and PINP following dosing of CT-P41 or US-denosumab in the current trial were similar to those in the FREEDOM trial, in which median s-CTX levels decreased by around 80%, and median serum PINP levels decreased by around 70%, from 6 months [23].
In general, PK parameters were similar across treatment groups during each treatment period, except for some observed differences for Ctrough in TPII (i.e. following switch to CT-P41, or maintenance of CT-P41 or US-denosumab). This was due, at least in part, to one patient in the “switch” group with unusually high Ctrough values.
CT-P41 was generally well tolerated, with a safety profile similar to US-denosumab. Safety findings were in line with the known safety profile of reference denosumab [7, 8]. The majority of TEAEs in all groups were Grade 1 or 2 in severity. In the present study, overall infection rates were impacted by COVID-19. Some between-group differences in infection rates occurred, but were likely due to chance and not considered clinically meaningful. Cases of hypocalcemia were observed in this study, in line with reports from denosumab clinical trials [7, 8], with comparable rates between the CT-P41 and US-denosumab arms.
Immunogenicity profiles were similar for CT-P41 and US-denosumab. As results of immunogenicity analyses can be impacted by methodologic differences, comparisons across studies should be made with caution. In the present study, proportions of ADA-positive patients were similar between groups but higher than those previously reported for reference denosumab [7, 8]. However, no positive NAb results were seen and most ADA titers were low in ADA-positive patients.
Strengths of this investigation include the multicenter, international, double-blind, randomized, active-controlled study design. A broad range of endpoints was assessed against well-established treatment outcomes, comprising efficacy, PD, PK, safety, and immunogenicity. One limitation is that, due to the sensitive ADA assay used, most patients were ADA positive and it was therefore not possible to assess the impact of ADAs on efficacy, PK, and safety. Other limitations included disruption caused by ongoing conflict in Ukraine, causing some patients to miss or delay study center visits. A small number of patients also required delayed dosing due to contracting COVID-19. Another limitation is the smaller number of patients in the US-denosumab maintenance and switch groups compared with the CT-P41 maintenance group in TPII.
Conclusion
In this Phase 3 study of postmenopausal women with osteoporosis, CT-P41 was equivalent to US-denosumab with respect to primary measures of efficacy (Week 52) and PD effects (Week 26). Comparable results were seen between groups for secondary efficacy and PD endpoints, and PK, safety, and immunogenicity. Similarity was maintained between groups up to Week 78, following CT-P41 maintenance, US-denosumab maintenance, or switch from US-denosumab to CT-P41. This study affirms CT-P41 as a promising candidate biosimilar of denosumab.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary information.
References
Salari N, Ghasemi H, Mohammadi L, Behzadi MH, Rabieenia E, Shohaimi S, Mohammadi M (2021) The global prevalence of osteoporosis in the world: a comprehensive systematic review and meta-analysis. J Orthop Surg Res 16:609
Borgström F, Karlsson L, Ortsäter G et al (2020) Fragility fractures in Europe: burden, management and opportunities. Arch Osteoporos 15:59
Tarantino U, Cariati I, Greggi C et al (2022) Gaps and alternative surgical and non-surgical approaches in the bone fragility management: an updated review. Osteoporos Int 33:2467–2478
Williams SA, Daigle SG, Weiss R, Wang Y, Arora T, Curtis JR (2021) Economic burden of osteoporosis-related fractures in the US Medicare population. Ann Pharmacother 55:821–829
Eastell R, O’Neill TW, Hofbauer LC, Langdahl B, Reid IR, Gold DT, Cummings SR (2016) Postmenopausal osteoporosis. Nat Rev Dis Primers 2:16069
Deeks ED (2018) Denosumab: a review in postmenopausal osteoporosis. Drugs Aging 35:163–173
Amgen Inc. (2024) Prolia®: prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/125320s216lbl.pdf. Accessed 26 Jun 2024
Amgen Europe BV (2020) Prolia®: summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/prolia-epar-product-information_en.pdf. Accessed 26 Jun 2024
Lai EC-C, Lin T-C, Lange JL, Chen L, Wong ICK, Sing CW, Cheung CL, Shao SC, Yang YK (2022) Effectiveness of denosumab for fracture prevention in real-world postmenopausal women with osteoporosis: a retrospective cohort study. Osteoporos Int 33:1155–1164
Chen Y, Zhu J, Zhou Y, Peng J, Wang B (2021) Efficacy and safety of denosumab in osteoporosis or low bone mineral density postmenopausal women. Front Pharmacol 12:588095
Bone HG, Wagman RB, Brandi ML et al (2017) 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol 5:513–523
Kvien TK, Patel K, Strand V (2022) The cost savings of biosimilars can help increase patient access and lift the financial burden of health care systems. Semin Arthritis Rheum 52:151939
Busse A, Lüftner D (2019) What does the pipeline promise about upcoming biosimilar antibodies in oncology? Breast Care (Basel) 14:10–16
Araújo FC, Gonçalves J, Fonseca JE (2019) Biosimilars in rheumatology. Pharmacol Res 149:104467
US Food and Drug Administration (2024) FDA approves first interchangeable biosimilars to Prolia and Xgeva to treat certain types of osteoporosis and prevent bone events in cancer. https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-interchangeable-biosimilars-prolia-and-xgeva-treat-certain-types-osteoporosis-and. Accessed 26 Jun 2024
European Pharmaceutical Review (2024) European Commission grants first-of-a-kind biosimilar approval By Catherine Eckford. News. https://www.europeanpharmaceuticalreview.com/news/229320/european-commission-grants-first-of-a-kind-biosimilar-approval/
Jamshidi A, Vojdanian M, Soroush M et al (2022) Efficacy and safety of the biosimilar denosumab candidate (Arylia) compared to the reference product (Prolia®) in postmenopausal osteoporosis: a phase III, randomized, two-armed, double-blind, parallel, active-controlled, and noninferiority clinical trial. Arthritis Res Ther 24:161
Zhang H, Gu J-M, Chao A-J et al (2023) A phase III randomized, double-blind, placebo-controlled trial of the denosumab biosimilar QL1206 in postmenopausal Chinese women with osteoporosis and high fracture risk. Acta Pharmacol Sin 44:446–453
Gu J, Zhang H, Xue Q et al (2022) Denosumab biosimilar (LY06006) in Chinese postmenopausal osteoporotic women: a randomized, double-blind, placebo-controlled, multicenter phase III study. J Orthop Translat 38:117–125
Kim A, Hong JH, Shin W et al (2024) A randomized, double-blind, single-dose, phase 1 study comparing the pharmacokinetics, pharmacodynamics, safety, and immunogenicity of denosumab biosimilar CT-P41 and reference denosumab in healthy males. Expert Opin Biol Ther 22:1–9
US Preventive Services Task Force (2018) Screening for osteoporosis to prevent fractures: US Preventive Services Task Force recommendation statement. JAMA 319:2521–2531
Kanis JA, Cooper C, Rizzoli R, Reginster J-Y (2019) European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int 30:3–44
Cummings SR, San Martin J, McClung MR et al (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Bone HG, Bolognese MA, Yuen CK, Kendler DL, Wang H, Liu Y, San Martin J (2008) Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 93:2149–2157
Eastell R, Christiansen C, Grauer A et al (2011) Effects of denosumab on bone turnover markers in postmenopausal osteoporosis. J Bone Miner Res 26:530–537
Gillett MJ, Vasikaran SD, Inderjeeth CA (2021) The role of PINP in diagnosis and management of metabolic bone disease. Clin Biochem Rev 42:3–10
Acknowledgements
We thank all patients and investigators involved in the study. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Samantha Booth, PhD, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea).
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium. This study was funded by Celltrion, Inc. (Incheon, Republic of Korea).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This study was conducted according to Good Clinical Practice, the principles of the Declaration of Helsinki, and national, state, and local laws or regulations. Study materials were approved by independent ethics committees/institutional review boards at each site (Supplementary Table 1).
Conflicts of interest
JY-R consults for Celltrion, Inc. EC has received funding from Celltrion, Inc. for his role as an investigator in this study and honoraria for lectures from Amgen. PB has received funding from Celltrion, Inc. for his role as an investigator in this study and honoraria for speaker events and authorship on patient materials from Amgen, AstraZeneca, and Pierre Fabre. KW, KS, AP, AS, JK, JS, MJM, SP, and TB have received funding from Celltrion, Inc. for their role as investigators in this study. SLS has received grants from Amgen and Tissuegene, and consults for Celltrion, Inc. JHS, NRH, NHK, and SHB are employees of and own stock in Celltrion, Inc. SHK is an employee of Celltrion, Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Reginster, JY., Czerwinski, E., Wilk, K. et al. Efficacy and safety of candidate biosimilar CT-P41 versus reference denosumab: a double-blind, randomized, active-controlled, Phase 3 trial in postmenopausal women with osteoporosis. Osteoporos Int (2024). https://doi.org/10.1007/s00198-024-07161-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00198-024-07161-x