
Abstract
The stratiform and vein-hosted Kapunda Cu deposit in South Australia contains a saprolitized hydrothermal vein with 12.37 wt.% total rare earth oxide (TREO). The vein was analyzed by X-ray diffraction, scanning electron microscopy, synchrotron-based X-ray fluorescence microscopy and electron backscatter diffraction to understand the controls that govern high-grade REE accumulation during periods of intense weathering. Petrological assessments indicate the transformation of an apatite-calcite-aluminosilicate-bearing protolith to a supergene assemblage of Fe-oxides, kaolinite and REE-phosphate minerals that include rhabdophane-(Ce), monazite-(Ce) and florencite-(Ce). This transformation was facilitated by progressive acidification of the weathering fluid, which is indicated by: 1) the increasing crystallinity of authigenic Fe-oxides and kaolinite, which led to REE desorption; 2) the textural evolution and increase in grain size of authigenic REE-phosphates from nanoscopic crystallites, to acicular needles, to micro-scale hexagonal prisms; 3) the late dissolution of REE-phosphates; and 4) the replacement of goethite by jarosite, whose sulfate component originated from the oxidation and weathering of proximal sulfide minerals. Alongside the depletion of pH-buffering carbonate minerals that are indicated by the preservation of calcite menisci, this sulfide dissolution also facilitated acid generation. Results illustrate how highly acidic weathering fluids might facilitate either REE mobilization or REE accumulation in regolith. High-grade REE accumulation under acidic supergene conditions is prioritized when the host-rock contains a significant source of depositional ligands (i.e., phosphate in the form of apatite) that can be readily leached during intense weathering. Exploration companies should therefore assay routinely for REEs in any heavily weathered phosphatic rock, due to the observed efficiency of phosphate minerals as geochemical traps for REE accumulation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The rare-earth elements (REEs, La – Lu) are defined as critical metals (Grohol & Veeh 2023; United States Geological Survey (USGS) 2022) for the development of renewable energy and defence technologies (Balaram 2019; Beard et al. 2023; Habib et al. 2020; Hayes & McCullough 2018; Jowitt 2022; Klimpel et al. 2021; Lewicka et al. 2021; Verma et al. 2022). Significant work has recently sought to understand the processes that govern REE mobilization, transport, and accumulation in nature (Anenburg et al. 2020a; Borst et al. 2020; Louvel et al. 2022; Migdisov et al. 2016; Strzelecki et al. 2022; Walsh & Spandler 2023). Particularly, there is an increasing focus on the processes that govern REE behaviour in supergene systems (Andersen et al. 2017; Borst et al. 2020; Cook et al. 2023; Hutchinson et al. 2022; Li et al. 2022a, b; Wu et al. 2023). This is due to the complexities introduced by chemical weathering, overprinting, primary mineral destruction and multiple REE fractionation processes that result from combinations of aqueous complexation with organic and inorganic ligands, pH-Eh variation, biota, climate, authigenic mineral precipitation, clay mineral adsorption, and groundwater flow (Andersen et al. 2017; Borst et al. 2020; Chapela-Lara et al. 2018; Cook et al. 2023; Kalintsev et al. 2021; Ondrejka et al. 2018; Ram et al. 2019; Wu et al. 2023; Zhukova et al. 2021). Supergene processes also enriched some of the world’s most important REE deposits, such as Mt. Weld, Australia (Cook et al. 2023; Lottermoser 1990; Zhukova et al. 2021), the Araxá deposit, Brazil (Braga & Biondi 2023; Neumann & Medeiros 2015), the Bear Lodge carbonatites, USA (Andersen et al. 2017; Hutchinson et al. 2022) and Chinese ion-adsorption deposits (IADs, Li & Zhou 2020; Wu et al. 2023).
Although REE transport by low temperature supergene fluids has received some investigation due to the importance of understanding REE distributions in seawater and groundwater (Byrne & Kim 1990; Johannesson et al. 1996; Leybourne & Johannesson 2008; Migdisov et al. 2016; Tanaka et al. 2004; Wood 1990), the geochemical and mineralogical controls on REE deposition during weathering are poorly understood. It is known that REE accumulation in regolith occurs via the adsorption of dissolved REE ions onto the surfaces of clay minerals (Borst et al. 2020; Li et al. 2022a, b; Wu et al. 2023) or via the precipitation of insoluble REE-bearing minerals (Andersen et al. 2017; Cook et al. 2023; Li et al. 2022a, b; Migdisov et al. 2016), but the local geochemical and mineralogical controls that govern the importance of either mechanism are not well constrained because there have been few focused studies. Such controls are important for defining the mineralogical and geochemical criteria that facilitate REE accumulation and mineralization, which can then be included in exploration models and approaches.
This paper focusses on an intensely weathered ferruginous vein with high REE contents (12.3 wt.% TREO) from the Kapunda Cu mine, South Australia. Localized saprolite-hosted REE enrichment offers an opportunity to understand, in detail, the mineral-scale processes responsible for such enrichment. Mineral-scale petrological analyses by synchrotron-based X-ray diffraction (XRD), X-ray fluorescence microscopy (XFM), scanning electron microscopy (SEM) and electron backscatter diffraction (EBSD) are combined with partial leaching and mineral replacement experiments to constrain the conditions of weathering, the primary mineralogy of the vein and how these governed REE accumulation. Results are then used to frame the importance of fluid pH and mineralogy for REE mineralization in regolith.
Geological background
Regional geology
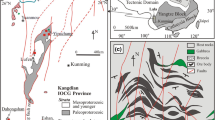
The Kapunda Cu mine is located 80 km north of Adelaide, South Australia, within the highly deformed metasediments of the Neoproterozoic to middle-Cambrian intracontinental Adelaide Rift Complex (Fig. 1) (Foden et al. 1999; Preiss 2000; Wingate et al. 1998). Early rift sediments of the Warrina Supergroup consist of dolomites, quartzites and sandstones, which make up the Callanna and Burra Groups (827 – 700 Ma). Glacial, interglacial and post-glacial sediments of the Umberatana and Wilpena Groups combine to form the younger Heysen Supergroup (700 – 556 Ma), while the Moralana Supergroup comprises all Cambrian sediments (Preiss 2000). These sediments underwent sub-greenschist metamorphism during the Delamerian Orogeny ca. 514-490 Ma (Foden et al. 2006), which was accompanied by the emplacement of basalts (522 ± 4 Ma), diorites (516 ± 2 Ma), I and S-type granites (515 – 505 Ma), adakites (495 ± 1 Ma) and A-type granites (490 – 470 Ma) across the complex (Foden et al. 2002, 2006, 2020; McKirdy et al. 1975). Basin evolution continued in the northern and eastern parts of the Adelaide Rift Complex following the cessation of the Delamerian Orogeny, though minimal evidence for sedimentation across the western Adelaide Rift Complex has led some authors to suggest that this area was instead subject to a sustained period (450 Ma) of weathering through the Phanerozoic (Milnes et al. 1985; Taylor & Shirtliff 2003). Palaeomagnetic data from various volcanic successions (Irving & Irving 1982; Li et al. 1990; Wellman et al. 1969) indicate northward movement of the Australian continent into sub-tropical latitudes towards the end of this period.

Geology of the Kapunda Mine Site. a Adelaie Rift Complex (modified from Lambert et al. 1980; Mahan et al. 2010). b Local geology (Government of South Australia 2023) with structures from Dentith and Stewart (2003). c Insert of the Kapunda mine site, showing lode and sample locations. d Kapunda cross-section, modified from Lambert et al. (1980)
Kapunda deposit geology
The Kapunda Cu deposit (Fig. 1d, Government of South Australia 2023) has a resource estimate of 119,000 tonnes Cu according to current tenant holders Enviro Copper Ltd. and belongs to a cluster of predominantly stratiform Cu deposits within the Burra and Umberatana groups (e.g., Brugger et al. 2006; Pring et al. 2006). The deposit is hosted within metamorphosed transgressive marine sediments of the Tapley Hill Formation; calcareous siltstones that grade upwards through dolosiltstones into calcareous mudstones of the Mount Caernarvon Greywacke Member (Lambert et al. 1980; Preiss 2000). Whole-rock Re-Os dating at various levels in the Tapley Hill Formation constrain its depositional age to between 657 ± 5 Ma and 592 ± 14 Ma (Kendall et al. 2006; Mahan et al. 2010; Schaefer & Burgess 2003). Copper mineralization is both stratiform and vein hosted (Dentith & Stuart 2003). Stratiform mineralization likely resulted from the interaction of Cu-rich groundwaters with biogenic sulfides (mainly pyrite) during sedimentation or diagenesis (Lambert et al. 1980). Secondary Cu-rich veins consisting of chalcopyrite, pyrite, pyrrhotite and carbonate (as well as minor chlorite, muscovite and apatite) crosscut the stratiform mineralization. These veins are suggested to have precipitated after oxidising and metal-rich fluids migrated into tensile fractures during sedimentary compaction (Lambert et al. 1980). Late-stage Cu remobilization during Delamerian orogenesis by higher temperature (300 °C) saline fluids is also indicated by fluid inclusion data from the vein carbonates (Lambert et al. 1980). The sulfide ores were subsequently transformed and enriched by supergene processes during periods of sub-tropical weathering, forming accumulations of Cu-carbonates and secondary sulfides whose discovery led to the birth of Cu mining in Australia (Lambert et al. 1980).
The REE accumulation studied herein occurs within a saprolite-hosted vein located in the eastern lode of the Stockyard Open Pit (Fig. 1e). The vein was marked as being REE-rich (relative to the Cu veins and host-rock) during routine XRF analysis of Cu veins in the deposit. A single sample of this vein (KV00X, Fig. 2) was provided by Terramin Australia Ltd. to determine the controls on REE enrichment.

Photographs of the REE-rich vein. a Vein structures and bedding in the Kapunda Stockyard Open Pit wall, facing direction 019°. b close-up of the REE-rich vein and sample location, S34°20’57.5” E138°55’08.6”. c Vein sample used for analysis (KV00X)
Materials and methods
Sample preparation
Specimen KV00X was sampled from the wall of the open-pit mine at a depth of ≈ 5 m (Fig. 2). Three polished thin sections (45 mm x 25 mm x 30 μm) of KV00X were prepared by Adelaide Petrographic Laboratories, two parallel and one orthogonal to the vein orientation to control for potential structural features. Thick sections (15 mm x 15 mm x 4 mm) were also prepared and fixed in epoxy mounts for SEM and synchrotron XFM analyses. All samples were Pt-coated (5 nm) for SEM imaging at the Centre for Microscopy, Characterisation and Analysis (CMCA), University of Western Australia. The Pt-coating was removed and the samples subsequently C-coated (5 nm) at CMCA for all other analyses. Thin sections were polished by colloidal silica at the Commonwealth Scientific and Industrial Research Organisation (CSIRO), Kensington site, Perth, prior to EBSD analysis only. For whole-rock analyses, a diamond wire saw was used to remove the silicious edges of KV00X from the ferruginous bulk of the vein (KV00X-BLK, Fig. 2), prior to ring mill comminution of the KV00X-BLK sample. This prevented the contamination of KV00X-BLK by silicate minerals from outside the vein boundary. The KV00X-BLK powder was riffle-sampled to produce six representative subsamples, which were subject to REE ion leaching experiments, whole-rock analysis and synchrotron XRD once sieved to a size fraction <20 μm.
Ferrihydrite was identified by its selective dissolution and comparison between the XRD peak counts of the reacted and fresh samples, a method referred to as differential XRD (DXRD, Dold 2003; Lee & Xu 2019; Schwertmann et al. 1982). 125 mg of one of three un-sieved KV00X-BLK powders was reacted with 250 ml of 0.2 M NH4-oxalate solution, brought to pH 3 with oxalic acid. Ferrihydrite was selectively dissolved by its reaction in the dark for 2 hours (Dold 2003). The reacted powder (KV00X-BLK-Rct) was filtered and dried at 105 °C for 2 hours, before being sieved to <20 μm for XRD analysis.
Synchrotron XRD and XFM
Synchrotron XRD was performed at the powder diffraction beamline, Australian Synchrotron, Melbourne. A beam energy of 21 keV (λ = 0.5909 Å), calibrated with a LaB6 standard (NIST SRM 660b), was used. Samples were loaded in a 0.3 mm glass capillary tube (Yao et al. 2021). Phase identification and pattern subtraction required for DXRD was conducted in Match! (v. 3.14) via the crystallographic open database (COD, Gražulis et al. 2009; Vaitkus et al. 2021). Synchrotron XFM was conducted on the XFM beamline at the Australian Synchrotron (Howard et al. 2020). Details of measurement and data reduction procedures can be found in Etschmann et al. (2014). The incident beam was set to 18.5 keV using a horizontal Si (111) monochromator and was focused to a ~2×2 µm2 spot size using Kirkpatrick-Baez mirrors. Full fluorescence data (rather than regions-of-interest) were collected with the Maia model D384 detector at an energy resolution of 240 eV (Ryan et al. 2014). Resin-embedded samples were mapped in continuous mode, at a scanning speed of 10 mm/s and dwell time of 0.2 ms/pixel. Data were analyzed using GeoPIXE II (Li et al. 2016; Ryan et al. 1996, 2010). The Dynamic Analysis Method (Ryan 2000) was used to subtract background, escape peaks and detector artefacts.
SEM analysis
SEM imaging of both the thin section and resin block samples were conducted on a FEI Verios XHR SEM at CMCA. A probe current of 100 pA and accelerating voltage of 15 kV were applied. The Verios was equipped with an Everhart-Thornley SE detector, Elstar in-column BSE detector and Oxford Instruments 80 mm2 X-Max silicon drift detector for energy dispersive X-ray spectroscopy (EDS).
EBSD analysis
Electron backscatter diffraction analysis was conducted at CSIRO on a Zeiss UltraPlus field emission SEM, equipped with an Oxford instrument symmetry EBSD detector and X-Max EDS detector. Maps were collected at a working distance of 7.7 mm, tilt angle of 70°, accelerating voltage of 20 kV and current of 12.1 nA. Large and small-scale maps used step sizes of 0.31 μm and 60.7 nm, respectively. Datasets were imported into Oxford Instruments-HKL CHANNEL5 (service pack 10) for processing.
Whole-rock geochemistry
Whole-rock concentrations of two KV00X-BLK subsamples were analyzed by lithium borate fusion with XRF and LA-ICP-MS finishes at Bureau Veritas (BV), Perth. Approximately 0.66 g of each sample was mixed with 7 g of flux and fused in an induction furnace at 1050 °C for 10 min. The melt was poured into a platinum mould to form a homogeneous borate glass disk. Major elements (wt.%) were measured by XRF using a PANalytical MagiX FAST XRF instrument equipped with a rhodium anode 4 kW tube. Trace elements (ppm-ppb) were measured by LA-ICP-MS analysis via a 193 nm New Wave Excimer Laser coupled to a 7900 Agilent ICP-MS. Ablation was conducted for 70 s using a laser spot of 150 µm pulsing at 13 Hz. A repeat analysis of subsample Bulk_1 was conducted for quality control and fell within 5% variability (Table 1). Data was calibrated against several certified reference materials (OREAS 465, NCSDC 86318, NCSDC 79002 and USZ-42-2006) as well as undisclosed in-house standards.
Partial leaching
Ion exchangeable fractions in KV00X were determined as per Estrade et al. (2019), using an ammonium sulfate solution that achieves up to 97% extraction of sorbed REEs (Moldoveanu & Papangelakis 2016). Three powder fractions (3 g each) from KV00X were dried in an oven at 90 °C until constant weight. One gram of each powder was then introduced to a 50 ml centrifuge tube for reaction with 40 ml 0.5 M (NH4)2SO4 solution brought to pH 4 with 0.1 M H2SO4. One gram of each sample was also reacted with 40 ml 1 M MgCl2 solution (pH 6) to determine REE variance that may result from the dissolution of carbonate minerals (Estrade et al. 2019). The suspensions were positioned laterally and mixed for 1 hour using a Griffin shaker to prevent settling. Solids were separated from the leachate by centrifuging at a temperature of 9 °C and relative centrifugal force of 3000 g for 20 minutes, prior to filtration through 0.2 μm Supor® PES membrane. Of each leachate, 8 ml was filtered. The syringe and the filter were then rinsed with 2 ml of MilliQ water, bringing the final solution to 10 ml. The leached REE concentrations were determined by solution ICP-MS at Bureau Veritas, where a single repeat analysis was conducted for quality control and fell within the 5% variability (Table 2).
Electron Probe Microanalysis (EPMA)
Major elements within the REPs were determined by EPMA and used exclusively to normalize the LA-ICP-MS results. Compositional analyses were acquired at CMCA on a JEOL 8530F Hyperprobe equipped with 5 tuneable wavelength dispersive spectrometers. Operating conditions were a 40 degrees take-off angle, a beam energy of 15 keV, a current of 100 nA and a beam diameter of 0 μm. The on-peak and off-peak counting times were 80 seconds for all elements, aside from P which was 40 seconds. Intensity data was corrected for Time Dependent Intensity (TDI) loss (or gain) using a self-calibrated correction for Y la, Ce la, Eu la and La la. The off-peak correction method was Linear for all WDS elements. Unknown and standard intensities were corrected for deadtime using the Normal (traditional single term) correction method (Donovan et al. 2023). Standard intensities were corrected for drift over time. Oxygen was calculated by cation stoichiometry and included in the matrix correction (Moy et al. 2023). The quantitative blank correction was utilized (Donovan et al. 2011). Other analytical details (including standards) are given in Electronic Supplementary Material 1 (ESM1).
Laser ablation inductively coupled mass spectroscopy (LA-ICP-MS)
The concentrations of 41 isotopes (7Li, 23Na, 24Mg, 27Al, 28Si, 31P, 34S, 39K, 43Ca, 44Ca, 45Sc, 47Ti, 51V, 52Cr, 55Mn, 57Fe, 59Co, 60Ni, 63Cu, 66Zn, 85Rb, 88Sr, 89Y, 90Zr, 139La, 140Ce, 141Pr, 146Nd, 147Sm, 153Eu, 157Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, 175Lu, 208Pb, 232Th, and 238U) were determined for rare-earth phosphate minerals by LA-ICP-MS at CSIRO, Perth. A Photon Machines ATLex 300si-x Excite 193-nm Excimer ArF laser, connected to an Agilent 7700 ICP-MS, was used. A beam diameter of either 20 μm or 50 μm was set depending on grain size. Standards were collected every 10 spots for drift correction. USGS GSD2g was used as a primary reference material due to its high P concentration, which returned a precision of < 0.44% relative standard deviation (RSD) for all but one element (7.54% RSD for S). Data exhibited < 0.02% variation from the accepted standard values for all elements aside from S (1% variation). The results were normalized to Ca (collected by EPMA) using the Iolite software v. 4.6 (Paton et al. 2011).
Results
Mineralogy
Synchrotron XRD and XFM
Synchrotron XRD analysis (Fig. 3a) identified quartz, goethite and rhabdophane-(Ce) as major phases in KV00X. Minor phases included monazite-(Ce), jarosite and hematite. Due to their inability to be differentiated outside of XRD analysis, rhabdophane (REEPO4 · nH2O, n = 0 – 1) and monazite (REEPO4) are grouped as the rare-earth phosphates (REPs) hereafter. Differential XRD (Fig. 3b) provided no evidence of ferrihydrite. Stitched SEM maps and synchrotron XFM maps of Ce, Nd, U, Fe, Y and Cu (Fig. 4) highlight the overarching textures of these minerals that include: 1) coarse (> 200 μm), euhedral grains of quartz and barite (SEM map, Fig. 4a); 2) coarse agglomerations of Fe-oxides (Fe map); 3) disseminated clusters of Fe-oxides; 4) linear REP agglomerations (Ce-Nd, Y map), and; 5) disseminated REP grains. These disseminated REP grains form an interstitial matrix with the disseminated Fe-oxides, which is referenced as the REE-Fe matrix (Mtx, Fig. 4). Other notable features of the XFM maps include Cu-rich patches around the Fe-oxide agglomerates and distinct zones of Ce-Nd fractionation (Fig. 4c).

Synchrotron XRD analysis of KV00X. a Whole-rock XRD analysis exhibiting major REE phases rhabdophane (R) and monazite (M) as well as goethite (G), quartz (Q), hematite (H) and jarosite (J) in grey. b Synchrotron differential X-ray diffraction (DXRD) pattern of the same powder sample, determined via subtracting the XRD pattern of the whole-rock that was reacted in an oxalate solution from that of the unreacted whole-rock and zoomed into where peaks would be expected of two-line (dashed blue lines) or six-line (all blue lines) ferrihydrite (Dold 2003; Lee and Xu 2019). The absence of peaks indicates no ferrihydrite precipitation within the sample

a Stitched SEM image and b, c synchrotron XFM maps of representative textures from KV00X. Notable features include the large masses of goethite (Gth-II) agglomeration with Cu-rich alteration channels (red-orange, represented by Fe and Cu in c), linear structures and masses of rare-earth phosphate (REP) material (represented by Ce, Nd and Y in b and c) and the formation of a pervasive interstitial matrix (Mtx) dominated by REPs and disseminated Gth-I material. Maximum concentrations of Ce, Nd, U, Y, Fe and Cu within the images are 33.5 wt%, 33.5 wt%, 0.64 wt%, 3.77 wt%, 6.66 wt% and 3.1 wt%, respectively
REE-poor minerals and textures
Iron-oxides in KV00X exhibit a diverse range of morphologies, including microscopic goethite (3-4 μm, Fig. 5a) and blocky hematite crystals (>20 μm, Fig. 5b). Goethite colloids are ubiquitous and contribute to the sample’s interstitial matrix alongside kaolinite and disseminated REPs (Fig. 4a). Goethite is either dispersed (Gth-I) or forms aggregates up to 2 mm in diameter (Gth-II, Fig. 5c). Backscatter electron contrast variations within the Gth-II aggregates correlate with higher Cu concentrations (Figs. 4, 5c, d) and Al concentrations (Electronic Supplementary Material 2, ESM2) towards the contacts, between granular masses and pore space and along fracture/alteration pathways. The only Al-rich mineral identified was kaolinite, which occurs as randomly oriented platy masses or, more commonly, vermicular platelets organized into ‘book-like’ aggregates (Fig. 5e).

Gangue mineral textures within the Kapunda vein. a Matrix-hosted colloidal goethite (Gth-I) associated with acicular REE phosphates (REP, likely rhabdophane). b uncommon crystalline and tetragonal hematite. c euhedral goethite agglomeration, exhibiting and Al-Cu-rich alteration rim (inset). d polished surface and cavity of a goethite agglomeration, demonstrating its formation by individual goethite colloids (inset) as well as dark Al-Cu alteration rims which conform to the grain boundaries of the colloids and preserve their botryoidal texture. e vermicular kaolinite embedded in the interstitial Fe-REE matrix, demonstrating ordering of the individual kaolinite platelets along their {001} axes (inset). f jarosite with REPs and kaolinite. g jarosite associated with goethite. The flat and well-polished zones within the image (dotted boundaries) are absent of jarosite and exhibit goethite only, indicating that the jarosite exists only as a fine outer layer and replacement product of that goethite. h dolomite in REP cluster and goethite agglomeration
Brecciated quartz that is often filled with REP material occurs on the edges of the vein. Individual quartz grains are large (>1 mm) and euhedral (Fig. 4). Large barite crystals (>500 μm) are also euhedral (Fig. 4). Late-stage jarosite is observed to have formed after every other phase (Fig. 5f) and associates primarily with goethite in the matrix (Fig. 5g). It is uniquely observable in small sections of the sample that, due to their topographies and the variable extents of resin percolation, received lesser amounts of polishing during sample preparation. Where highly polished surfaces are observed, the jarosite disappears (Fig. 5g). This suggests that jarosite formed predominantly as a surface layer and possibly as a replacement product of goethite. Lastly, trace amounts of dolomite were identified (Dol, Fig. 5h).
REE-rich minerals and textures
X-ray diffraction data frame rhabdophane as the major REP in KV00X, though minor monazite was also identified (Fig. 3). These REPs present in three distinct morphologies: 1) REP-I is represented by thin (5–10 µm) layers of nanoscale crystallites formed along paleo-mineral-fluid interfaces (Fig. 6a, see discussion); 2) REP-II occurs as acicular needles, < 100 nm in width and < 5 µm in length (Fig. 6b); and 3) REP-III occurs as prismatic grains > 5 µm wide (Fig. 6c). These morphologies group in various ways to form larger textural features. Lenticular REP clusters up to hundreds of micrometres in diameter (Fig. 6g) are observed to have developed from REP-III grains. These clusters exhibit variable extents of infill, from complete infilling by REP-II and REP-III (Fig. 6d), to minor infilling by REP-II (Fig. 6e), to negligible infilling associated with REP-I/REP-II. The lenses themselves are either doubly terminated (Fig. 6f) or truncated by linear terminations (Fig. 6g). Most exhibit REP rims, separated from the main infill material by gaps of 5 to 20 µm (Fig. 6d, g). These rims conform to the edges of the lenses, whether curved (Fig. 6f) or linear (Fig. 6g), and either transition between REP-I and REP-III (Fig. 6f, g) or are, less commonly, exclusively REP-III (Fig. 6d). Spherical structures of REP-I material are observed in some rims (Fig. 6h). REP-III grains are confirmed as rhabdophane via EBSD (Fig. 7a – c). EBSD data reveal two preferential REP-III orientations that run perpendicular to each other (Fig. 7d); one with a point maximum of the basal planes (0001) that is interpreted to represent the growth direction (Subset A, Ortn-I), and the other that defines a girdle across the {01-10} direction (Subset B, Ortn-II). These coexist as infill and alternate depending on whether the edge is linear or convex (Fig. 7b-d).

Rare earth phosphate (REP) morphologies associated with stacked lenticular textures in KV00X. a nanocrystalline layers of REP-I, developing outwards into prismatic REP-III. b acicular REP-II, developing around the edges of the lenticular textures and infilling them. c prismatic REP-III grains (center and right) in the interstitial matrix, as well as REP-II grains associated with goethite (top left). d truncated and stacked lenticular textures, fully infilled by REP-II and REP-III grains and surrounded by a REP-III rim (left hand side, lhs) that develops into a combined REP-I and REP-III rim (top right). e comparison of complete lenticular infilling by REP-II and REP-III (lhs) with partial lenticular infilling by REP-II and matrix material (right hand side, rhs). Outer REP-III rims are observed to form on both. f large-scale and lenticular REP agglomerations, exhibiting multiple lens orientations (with some doubly-terminated, inset) and surrounded by the interstitial Fe-REP matrix. g similar large-scale textures, this time with sharp and linear truncations of the lenses that are preserved by REP-I/REP-II material rime overgrowth (lhs). h outer REP-I + REP-III rim and the preservation of pseudospherical structures by REP-I growth. Spherical structures (labelled as Menisci) formed through the entrapment of gas bubbles during carbonate dissolution and subsequent REP growth around these entrapments (see text)

EBSD analysis (IPF Colouring II X0) of lenticular structures in KV00X, and their relation to the formation of rhabdophane aggregates. a SEM image. b orientations of prismatic rhabdophane aggregates, exhibiting the two preferential orientations in colours of green/blue (Subset A, Ortn-I) and red/orange (Subset B, Ortn-II). c representative SEM image of hexagonal REP prisms in Ortn-I (rhs) and Ortn-II (lhs). d pole figure maps of the sample area, displaying an orthogonal relationship between the two subsets. Note the preservation of curvature by Ortn-I grains and intergrowth by both orientations (inset)
Minor REP-II and dominant REP-III exist in the matrix (Fig. 8a). Acicular REP-II also grows on the surface of goethite (Fig. 8b), while matrix-hosted REP-III grains associate primarily with kaolinite (Fig. 8c). The grains appear randomly orientated (Fig. 8a) and may form radial clusters (Fig. 8d). A few REP-III grains display hexagonal dissolution pits (Fig. 8e). Florencite-Ce (REEAl3(PO4)2(OH)6), identified by high Al-to-P EDS ratios (ESM2), occurs sparingly across KV00X in zones of Al-Cu-rich goethite alteration (Fig. 8f).

REP textures within the matrix and associated with gangue minerals. a coexistence of acicular REP-II and prismatic REP-III within the interstitial matrix. b precipitation of REP-II grains upon REP-III and in association with colloidal goethite. c randomly oriented REP-III grains in association with matrix-hosted vermicular kaolinite. d radial aggregates of REP-III developing at the boundaries of quartz grains. e hexagonal dissolution pit within an REP-III grain. f Gth-II agglomerations associated with interstitial REPs (lhs) and exhibiting Al-Cu-rich alteration (dark patches). Florencite (inset) was determined to have precipitated uniquely within these Al-rich alteration zones, while the REPs precipitate outside of them
Geochemistry
Whole-rock compositions
Results of whole-rock analyses are provided in Table 1. Lambda shape parameters were derived from the analysis of chondrite normalized REE patterns (O’Neill 2016) using the web application BLambdaR (Anenburg & Williams 2022). Their definitions are provided in ESM2, alongside the equations used for calculating Ce, Eu and Yb anomalies. The whole-rock REE profile (Fig. 9) is characterized by LREE enrichment (\({\uplambda }_{1}\) = 22.51), convex La – Sm distributions (\({\uplambda }_{2}\) = - 90.22), concave Dy – Tm distributions, negative Ce anomalies (Ce/Ce* = 0.93), negative Eu anomalies (Eu/Eu* = 0.69) and positive Yb anomalies (Yb/Yb* = 1.22). The vein is Fe-rich (56.84 wt.% Fe2O3), with low SiO2 (5.07 wt.%) and Al2O3 (2.80 wt.%) and high REE (12.37 wt.% TREO). Other notable geochemical features (Table 1) include high P2O5 (6.65 wt.%), high BaO (2343 ppm) and high UO2 (1110 ppm) coupled with low ThO2 (< 20 ppm).

Chondrite-normalized REE distributions (O’Neill 2016) of the whole-rock, leachate solutions and REE-phosphate minerals in KV00X as analyzed by (LA)-ICP-MS, as well as the calculated recovery of REE metals in the (NH4)2SO4 leachate (in %)
Leachate compositions
All REE concentrations were below detection limit (BDL) in the MgCl2 leach. The (NH4)2SO4 leach results are given in Table 2. Average REE concentrations in the (NH4)2SO4 leachate were between 337 ppb (Ce) and 1 ppb (Lu), corresponding to a maximum recovery of 0.162% for Lu and a minimum recovery of 0.033% for Pr (Fig. 9). The leachate was LREE-rich (\({\uplambda }_{1}\) = 14.75), but less so than the whole-rock. The leachate exhibited negative Eu anomalies (Eu/Eu* = 0.55) and negative Ce anomalies (Ce/Ce* = 0.79).
Rare-earth phosphate REE distributions
An average composition of (La0.09Ce0.42Pr0.07Nd0.26Sm0.05Y0.03)PO4·xH2O was derived from 35 LA-ICP-MS analyses of the REPs. Chondrite-normalized distributions exhibit consistent LREE enrichment (average \({\uplambda }_{1}\) = 22.18), convex La – Sm distributions (\({\uplambda }_{2}\) = - 103.67), positive Ce anomalies (average Ce/Ce* = 1.07), negative Eu anomalies (average Eu/Eu* = 0.71) and positive Yb anomalies (average Yb/Yb* = 1.25). These largely correspond with the whole-rock REE distribution (Fig. 9). The mean concentration of Y within the REPs (8500 ppm) generated an average Y/Ho ratio of 13.73 (Table 1). Synchrotron XFM maps indicate some degree of heterogeneous LREE (Ce, Nd) distribution across the sample (Fig. 4b), though heterogeneous distributions are not readily observed within the REPs measured by LA-ICP-MS.
Discussion
Owing to the fact that the KV00X sample contains significant concentrations of the REEs, it provides a good opportunity to study the conditions and mechanisms under which REEs accumulate in regolith. Once those conditions (such as the geochemical nature of weathering) and processes (such as the mineral-scale mechanisms of REE accumulation) are determined, they are then applied to develop a formation model for KV00X which provides specific criteria for REE exploration in weathered terrains.
Nature of the weathering fluid
Factors that control how the REEs are mobilized, transported or deposited in geological fluids include pH, temperature, ligand activities (i.e., of chloride, sulfate, fluoride, carbonate, phosphate and organic species), the availability of oxidising or reducing agents, crystallographic controls and adsorption onto clays or suspended particulate matter (Aubert et al. 2001; Borst et al. 2020; Li et al. 2022a, b; Liu et al. 2018; Liu et al. 2017; Louvel et al. 2022; Migdisov et al. 2016; Nelson et al. 2003). Many of these factors are challenging to constrain in regolith due to the poor preservation of fluid inclusions, small mineral grain-sizes and dynamic (bio)geochemical processes (Kalintsev et al. 2021). Nevertheless, certain minerals and textures can provide insight into the conditions of weathering. Jarosite is known to be stable in Fe-rich regolith subject to acidic and sulfate-rich weathering (Baron & Palmer 1996; Brown 1971; Norlund et al. 2010). It commonly forms at the expense of goethite at pH < 3 (Brown 1971) following the oxidative dissolution of pyrite (Bladh 1982; Kartal et al. 2020; Zolotov & Shock 2005). The presence of late-stage jarosite (Fig. 5f, g) therefore indicates that the fluid was highly acidic and S-rich in the final stages of weathering. Barite was the only other sulfate mineral observed in KV00X.
Sulfate was most likely sourced following the oxidative weathering of proximal sulfide minerals (i.e., pyrite and chalcopyrite), located within the vein or wall-rock (Lambert et al. 1980). Pervasive weathering of the Kapunda Cu deposit at and below the shallow water table (currently ~20 m depth) is supported by its supergene mineralogy that includes covellite, cuprite, atacamite and chrysocolla (Wang et al. 2022). This associated remobilisation of Cu during weathering is also supported by the Cu alteration zones that developed in Gth-II agglomerations during late-stage weathering (Fig. 4c). As well as being a source of S, oxidative sulfide mineral weathering represents a likely source of acidity and ferric ions to the weathering fluid which may have contributed to the formation of goethite in KV00X.
The dominance of this goethite (relative to ferrihydrite or hematite) also alludes to a gradual decrease in weathering fluid pH. Ferrihydrite was not detected by DXRD (Fig. 3), and goethite aggregates formed prior to the interstitial matrix and increase in acidity that is indicated by jarosite precipitation. The potential for direct goethite precipitation from solution under acidic conditions (Schwertmann & Murad 1983) is therefore voided by the circumneutral conditions that goethite is interpreted to have precipitated under. Adjacent crystalline hematite occurs sparingly and shows no evidence of alteration, such that it was likely the last Fe-oxide to develop (Fig. 5b). All observations are consistent with the authigenic precipitation of ferrihydrite from a Fe-rich fluid, followed by its recrystallization to goethite and then hematite (Das et al. 2011; Jambor & Dutrizac 1998; Schwertmann et al. 1999; Zhu et al. 2016). If each of the Fe-oxides had instead developed via the weathering and crystallographic devolution of primary hematite to goethite and ferrihydrite (Ramanaidou et al. 1996) or via the direct precipitation of goethite from solution, then it is more likely that the goethite agglomerations would have developed during the later acidic stages of weathering that are more favourable for hematite dissolution (Xu & Gao 2008) and ferric ion polymerization (Schwertmann & Murad 1983).
Ferrihydrite (as a precursor to goethite) is therefore likely to have precipitated under circumneutral conditions from ferric ions provided by the early oxidative weathering of proximal sulphide minerals (Bladh 1982; Jambor & Dutrizac 1998; Savage et al. 2000). The dominance of goethite relative to ferrihydrite can then be explained by rapid transformation catalyzed by high Fe2+(aq) activities and low Si (aq) activities over long periods of weathering (Das et al. 2011; Jambor & Dutrizac 1998; Pedersen et al. 2005; Schwertmann et al. 1999). The transition from Gth-I agglomerations to a disseminated goethite-REP matrix also aligns with the proposed transition from circumneutral to acidic fluids, since goethite clusters deagglomerate at low pH (Carstens et al. 2021; Celi et al. 2001; Ding & Pacek 2008).
Evidence for primary carbonate minerals, such as the preservation of minor dolomite (Fig. 5h) and the identification of pseudo-spherical void structures (Fig. 6h), also indicate a gradual decrease in pH. The pseudo-spherical voids preserved by REP-I layers are interpreted to represent menisci that developed via the entrapment of CO2 bubbles during carbonate dissolution (Andrieu et al. 2018; Du Bernard & Carrio-Schaffhauser 2003; Hillgärtner et al. 2001). This early dissolution of primary carbonates (i.e., dolomite ± calcite) is likely to have buffered fluid pH during the onset of weathering, prior to a gradual decrease in pH throughout their destruction. This decrease in pH then caused simultaneous textural evolution of the REPs. Since REP minerals are less soluble under circumneutral conditions (Gausse et al. 2018; Lin et al. 2021), the weathering fluids were most REP-saturated during early carbonate dissolution. This caused rapid REP nucleation and the formation of nanocrystalline REP-I layers around CO2 menisci. As the pH decreased, these nanocrystalline layers then evolved into coarse-grained and re-dissolved REP-III layers (Figs. 6f – h, 8e), due to their increasing solubility under acidic conditions.
Kaolinite morphologies provide further insights into the geochemical evolution of weathering fluids. Vermicular kaolinite (Fig. 5e) forms pseudomorphs after phyllosilicates (Jeong 1998a; Li & Zhou 2020; Rebertus et al. 1986) and feldspars (Jeong 1998b; Li & Zhou 2020; Papoulis et al. 2004) during chemical weathering. The ‘fanned’ morphologies of vermicular kaolinite in KV00X are typical of phyllosilicate weathering rather than feldspar weathering (Jeong 1998a, 1998b; Kretzschmar et al. 1998), though the precursor mineral cannot be confidently assigned. Even so, the presence of vermicular kaolinite supports interpretations of highly acidic weathering, since more intense periods of chemical alteration favour the progressive stabilisation of such highly crystalline kaolinite morphologies (Papoulis et al. 2004). The early crystallographic evolution of kaolinite also coincides with decreasing Al/Fe ratios (Papoulis et al. 2004), such that kaolinite evolution may have contributed Al to the weathering fluid which then facilitated the Al-Cu-rich alteration of goethite and stabilized florencite (Figs. 4, 8f, ESM2).
Mechanisms and textures of REE accumulation
Ion adsorption
Rare-earth element accumulation in regolith occurs either via ion adsorption onto the negatively charged surfaces of clays minerals (i.e., kaolinite, Borst et al. 2020; Estrade et al. 2019) or the precipitation of insoluble phases like rhabdophane and monazite (Lottermoser 1990). Ion species adsorbed onto the surfaces of kaolinite tend to desorb as kaolinite crystallinity increases (Li and Zhou 2020; Parfitt 1989). Resultingly, the presence of highly crystalline, vermicular kaolinite at Kapunda theoretically limits the potential for ion adsorbed REE accumulation within KV00X. This hypothesis is supported by partial leach experiments, which show extremely low REE recoveries in the (NH4)2SO4 leachate (Fig. 9, 0.033% [Pr] - 0.162% [Lu]) and negligible recovery in the MgCl2 leachate. Such negligible recovery in the MgCl2 leachate, relative to minimal recovery in the (NH4)2SO4 leachate, indicates that acid-soluble minerals (like carbonates) or even REPs cannot be discounted as a source of REEs to the (NH4)2SO4 leachate (Estrade et al. 2019). Either way, the source of REEs is somewhat irrelevant relative to the extremely low concentrations that are observed. These indicate that the vast majority of REE accumulation is not ion adsorbed. Since the crystallographic evolution of kaolinite is promoted over intense periods of weathering (Ling et al. 2018; Papoulis et al. 2004; Proust et al. 2006), this in turn provides evidence that the late-stage and acidic nature of weathering at Kapunda likely facilitated REE desorption from early clay phases prior to their preferential uptake into late-stage and ‘high-grade’ REP minerals. This is emphasized by studies which demonstrate that both the adsorptive capacity (Coppin et al. 2002; Yang et al. 2019) and surface area of kaolinite decrease as pH decreases, where the latter is due to increased kaolinite coagulation (Gupta et al. 2011; Kretzschmar et al. 1998).
Though there is subsequently no evidence of REE adsorption onto kaolinite in KV00X, SEM Images document an intimate and ubiquitous relationship between Gth-I colloid surfaces and REP-II precipitation (Fig. 8b). The REEs and P sorb readily onto goethite in low-temperature hydrothermal systems (Borst et al. 2020; Geelhoed et al. 1997; Hsu et al. 2020; Parfitt & Atkinson 1976), such that their co-adsorption may have catalyzed REP formation (i.e., Ichimura et al. 2020). Like kaolinite, the REE adsorption capacity of goethite decreases with a decrease in pH (De Carlo et al. 1997; Quinn et al. 2006). Unlike kaolinite, goethite coagulation also decreases with a decrease in pH (Celi et al. 2001) such that an increase in acidity likely facilitated an increase in adsorptive area for PO42- and REE ions.
Authigenic REE precipitation and the preservation of apatite dissolution textures
Metal accumulation at Kapunda is thus expressed exclusively by REP precipitation. Analysis by SXRD and SEM show the presence of major rhabdophane with minor monazite and florencite (Fig. 3), as are observed in other supergene REE deposits (Li & Zhou 2020; Lottermoser 1990; Sanematsu et al. 2015). Florencite-(Ce) precipitation was limited to Al-Cu-rich micro-environments in goethite (Fig. 8f, ESM2), suggesting that the weathering fluid was Al-poor and only stabilized florencite-(Ce) where; 1) bulk-fluid phosphate activities decreased following earlier rhabdophane and monazite precipitation, and/or; 2) the local geochemical conditions in the goethite fractures (i.e., low fluid-rock ratios and high Al activities) were favourable for florencite-(Ce) precipitation. Conversely, rhabdophane-Ce and monazite-Ce pervasiveness through KV00X indicates that fluid phosphate activities were consistently high. Phosphate contributes heavily to REE immobilization in low temperature systems (Migdisov et al. 2016), though the source of P in KV00X is not apparent in the absence of primary phases. Apatite (Ca5[PO4]3[F,Cl,OH]) is the most abundant phosphate mineral on the planet, undergoes replacement by REPs in regolith (Banfield & Eggleton 1989; Braun et al. 1990; Stille et al. 2009), forms doubly or linearly-terminated lenticular dissolution pits (Figs. 6d – g, 7a, Harouiya et al. 2007) depending on the weathering conditions (Phillips-Lander et al. 2017; Velbel 1989, 2007), and occurs at Kapunda in association with both the stratiform sulfides and secondary veins (Lambert et al. 1980). We therefore hypothesize that the lenticular clusters in KV00X formed through the replacement of apatite by REPs.
To validate this hypothesis, we experimentally investigated apatite as a possible precursor to lenticular REP precipitation (Fig. 6). Hydroxyapatite from the Mud Tank carbonatite complex 100km northeast of Alice Springs, Australia, was reacted with a 0.01 M HCl, 0.02 M CeCl3 solution at a ratio of 0.05 g of apatite (150-250 μm) to 20 ml of fluid for 72 hours at 30°C. Subsequent SEM investigation shows that: 1) stacks of lenticular etch pits and denticular arrays developed on the apatite (Fig. 10b-f); 2) the release of phosphate from apatite induced rhabdophane-(Ce) precipitation following its reaction with dissolved Ce (Fig. 10); 3) rhabdophane-(Ce) preserved the internal morphologies of the apatite denticles, mimicking the complex textures that are observed at Kapunda (Fig. 10h-j), and; 4) apatite dissolution outpaced rhabdophane reprecipitation, as indicated by the voids that developed between each mineral (Xia et al. 2009). These experiments indicate that: 1) apatite is the probable source of P and precursor to pervasive REP precipitation at Kapunda; 2) the preservation of dissolution features indicates acidic and/or prolonged weathering conditions; and 3) fluid acidity likely expedited apatite dissolution and P liberation, which is interpreted to have been vital for high-grade REE accumulation.

Back scatter electron images of products from hydroxyapatite reaction in a 0.01M HCl, 0.02M CeCl3 solution for 72 hours at 30°C, as well as comparison with natural textures. a XRD pattern of experimental products apatite (A) and rhabdophane (R). b epitaxial growth of experimental rhabdophane clusters (white) upon the surface of the apatite (grey). c close up of lenticular dissolution features on apatite after acidic dissolution. Notice their association with broader and linear dissolution steps that truncate some of the lenses (top of image), the formation of double-terminated lenses (bottom of image) and the ‘stacking’ effect created by layered dissolution steps. d denticles at the edge of an apatite grain, formed by the stacking of lenticular terminations. Notice the fossil preservation of denticle terminations at the rhabdophane interface. e close-up of lenticular dissolution pits in apatite. f coalescence of lenticular dissolution pits and steps (rhs) to form denticles (lhs). g detailed image of radial and acicular rhabdophane clusters formed during experimental apatite replacement. h cross section of an experimental grain, showing how the terminations of the primary denticles (green dashed lines) are preserved by epitaxial rhabdophane overgrowth in both instances to form fossil lens structures (red dashed lines). i the same textures as in h, but within KV00X. Within the natural sample, stacked primary denticles are also preserved by rhabdophane overgrowth, even after complete dissolution of the apatite precursor. j three-dimensional representation of lens stacking (rhs) at natural dissolution steps during rhabdophane replacement (entire image). Prismatic rhabdophane is observed to develop outward from the fossil lens interface, like textures which are observed in i
In KV00X, the REP-III grains that preserve apatite dissolution textures exhibit additional characteristics: 1) they precipitate perpendicular to each other (Fig. 7d); 2) their morphologies are truncated by the curvature of the lenses (Fig. 7), and; 3) their development occurred both away from the outside of the lenses (Fig. 6d) and inwards towards their center (Fig. 6b). These textures are interpreted to represent REP overgrowth from the base and walls of lenticular etch pits in apatite, as well as upon the dissolution steps and denticles that form during pit coalescence (Velbel 2007). Rhabdophane-(Ce) growth from the base (Ortn-I) and the walls (Ortn-II) of the dissolution pits resulted in perpendicular grain orientations, facilitated inwards growth, and led to truncation of hexagonal grain morphologies by the pit walls (Fig. 7b). Outwards REP development represents epitaxial rhabdophane overgrowth from the palaeo-apatite dissolution steps and preserved denticle morphologies in the rhabdophane-(Ce) clusters (Fig. 10h, i). Growth upon these subsequent dissolution steps (Fig. 10c) then caused secondary REP denticles to appear ‘overlapped’ in two and three dimensions (Fig. 10 i, j).
Paragenesis and evolution
Interpretations as to the mechanisms of mineralogical evolution at Kapunda were used to develop a paragenetic model for KV00X. The model tracks Fe-oxide (Fig. 11, top) and REP-apatite-calcite evolution (Fig. 11, bottom) from periods of low intensity, high-pH weathering (during mineral buffering, left) to those of high intensity, low-pH weathering (after the destruction of mineral buffers, right). The primary mineralogy of KV00X likely included apatite, calcite and aluminosilicates (Fig. 11a). At the onset of weathering (Fig. 11a) and under circumneutral conditions, dissolved ferrous ions were introduced to the vein which stimulated ferrihydrite precipitation. Goethite agglomerations (Gth-II) then developed from that ferrihydrite and dissociated goethite (Gth-I) under high pH, low Si and highly ferrous fluid conditions. Apatite dissolution formed denticular textures, while nanocrystalline REP-I precipitated at the apatite-calcite-fluid interface to encompass calcitic menisci (Fig. 11b). As the pH decreased due to the lessening impact of mineral buffering (Fig. 11c), Gth-II agglomerations dissociated. Increasing kaolinite crystallinities released Al into the fluid and caused REE desorption. Apatite was fully replaced by REPs and calcite was destroyed. Finally, REP textures transitioned from REP-I to REP-III. Under the most acidic conditions (Fig. 11d), jarosite (Jrs) superseded goethite while fluids facilitated the alteration of Gth-II to form botryoidal, Al-Cu-rich zones. Goethite development resulted in the formation of minor tetragonal hematite. Remaining desorbed and dissolved REEs and P combined with Gth-I to form an interstitial REE-Fe matrix, and with Al to stabilize florencite in Gth-II alteration zones. The adsorption capacity of Gth-I increased, adsorbing REEs and P to catalyse the formation of REP-II grains on goethite colloids. Dissolution pits eventually developed in REP-III grains.

Mineralogical and textural model of KV00X in response to increasing fluid acidity over time. a remnant textures in the vein indicate a primary mineral assemblage that includes apatite (ap), calcite (Clc) and aluminosilicates (ASC). At the onset of weathering, dissolved ferrous ions either form or were introduced to the vein prior to the precipitation of ferrihydrite (Fhy) under circumneutral conditions. b the development of goethite agglomerations (Gth-II) from ferrihydrite and dissociated goethite (Gth-I) is supported by relatively high pH levels, low Si and high ferrous concentrations. Apatite dissolves to denticular textures, while nanocrystalline REP-I develops ay the apatite-calcite interface to encompass calcitic menisci (Mnsc). c as the pH decreases due to the lessening impact of mineral buffering, Gth-II agglomerations begin to dissociate. The increasing crystallinity of kaolinite releases Al into the weathering fluid and facilitates REE desorption. Apatite is fully replaced by REPs and calcite is destroyed. REP textures transition from REP-I to REP-III. d under the most acidic conditions, jarosite (Jrs) replaces goethite while fluids facilitate the alteration of goethite agglomerations to form botryoidal, Al-rich alteration zones. Increasing goethite crystallinity results in the development of minor tetragonal hematite (Hem). Remaining desorbed and dissolved REEs and P combine with Gth-I to form an interstitial REE-Fe matrix, and with Al to precipitate florencite (Flc) in Gth-II alteration zones. The adsorption capacity of Gth-I increases, thus adsorbing free REE and P ions to catalyse the formation of REP-II grains on individual goethite colloids. Dissolution pits (DPs) form within REP-III grains
Composition and formation environment of the KV00X protolith
This section summarises available evidence for the primary mineralogy of the KV00X protolith, before discussing formation environments that may have facilitated the development of such an assemblage. The primary source of phosphate in KV00X is interpreted to have been vein-hosted apatite. Although apatite is not preserved within the highly weathered vein sample, this interpretation is based on several mineralogical factors that are described in previous sections. Primary dolomite ± calcite is also indicated through the textural preservation of calcitic menisci, though aside from these minerals the primary composition of the highly weathered KV00X vein remains practically undefinable. Additional mineralogical interpretations are instead based on prior descriptions of fresh vein samples from drill cores, which align well with the textural observations made herein. Fresh veins are reportedly calcite-rich and contain accessory apatite, allanite, monazite and rare-earth oxides (Lambert et al. 1980). Therefore, in-situ sources of the REEs from minerals like allanite, monazite, apatite and REE-oxides, as well as the presence of calcite as a precursor to the formation of calcitic menisci, are considered likely.
If this assumption is correct, one might expect that other weathered veins at Kapunda would also be REE-rich. However, a comparative assessment of several unweathered vein samples has not been done. Based on isotopic evidence (Lambert et al. 1980) and observations of brecciated quartz (Fig. 4), vein-hosted REE-bearing minerals and apatite are interpreted to have been derived from sedimentary fluid exsolution and are thus a product of hydrothermal processes. It is therefore possible that multiple generations of hydrothermal veins may have formed with distinct geochemical and mineralogical characteristics. Lambert et al. (1980) note how Cu-mineralized units around Kapunda are enriched in carbonate minerals relative to Cu-barren units, such that carbonate complexation might have been vital for hydrothermal (< 300 °C) REE (± Cu) mobilization during sedimentary compaction. Carbonate is an important ligand for REE mobilization in low temperature systems (especially in alkaline fluids) and commonly competes with phosphate for control over REE behaviour (Johannesson et al. 1996; Louvel et al. 2022; Migdisov et al. 2016). One possible control on REE-enrichment in KV00X could therefore relate to timing and the availability of carbonate complexes, which mobilized more REEs from the sediments into certain veins that today exhibit a higher abundance of secondary REE phases.
Another explanation for localized REE enrichment is that the vein formed via discordant hydrothermal processes unrelated to sedimentary compaction. Evolution of the Adelaide Rift Complex is characterized by sub-greenschist facies metamorphism, widespread granitic magmatism (Foden et al. 2006; Turner et al. 2022) and some evidence of alkaline magmatism (Lottermoser 1988; Tucker & Collerson 1972). Each of these can induce sedimentary or magmatic devolatization with associated element migration and deposition in phosphate-rich veins (Andersson et al. 2018; Campbell & Henderson 1997; Vapnik et al. 2007). This is especially true for fluids that are associated with peralkaline granites or carbonatites (Campbell & Henderson 1997; Ferenc et al. 2021; Ren et al. 2019) that form in post-collisional extensional settings (Dostal 2017; Goodenough et al. 2016; Yaxley et al. 2022). The primary ore minerals in hydrothermal REE deposits that are associated with extensional settings, which include monazite, bastnaesite and less commonly apatite, correspond well with those assigned to the KV00X protolith. In particular, the apatite-rich and vein-hosted Nolans Bore REE deposit in Northern Territory, Australia (Anenburg et al. 2020b; Schoneveld et al. 2015) represents an interesting analogue to the interpreted protolith at Kapunda. Huston et al. (2016) invoke a syn-genetic extensional regime (like the Adelaide Rift Complex) as being essential for the formation of Nolan’s Bore, while the REE profiles in vein-hosted apatite (convex La – Sm distributions, minor negative Eu anomalies) imitate those of the KV00X rhabdophane (Anenburg et al. 2020b; Schoneveld et al. 2015).
Alongside its formation as an apatite-rich hydrothermal vein emplaced within an extensional tectonic regime, these seemingly unique REE distributions could be argued as evidence for the KV00X protolith being an analogue to Nolan’s Bore. However, these REE distributions are not unique enough to make this interpretation with any degree of confidence. Convex La – Sm distributions can also be attributed to unrelated weathering processes or hydrothermal processes such as: (1) fluid evolution following the initial precipitation of La-Ce-enriched minerals and the subsequent precipitation of La and Ce-poor minerals (Le Bas et al. 1992; Moore et al. 2015; Xu et al. 2012) or; (2) the late-stage metasomatism of primary minerals, with concurrent La/Ce mobilization and uptake into secondary phases (Andersen et al. 2017; Anenburg et al. 2020b; Le Bas et al. 1992). Weathering-related REE fractionation certainly impacted KV00X to at least some extent, as indicated by heterogeneous Ce-Nd distributions in the sample (Fig. 4b) which may relate to Ce oxidation and cerianite precipitation (resulting in adjacent Nd enrichment) or some adsorptive fractionation process. Moreover, external hydrothermal processes were previously disregarded as being unimportant for vein formation at Kapunda, due to a lack of magmatic sources in the area (Lambert et al. 1980). A calcite-apatite-monazite-allanite-rich vein formed by sedimentary compaction and exsolution thus remains as the preferred protolith for KV00X.
Implications for REE exploration
Acidic weathering fluids: REE transporters or depositors?
Increases in pH can result in the removal of REEs from weathering fluids through the precipitation of increasingly insoluble phosphate minerals like monazite, rhabdophane and florencite (Migdisov et al. 2016; Wood & Williams-Jones 1994). It is therefore unsurprising that pH-related depositional processes find their place in most models that describe the formation of regolith-hosted REE deposits (Lottermoser 1990; Jowitt et al. 2017). Nevertheless, a generalized attribution of high-pH fluids to REE ore deposition – and in turn, the attribution of low-pH fluids to REE mobilization and transport – considers only half of the story. Although it is accepted that REE ore minerals may precipitate from weathering fluids due to their lower solubilities as the pH increases (Gausse et al. 2018; Lin et al. 2021), it is also true that the solubility of apatite decreases as pH increases (Guidry & Mackenzie 2003). If low REP solubilities prevent the co-transport of REEs and phosphate in weathering fluids (Migdisov et al. 2016), these solubility features of the REPs and apatite then create a theoretical paradox. Rare-earth phosphate deposition is limited at low pH by higher REP solubilities (i.e., greater REE mobilization) and at high pH by the lesser solubilities (and lesser dissolution) of phosphate mineral traps like apatite (i.e., lesser REE immobilization through secondary phosphate precipitation). This paradox must be resolved to determine whether acidic weathering fluids are favourable or unfavourable for REE accumulation.
The dissolution of abundant primary apatite at Kapunda facilitated a significant increase in phosphate activities within the weathering fluid, to the point where rhabdophane and monazite precipitated even under highly acidic conditions. In other words, the KV00X sample is one where acidic weathering fluids were favourable – or perhaps essential – for ensuring a supply of depositional ligands which facilitated high-grade REE accumulation. The implication is that, where low pH weathering fluids can leach significant concentrations of depositional ligands (i.e., phosphate, Migdisov et al. 2016) from the surrounding host-rocks, those depositional ligands can outcompete acidity and induce REE deposition (Fig. 12a). The more available the depositional ligand, the more locally the REEs will precipitate and the higher the grade of the deposit will be. Where depositional ligands are less available (such as in instances where the pH is higher or there are lower P concentrations in the host rock), REE deposition extends over greater distances and grades decrease (Fig. 12b). Another example of where this might occur is following initial REP precipitation, where the depositional ligand (PO42-) is used up such that the weathering fluids again start mobilising the REEs (Fig. 12c, d). Evidence for this at Kapunda is found in the formation of dissolution pits in late stage rhabdophane (Fig. 8e). The implications of this for REE exploration are that high-grade REE accumulation in regolith is likely to occur nearer to the surface (or otherwise in horizons where the fluid can maintain a low pH), provided that those horizons can contribute depositional ligands (i.e., phosphate from apatite). These implications offer nuance as to how the pH of the weathering fluid influences REE mobilization or deposition in supergene systems.

Interpreted influence of weathering fluid acidity on REE accumulation in phosphatic host-rocks. a more acidic weathering fluids will liberate higher concentrations of phosphate from the host rock, resulting in increased REP precipitation and higher REE grades over smaller distances. b less acidic weathering fluids liberate phosphate from the host-rock less efficiently, culminating in decreased REE deposition, increased transport and lower REE grades over larger distances. c as phosphate becomes depleted in the system, highly acidic weathering fluids are more likely to facilitate secondary REP dissolution, mobilization and deposition at lower stratigraphic positions as a function of fluid-rock interaction and increase in pH. d less acidic fluids are less likely to facilitate REP mineral dissolution and remobilization
Suitable settings for supergene enrichment and high grade REE accumulation
Despite the KV00X protolith not having been assuredly identified, the discussion surrounding its potential composition and subsequent conditions of formation highlights the range of geological environments (i.e., hydrothermal, diagenetic, and supergene) that may facilitate the formation of phosphate-rich assemblages that make excellent geochemical traps for the REEs during weathering. The outcome of this for REE exploration is that, despite the tendency for explorers to target primary lithologies that are typified by REE enrichment (i.e., carbonatites and peralkaline complexes), mining companies should also consider assaying for REEs in regions and lithologies that are prone to any form of phosphate mineralization. This is especially true if those regions may have also been subject to sustained periods of tropical weathering, since a growing body of evidence suggests that the protolith itself need not be ‘REE-rich’ for significant fluid volumes to mobilise the REEs necessary for economic mineralization upon contact with phosphate-enriched fluids or rock-types (Walsh and Spandler 2023).
Conclusions
Based on detailed XRD, SEM, XFM, EBSD and whole rock geochemical analysis of an intensely weathered vein sample from the Kapunda Cu mine, it is shown that rare-earth element accumulation within the vein was facilitated by highly acidic (pH < 3) supergene fluids that evolved through the weathering of proximal sulfides. Acids expedited the dissolution of vein apatite and the liberation of phosphate into the weathering fluid, which caused immobilization of the REEs (likely sourced from primary moanzite, apatite and allanite) and their high-grade, in-situ accumulation through the precipitation of highly insoluble REE-phosphate minerals like rhabdophane, monazite and florencite.
These results demonstrate how acidic supergene fluids can facilitate either REE mobilization or accumulation in regolith, depending on the local mineralogy of the unweathered protolith and its capacity to provide depositional ligands. The main implications are that high-grade REE accumulation in regolith is more likely to occur close to the surface or in stratigraphic layers where the fluid can maintain a low pH, provided there is a notable source of depositional ligands (i.e., phosphate). Mining companies should therefore assay for REEs in rocks that are prone to phosphate mineralization and that may have undergone intense weathering. These conditions are likely to facilitate the formation of supergene phosphatic assemblages that make excellent geochemical traps for REE accumulation.
References
Andersen AK, Clark JG, Larson PB, Donovan JJ (2017) REE fractionation, mineral speciation, and supergene enrichment of the Bear Lodge carbonatites, Wyoming, USA. Ore Geol Rev 89:780–807. https://doi.org/10.1016/j.oregeorev.2017.06.025
Andersson SS, Wagner T, Jonsson E et al (2018) Origin of the high-temperature Olserum-Djupedal REE-phosphate mineralisation, SE Sweden: A unique contact metamorphic-hydrothermal system. Ore Geol Rev 101:740–764. https://doi.org/10.1016/j.oregeorev.2018.08.018
Andrieu S, Brigaud B, Barbarand J, Lasseur E (2018) The complex diagenetic history of discontinuities in shallow-marine carbonate rocks: New insights from high-resolution ion microprobe investigation of δ 18 O and δ 13 C of early cements. Sedimentology 65:360–399. https://doi.org/10.1111/sed.12384
Anenburg M, Williams MJ (2022) Quantifying the Tetrad Effect, Shape Components, and Ce–Eu–Gd Anomalies in Rare Earth Element Patterns. Math Geosci 54:47–70. https://doi.org/10.1007/s11004-021-09959-5
Anenburg M, Mavrogenes JA, Bennett VC (2020a) The Fluorapatite P-REE-Th Vein Deposit at Nolans Bore: Genesis by Carbonatite Metasomatism. J Petrol 61:egaa003. https://doi.org/10.1093/petrology/egaa003
Anenburg M, Mavrogenes JA, Frigo C, Wall F (2020b) Rare earth element mobility in and around carbonatites controlled by sodium, potassium, and silica. Sci Adv 6:eabb6570. https://doi.org/10.1126/sciadv.abb6570
Aubert D, Stille P, Probst A (2001) REE fractionation during granite weathering and removal by waters and suspended loads: Sr and Nd isotopic evidence. Geochim Cosmochim Acta 65:387–406. https://doi.org/10.1016/S0016-7037(00)00546-9
Balaram V (2019) Rare earth elements: a review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci Front 10:1285–1303. https://doi.org/10.1016/j.gsf.2018.12.005
Banfield JF, Eggleton RA (1989) Apatite replacement and rare earth mobilization, fractionation, and fixation during weathering. Clays Clay Miner 37:113–127. https://doi.org/10.1346/CCMN.1989.0370202
Baron D, Palmer CD (1996) Solubility of jarosite at 4–35°C. Geochim Cosmochim Acta 60:185–195. https://doi.org/10.1016/0016-7037(95)00392-4
Beard CD, Goodenough KM, Borst AM et al (2023) Alkaline-Silicate REE-HFSE Systems. Econ Geol 118:177–208. https://doi.org/10.5382/econgeo.4956
Bladh KW (1982) The formation of goethite, jarosite, and alunite during the weathering of sulfide-bearing felsic rocks. Econ Geol 77:176–184. https://doi.org/10.2113/gsecongeo.77.1.176
Borst AM, Smith MP, Finch AA et al (2020) Adsorption of rare earth elements in regolith-hosted clay deposits. Nat Commun 11:4386. https://doi.org/10.1038/s41467-020-17801-5
Braga JM, Biondi JC (2023) Geology, geochemistry, and mineralogy of saprolite and regolith ores with Nb, P, Ba, REEs (+ Fe) in mineral deposits from the Araxá alkali-carbonatitic complex, Minas Gerais state. Brazil. J South Am Earth Sci 125:104311. https://doi.org/10.1016/j.jsames.2023.104311
Braun JJ, Pagel M, Muller JP et al (1990) Cerium anomalies in lateritic profiles. Geochim Cosmochim Acta 54:781–795. https://doi.org/10.1016/0016-7037(90)90373-S
Brown JB (1971) Jarosite-geoethite stabilities at 25 °C, 1 ATM. Miner Depos 6:245–252. https://doi.org/10.1007/BF00208032
Brugger J, Ogierman J, Pring A et al (2006) Origin of the secondary REE -minerals at the Paratoo copper deposit near Yunta, South Australia. Mineral Mag 70:609–627. https://doi.org/10.1180/0026461067060361
Byrne RH, Kim KH (1990) Rare earth element scavenging in seawater. Geochim Cosmochim Acta 54:2645–2656. https://doi.org/10.1016/0016-7037(90)90002-3
Campbell LS, Henderson P (1997) Apatite paragenesis in the Bayan Obo REE-Nb-Fe ore deposit, Inner Mongolia, China. Lithos 42:89–103. https://doi.org/10.1016/s0024-4937(97)00038-8
Carstens JF, Bachmann J, Guggenberger G (2021) Aggregation and transport behavior of goethite colloids as affected by dissolved organic matter and pH: Electrostatic vs. hydrophilic interactions. Colloids Surf A Physicochem Eng Asp 609:125639. https://doi.org/10.1016/j.colsurfa.2020.125639
Celi L, Presta M, Ajmore-Marsan F, Barberis E (2001) Effects of pH and Electrolytes on Inositol Hexaphosphate Interaction with Goethite. Soil Sci Soc Am J 65:753–760. https://doi.org/10.2136/sssaj2001.653753x
Chapela Lara M, Buss HL, Pett-Ridge JC (2018) The effects of lithology on trace element and REE behavior during tropical weathering. Chem Geol 500:88–102. https://doi.org/10.1016/j.chemgeo.2018.09.024
Cook NJ, Ciobanu CL, Wade BP et al (2023) Mineralogy and distribution of REE in Oxidised Ores of the Mount Weld Laterite deposit, Western Australia. Minerals 13:656. https://doi.org/10.3390/min13050656
Coppin F, Berger G, Bauer A et al (2002) Sorption of lanthanides on smectite and kaolinite. Chem Geol 182:57–68. https://doi.org/10.1016/S0009-2541(01)00283-2
Das S, Hendry MJ, Essilfie-Dughan J (2011) Transformation of two-line ferrihydrite to goethite and hematite as a function of pH and temperature. Environ Sci Technol 45:268–275. https://doi.org/10.1021/es101903y
De Carlo EH, Wen XY, Irving M (1997) The influence of redox reactions on the uptake of dissolved Ce by suspended Fe and Mn oxide particles. Aquat Geochem 3:357–389. https://doi.org/10.1023/A:1009664626181
Dentith M, Stuart R (2003) Sediment-hosted stratiform copper deposits in the Adelaide Geosyncline, South Australia: geophysical responses of mineralisation and the mineralised environment. ASEG Extended Abstracts 2003:169–196. https://doi.org/10.1071/asegspec12_14
Ding P, Pacek AW (2008) Effect of pH on deagglomeration and rheology/morphology of aqueous suspensions of goethite nanopowder. J Colloid Interface Sci 325:165–172. https://doi.org/10.1016/j.jcis.2008.04.077
Dold B (2003) Dissolution kinetics of schwertmannite and ferrihydrite in oxidized mine samples and their detection by differential X-ray diffraction (DXRD). Appl Geochem 18:1531–1540. https://doi.org/10.1016/S0883-2927(03)00015-5
Donovan JJ, Lowers HA, Rusk BG (2011) Improved electron probe microanalysis of trace elements in quartz. American Mineralogist 96:274–282. https://doi.org/10.2138/am.2011.3631
Donovan JJ, Moy A, Von Der Handt A et al (2023) a new method for dead time calibration and a new expression for correction of WDS Intensities for microanalysis. Microscopy and Microanalysis 29:1096–1110. https://doi.org/10.1093/micmic/ozad050
Dostal J (2017) Rare earth element deposits of alkaline igneous rocks. Resources 6:34. https://doi.org/10.3390/resources6030034
Du Bernard X, Carrio-Schaffhauser E (2003) Kaolinitic meniscus bridges as an indicator of early diagenesis in Nubian sandstones, Sinai, Egypt. Sedimentology 50:1221–1229. https://doi.org/10.1111/j.1365-3091.2003.00602.x
Estrade G, Marquis E, Smith M et al (2019) REE concentration processes in ion adsorption deposits: evidence from the Ambohimirahavavy alkaline complex in Madagascar. Ore Geol Rev 112:103027. https://doi.org/10.1016/j.oregeorev.2019.103027
Etschmann BE, Donner E, Brugger J et al (2014) Speciation mapping of environmental samples using XANES imaging. Environ Chem 11:341–350. https://doi.org/10.1071/EN13189
Ferenc Š, Števko M, Mikuš T et al (2021) Primary minerals and age of the hydrothermal quartz veins containing u-mo-(Pb, bi, te) mineralization in the majerská valley near Čučma (gemeric unit, spišsko-gemerské rudohorie mts., slovak republic). Minerals 11:629. https://doi.org/10.3390/min11060629
Foden J, Sandiford M, Dougherty-Page J, Williams I (1999) Geochemistry and geochronology of the Rathjen Gneiss: Implications for the early tectonic evolution of the Delamerian Orogen. Aust J Earth Sci 46:377–389. https://doi.org/10.1046/j.1440-0952.1999.00712.x
Foden J, Elburg MA, Turner SP et al (2002) Granite production in the Delamerian Orogen, South Australia. J Geol Soc London 159:557–575. https://doi.org/10.1144/0016-764901-099
Foden J, Elburg MA, Dougherty-Page J, Burtt A (2006) The timing and duration of the Delamerian orogeny: correlation with the Ross Orogen and implications for Gondwana assembly. J Geol 114:189–210. https://doi.org/10.1086/499570
Foden J, Elburg M, Turner S et al (2020) Cambro-Ordovician magmatism in the Delamerian orogeny: implications for tectonic development of the southern Gondwanan margin. Gondwana Res 81:490–521. https://doi.org/10.1016/j.gr.2019.12.006
Gausse C, Szenknect S, Mesbah A et al (2018) Dissolution kinetics of monazite LnPO4 (Ln = La to Gd): A multiparametric study. Appl Geochem 93:81–93. https://doi.org/10.1016/j.apgeochem.2018.04.005
Geelhoed JS, Hiemstra T, Van Riemsdijk WH (1997) Phosphate and sulfate adsorption on goethite: Single anion and competitive adsorption. Geochim Cosmochim Acta 61:2389–2396. https://doi.org/10.1016/S0016-7037(97)00096-3
Goodenough KM, Schilling J, Jonsson E et al (2016) Europe’s rare earth element resource potential: an overview of REE metallogenetic provinces and their geodynamic setting. Ore Geol Rev 72:838–856. https://doi.org/10.1016/j.oregeorev.2015.09.019
Government of South Australia (2023) South Australian Resource Information Gateway (SARIG). Available at: https://map.sarig.sa.gov.au. Accessed 18 Oct 2023
Gražulis S, Chateigner D, Downs RT et al (2009) Crystallography Open Database - An open-access collection of crystal structures. J Appl Crystallogr 42:726–729. https://doi.org/10.1107/S0021889809016690
Grohol M, Veeh C (2023) Study on the critical raw materials for the EU 2023 – Final report, Publications Office of the European Union, 2023. https://doi.org/10.2873/725585
Guidry MW, Mackenzie FT (2003) Experimental study of igneous and sedimentary apatite dissolution: control of pH, distance from equilibrium, and temperature on dissolution rates. Geochim Cosmochim Acta 67:2949–2963. https://doi.org/10.1016/S0016-7037(03)00265-5
Gupta V, Hampton MA, Stokes JR et al (2011) Particle interactions in kaolinite suspensions and corresponding aggregate structures. J Colloid Interface Sci 359:95–103. https://doi.org/10.1016/j.jcis.2011.03.043
Habib K, Hansdóttir ST, Habib H (2020) Critical metals for electromobility: global demand scenarios for passenger vehicles, 2015–2050. Resour Conserv Recycl 154:104603. https://doi.org/10.1016/j.resconrec.2019.104603
Harouiya N, Chaïrat C, Köhler SJ et al (2007) The dissolution kinetics and apparent solubility of natural apatite in closed reactors at temperatures from 5 to 50 °C and pH from 1 to 6. Chem Geol 244:554–568. https://doi.org/10.1016/j.chemgeo.2007.07.011
Hayes SM, McCullough EA (2018) Critical minerals: a review of elemental trends in comprehensive criticality studies. Resources Policy 59:192–199. https://doi.org/10.1016/j.resourpol.2018.06.015
Hillgärtner H, Dupraz C, Hug W (2001) Microbially induced cementation of carbonate sands: are micritic meniscus cements good indicators of vadose diagenesis? Sedimentology 48:117–131. https://doi.org/10.1046/j.1365-3091.2001.00356.x
Howard DL, Jonge MDD, Afshar N et al (2020) The XFM beamline at the Australian Synchrotron. J Synchrotron Radiat 27:1447–1458. https://doi.org/10.1107/S1600577520010152
Hsu LC, Tzou YM, Ho MS et al (2020) Preferential phosphate sorption and Al substitution on goethite. Environ Sci Nano 7:3497–3508. https://doi.org/10.1039/c9en01435g
Huston DL, Maas R, Cross A et al (2016) The Nolans Bore rare-earth element-phosphorus-uranium mineral system: geology, origin and post-depositional modifications. Miner Depos 51:797–822. https://doi.org/10.1007/s00126-015-0631-y
Hutchinson M, Slezak P, Wendlandt R, Hitzman M (2022) Rare earth element enrichment in the weathering profile of the bull hill carbonatite at bear lodge, Wyoming, USA. Econ Geol 117:813–831. https://doi.org/10.5382/econgeo.4900
Ichimura K, Sanematsu K, Kon Y et al (2020) REE redistributions during granite weathering: Implications for Ce anomaly as a proxy for paleoredox states. American Mineralogist 105:848–859. https://doi.org/10.2138/am-2020-7148
Irving E, Irving GA (1982) Apparent polar wander paths carboniferous through cenozoic and the assembly of Gondwana. Geophysical Surveys 5:141–188. https://doi.org/10.1007/BF01453983
Jambor JL, Dutrizac JE (1998) Occurrence and constitution of natural and synthetic ferrihydrite, a widespread iron oxyhydroxide. Chem Rev 98:2549–2586. https://doi.org/10.1021/cr970105t
Jeong GY (1998) Vermicular kaolinite epitactic on primary phyllosilicates in the weathering profiles of anorthosite. Clays Clay Miner 46:509–520. https://doi.org/10.1346/ccmn.1998.0460504
Jeong GY (1998) Formation of vermicular kaolinite from halloysite aggregates in the weathering of plagioclase. Clays Clay Miner 46:270–279. https://doi.org/10.1346/ccmn.1998.0460306
Johannesson KH, Stetzenbach KJ, Hodge VF, Lyons WB (1996) Rare earth element complexation behavior in circumneutral pH groundwaters: assessing the role of carbonate and phosphate ions. Earth Planet Sci Lett 139:305–319. https://doi.org/10.1016/0012-821x(96)00016-7
Jowitt SM (2022) Mineral economics of the rare-earth elements. MRS Bull 47:276–282. https://doi.org/10.1557/s43577-022-00289-3
Jowitt SM, Wong VNL, Wilson SA, Gore O (2017) Critical metals in the critical zone: controls, resources and future prospectivity of regolith-hosted rare earth elements. Aust J Earth Sci 64:1045–1054. https://doi.org/10.1080/08120099.2017.1380701
Kalintsev A, Brugger J, Etschmann B, Ram R (2021) An in situ, micro-scale investigation of inorganically and organically driven rare-earth remobilisation during weathering. Mineral Mag 85:105–116. https://doi.org/10.1180/mgm.2021.4
Kartal M, Xia F, Ralph D et al (2020) Enhancing chalcopyrite leaching by tetrachloroethylene-assisted removal of sulphur passivation and the mechanism of jarosite formation. Hydrometallurgy 191:105192. https://doi.org/10.1016/j.hydromet.2019.105192
Kendall B, Creaser RA, Selby D (2006) Re-Os geochronology of postglacial black shales in Australia: constraints on the timing of “Sturtian” glaciation. Geology 34:729–732. https://doi.org/10.1130/G22775.1
Klimpel F, Bau M, Graupner T (2021) Potential of garnet sand as an unconventional resource of the critical high-technology metals scandium and rare earth elements. Sci Rep 11:5306. https://doi.org/10.1038/s41598-021-84614-x
Kretzschmar R, Holthoff H, Sticher H (1998) Influence of pH and humic acid on coagulation kinetics of kaolinite: a dynamic light scattering study. J Colloid Interface Sci 202:95–103. https://doi.org/10.1006/jcis.1998.5440
Lambert IB, Donnelly TH, Rowlands NJ (1980) Genesis of upper proterozoic stratabound copper mineralization, Kapunda, South Australia. Miner Depos 15:1–18. https://doi.org/10.1007/BF00202840
Le Bas MJ, Kellere J, Kejie T et al (1992) Carbonatite dykes at bayan Obo, inner Mongolia, China. Mineral Petrol 46:195–228. https://doi.org/10.1007/BF01164647
Lee S, Xu H (2019) One-step route synthesis of siliceous six-line ferrihydrite: implication for the formation of natural ferrihydrite. ACS Earth Space Chem 3:503–509. https://doi.org/10.1021/acsearthspacechem.8b00179
Lewicka E, Guzik K, Galos K (2021) On the possibilities of critical raw materials production from the eu’s primary sources. Resources 10:50. https://doi.org/10.3390/RESOURCES10050050
Leybourne MI, Johannesson KH (2008) Rare earth elements (REE) and yttrium in stream waters, stream sediments, and Fe-Mn oxyhydroxides: Fractionation, speciation, and controls over REE + Y patterns in the surface environment. Geochim Cosmochim Acta 72:5962–5983. https://doi.org/10.1016/j.gca.2008.09.022
Li YHM, Zhou MF (2020) The role of clay minerals in formation of the regolith-hosted heavy rare earth element deposits. Am Mineral 105:92–108. https://doi.org/10.2138/am-2020-7061
Li ZX, Powell CMA, Thrupp GA, Schmidt PW (1990) Australian Palaeozoic palaeomagnetism and tectonics-II. A revised apparent polar wander path and palaeogeography. J Struct Geol 12:567–575. https://doi.org/10.1016/0191-8141(90)90074-9
Li K, Etschmann B, Rae N et al (2016) Ore petrography using megapixel X-ray imaging: Rapid insights into element distribution and mobilization in complex Pt and U-Ge-Cu ores. Econ Geol 111:487–501. https://doi.org/10.2113/econgeo.111.2.487
Li X, Liang X, He H et al (2022a) Microorganisms Accelerate REE Mineralization in Supergene Environments. Appl Environ Microbiol 88:e00632-22. https://doi.org/10.1128/aem.00632-22
Li YHM, Kwong HT, Williams-Jones AE, Zhou MF (2022b) The thermodynamics of rare earth element liberation, mobilization and supergene enrichment during groundwater-regolith interaction. Geochim Cosmochim Acta 330:258–277. https://doi.org/10.1016/j.gca.2021.05.002
Lin P, Yang X, Werner JM, Honaker RQ (2021) Application of Eh-pH diagrams on acid leaching systems for the recovery of REEs from Bastnaesite, Monazite and Xenotime. Metals (Basel) 11:734. https://doi.org/10.3390/met11050734
Ling S, Wu X, Zhao S, Liao X (2018) Evolution of porosity and clay mineralogy associated with chemical weathering of black shale: A case study of Lower Cambrian black shale in Chongqing, China. J Geochem Explor 188:326–339. https://doi.org/10.1016/j.gexplo.2018.02.002
Liu W, Etschmann B, Migdisov A et al (2017) Revisiting the hydrothermal geochemistry of europium(II/III) in light of new in-situ XAS spectroscopy results. Chem Geol 459:61–74. https://doi.org/10.1016/j.chemgeo.2017.04.005
Liu H, Pourret O, Guo H et al (2018) Impact of hydrous manganese and ferric oxides on the behavior of aqueous rare earth elements (REE): Evidence from a modeling approach and implication for the sink of REE. Int J Environ Res Public Health 15:2837. https://doi.org/10.3390/ijerph15122837
Lottermoser BG (1988) A carbonatitic diatreme from Umberatana, South Australia. J - Geol Soc (London) 145:505–513. https://doi.org/10.1144/gsjgs.145.3.0505
Lottermoser BG (1990) Rare-earth element mineralisation within the Mt. Weld carbonatite laterite, Western Australia. Lithos 24:151–167. https://doi.org/10.1016/0024-4937(90)90022-S
Louvel M, Etschmann B, Guan Q et al (2022) Carbonate complexation enhances hydrothermal transport of rare earth elements in alkaline fluids. Nat Commun 13:1456. https://doi.org/10.1038/s41467-022-28943-z
Mahan KH, Wernicke BP, Jercinovic MJ (2010) Th-U-total Pb geochronology of authigenic monazite in the Adelaide rift complex, South Australia, and implications for the age of the type Sturtian and Marinoan glacial deposits. Earth Planet Sci Lett 289:76–86. https://doi.org/10.1016/j.epsl.2009.10.031
McKirdy DM, Sumartojo J, Tucker DH, Gostin V (1975) Organic, mineralogic and magnetic indications of metamorphism in the Tapley Hill Formation, Adelaide Geosyncline. Precambrian Res 2:345–373. https://doi.org/10.1016/0301-9268(75)90005-4
Migdisov A, Williams-Jones AE, Brugger J, Caporuscio FA (2016) Hydrothermal transport, deposition, and fractionation of the REE: experimental data and thermodynamic calculations. Chem Geol 439:13–42. https://doi.org/10.1016/j.chemgeo.2016.06.005
Milnes AR, Bourman RP, Northcote KH (1985) Field relationships of ferricretes and weathered zones in southern south australia: A contribution to ‘laterite’ studies in australia. Aust J Soil Res 23:441–465. https://doi.org/10.1071/SR9850441
Moldoveanu GA, Papangelakis VG (2016) An overview of rare-earth recovery by ion-exchange leaching from ion-adsorption clays of various origins. Min Mag 80:63–76. https://doi.org/10.1180/minmag.2016.080.051
Moore M, Chakhmouradian AR, Mariano AN, Sidhu R (2015) Evolution of rare-earth mineralization in the Bear Lodge carbonatite, Wyoming: mineralogical and isotopic evidence. Ore Geol Rev 64:499–521. https://doi.org/10.1016/j.oregeorev.2014.03.015
Moy A, Fournelle J, Nachlas W et al (2023) On the Importance of Including all elements in the EPMA matrix correction. Microsc Microanal 29:855–856. https://doi.org/10.1093/micmic/ozad067.424
Nelson BJ, Wood SA, Osiensky JL (2003) Partitioning of REE between solution and particulate matter in natural waters: a filtration study. J Solid State Chem 171:50–56. https://doi.org/10.1016/S0022-4596(02)00145-7
Neumann R, Medeiros EB (2015) Comprehensive mineralogical and technological characterisation of the Araxá (SE Brazil) complex REE (Nb-P) ore, and the fate of its processing. Int J Miner Process 144:1–10. https://doi.org/10.1016/j.minpro.2015.08.009
Norlund KLI, Baron C, Warren LA (2010) Jarosite formation by an AMD sulphide-oxidizing environmental enrichment: Implications for biomarkers on Mars. Chem Geol 275:235–242. https://doi.org/10.1016/j.chemgeo.2010.05.014
O’Neill HSC (2016) The smoothness and shapes of chondrite-normalized rare earth element patterns in basalts. J Petrol 57:1463–1508. https://doi.org/10.1093/petrology/egw047
Ondrejka M, Bačík P, Sobocký T et al (2018) Minerals of the rhabdophane group and the alunite supergroup in microgranite: products of low-temperature alteration in a highly acidic environment from the Velence Hills, Hungary. Mineral Mag 82:1277–1300. https://doi.org/10.1180/mgm.2018.137
Papoulis D, Tsolis-Katagas P, Katagas C (2004) Progressive stages in the formation of kaolin minerals of different morphologies in the weathering of plagioclase. Clays Clay Miner 52:275–286. https://doi.org/10.1346/CCMN.2004.0520303
Parfitt RL (1989) Phosphate reactions with natural allophane, ferrihydrite and goethite. J Soil Sci 40:359–369. https://doi.org/10.1111/j.1365-2389.1989.tb01280.x
Parfitt RL, Atkinson RJ (1976) Phosphate adsorption on goethite (α-FeOOOH). Nature 264:740–742. https://doi.org/10.1038/264740a0
Paton C, Hellstrom J, Paul B et al (2011) Iolite: Freeware for the visualisation and processing of mass spectrometric data. J Anal At Spectrom 26:2508–2518. https://doi.org/10.1039/c1ja10172b
Pedersen HD, Postma D, Jakobsen R, Larsen O (2005) Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II). Geochim Cosmochim Acta 69:3967–3977. https://doi.org/10.1016/j.gca.2005.03.016
Phillips-Lander CM, Legett C, Elwood Madden AS, Elwood Madden ME (2017) Can we use pyroxene weathering textures to interpret aqueous alteration conditions? Yes and No. Am Min 102:1915–1921. https://doi.org/10.2138/am-2017-6155
Preiss WV (2000) The Adelaide Geosyncline of South Australia and its significance in Neoproterozoic continental reconstruction. Precambrian Res 100:21–63. https://doi.org/10.1016/S0301-9268(99)00068-6
Pring A, Wallwork K, Brugger J, Kolitsch U (2006) Paratooite-(La), a new lanthanum-dominant rare-earth copper carbonate from Paratoo, South Australia. Min Mag 70:131–138. https://doi.org/10.1180/0026461067010319
Proust D, Caillaud J, Fontaine C (2006) Clay minerals in early amphibole weathering: Tri- to dioctahedral sequence as a function of crystallization sites in the amphibole. Clays Clay Miner 54:351–362. https://doi.org/10.1346/CCMN.2006.0540306
Quinn KA, Byrne RH, Schijf J (2006) Sorption of yttrium and rare earth elements by amorphous ferric hydroxide: Influence of pH and ionic strength. Mar Chem 99:128–150. https://doi.org/10.1016/j.marchem.2005.05.011
Ram R, Becker M, Brugger J et al (2019) Characterisation of a rare earth element- and zirconium-bearing ion-adsorption clay deposit in Madagascar. Chem Geol 522:93–107. https://doi.org/10.1016/j.chemgeo.2019.05.011
Ramanaidou E, Nahon D, Decarreau A, Melfi AJ (1996) Hematite and goethite from duricrusts developed by lateritic chemical weathering of precambrian banded iron formations, Minas Gerais, Brazil. Clays Clay Miner 44:22–31. https://doi.org/10.1346/CCMN.1996.0440102
Rebertus RA, Weed SB, Buol SW (1986) Transformations of Biotite to Kaolinite During Saprolite-Soil Weathering. Soil Sci Soc Am J 50:810–819. https://doi.org/10.2136/sssaj1986.03615995005000030049x
Ren Y, Yang X, Wang S, Öztürk H (2019) Mineralogical and geochemical study of apatite and dolomite from the Bayan Obo giant Fe-REE-Nb deposit in Inner Mongolia: New evidences for genesis. Ore Geol Rev 109:381–406. https://doi.org/10.1016/j.oregeorev.2019.04.020
Ryan CG (2000) Quantitative trace element imaging using PIXE and the nuclear microprobe. Int J Imaging Syst Technol 11:219–230. https://doi.org/10.1002/ima.1007
Ryan CG, Van Achterbergh E, Jamieson DN, Churms CL (1996) Overlap corrected on-line PIXE imaging using the proton microprobe. Nucl Instrum Methods Phys Res B 109–110:154–160. https://doi.org/10.1016/0168-583X(95)00898-5
Ryan CG, Kirkham R, Hougha RM et al (2010) Elemental X-ray imaging using the Maia detector array: The benefits and challenges of large solid-angle. Nucl Instrum Methods Phys Res A 619:37–43. https://doi.org/10.1016/j.nima.2009.11.035
Ryan CG, Siddons DP, Kirkham R et al (2014) Maia X-ray fluorescence imaging: capturing detail in complex natural samples. J Physics: Conf Series 499:012002. https://doi.org/10.1088/1742-6596/499/1/012002. IOP Publishing
Sanematsu K, Kon Y, Imai A (2015) Influence of phosphate on mobility and adsorption of REEs during weathering of granites in Thailand. J Asian Earth Sci 111:14–30. https://doi.org/10.1016/j.jseaes.2015.05.018
Savage KS, Tingle TN, O’Day PA et al (2000) Arsenic speciation in pyrite and secondary weathering phases, Mother Lode Gold District, Tuolumne County, California. Appl Geochem 15:1219–1244. https://doi.org/10.1016/S0883-2927(99)00115-8
Schaefer BF, Burgess JM (2003) Re-Os isotopic age constraints on deposition in the Neoproterozoic Amadeus Basin: Implications for the “Snowball Earth.” J Geol Soc London 160:825–828. https://doi.org/10.1144/0016-764903-050
Schoneveld L, Spandler C, Hussey K (2015) Genesis of the central zone of the Nolans Bore rare earth element deposit, Northern Territory, Australia. Contrib Mineral Petrol 170:1–22. https://doi.org/10.1007/s00410-015-1168-x
Schwertmann U, Murad E (1983) Effect of pH on the formation of goethite and hematite from ferrihydrite. Clays Clay Miner 31:277–284. https://doi.org/10.1346/CCMN.1983.0310405
Schwertmann U, Schulze DG, Murad E (1982) Identification of Ferrihydrite in Soils by Dissolution Kinetics, Differential X-ray Diffraction, and Mössbauer Spectroscopy. Soil Sci Soc Am J 46:869–875. https://doi.org/10.2136/sssaj1982.03615995004600040040x
Schwertmann U, Friedl J, Stanjek H (1999) From Fe(III) ions to ferrihydrite and then to hematite. J Colloid Interface Sci 209:215–223. https://doi.org/10.1006/jcis.1998.5899
Stille P, Pierret MC, Steinmann M et al (2009) Impact of atmospheric deposition, biogeochemical cycling and water-mineral interaction on REE fractionation in acidic surface soils and soil water (the Strengbach case). Chem Geol 264:173–186. https://doi.org/10.1016/j.chemgeo.2009.03.005
Strzelecki AC, Migdisov A, Boukhalfa H et al (2022) Fluocerite as a precursor to rare earth element fractionation in ore-forming systems. Nat Geosci 15:327–333. https://doi.org/10.1038/s41561-022-00921-6
Tanaka K, Ohta A, Kawabe I (2004) Experimental REE partitioning between calcite and aqueous solution at 25°C and 1 atm: Constraints on the incorporation of seawater REE into semount-type limestones. Geochem J 38:19–32. https://doi.org/10.2343/geochemj.38.19
Taylor G, Shirtliff G (2003) Weathering: Cyclical or continuous? An Australian perspective. Aust J Earth Sci 50:9–17. https://doi.org/10.1046/j.1440-0952.2003.00970.x
Tucker DH, Collerson KD (1972) Lamprophyric intrusions of probable carbonatitic affinity from South Australia. J Geol Soc Aust 19:387–391. https://doi.org/10.1080/00167617208728808
Turner S, Ireland T, Foden J et al (2022) A comparison of granite genesis in the adelaide fold belt and glenelg river complex using U-Pb, Hf and O isotopes in zircon. J Petrol 63:egac102. https://doi.org/10.1093/petrology/egac102
United States Geological Survey (USGS) (2022) Final list of critical minerals, 2022. https://www.federalregister.gov/documents/2022/02/24/2022-04027/2022-final-list-of-critical-minerals. Accessed 18 May 2024
Vaitkus A, Merkys A, Grazulis S (2021) Validation of the crystallography open database using the crystallographic information framework. J Appl Crystallogr 54:661–672. https://doi.org/10.1107/S1600576720016532
Vapnik Y, Bushmin S, Chattopadhyay A, Dolivo-Dobrovolsky D (2007) Fluid inclusion and mineralogical study of vein-type apatite ores in shear zones from the Singhbhum metallogenetic province, West Bengal, India. Ore Geol Rev 32:412–430. https://doi.org/10.1016/j.oregeorev.2006.11.002
Velbel MA (1989) Weathering of hornblende to ferruginous products by a dissolution-reprecipitation mechanism Petrography and stoichiometry. Clays Clay Miner 37:515–524. https://doi.org/10.1346/CCMN.1989.0370603
Velbel MA (2007) Chapter 4 surface textures and dissolution processes of heavy minerals in the sedimentary cycle: examples from pyroxenes and amphiboles. Dev Sedimentol 58:113–150. https://doi.org/10.1016/S0070-4571(07)58004-0
Verma S, Paul AR, Haque N (2022) Assessment of materials and rare earth metals demand for sustainable wind energy growth in India. Minerals 12:647. https://doi.org/10.3390/min12050647
Walsh JMJ, Spandler C (2023) The role of zircon in hydrothermal heavy REE mineralisation: the case for unconformity-related ore deposits of north-west Australia. Chem Geol 629:121493. https://doi.org/10.1016/j.chemgeo.2023.121493
Wang H, Xu C, Dowd PA et al (2022) Modelling in-situ recovery (ISR) of copper at the Kapunda mine, Australia. Miner Eng 186:107752. https://doi.org/10.1016/j.mineng.2022.107752
Wellman P, McElhinny MW, McDougall I (1969) On the Polar-Wander Path for Australia during the Cenozoic. Geophys J R Astron Soc 18:371–395. https://doi.org/10.1111/j.1365-246X.1969.tb03575.x
Wingate MTD, Campbell IH, Compston W, Gibson GM (1998) Ion microprobe U-Pb ages for Neoproterozoic basaltic magmatism in south-central Australia and implications for the breakup of Rodinia. Precambrian Res 87:135–159. https://doi.org/10.1016/s0301-9268(97)00072-7
Wood SA (1990) The aqueous geochemistry of the rare-earth elements and yttrium. 1. Review of available low-temperature data for inorganic complexes and the inorganic REE speciation of natural waters. Chem Geol 82:159–186. https://doi.org/10.1016/0009-2541(90)90080-Q
Wood SA, Williams-Jones AE (1994) The aqueous geochemistry of the rare-earth elements and yttrium 4. Monazite solubility and REE mobility in exhalative massive sulfide-depositing environments. Chem Geol 115:47–60. https://doi.org/10.1016/0009-2541(94)90144-9
Wu Z, Chen Y, Wang Y et al (2023) Review of rare earth element (REE) adsorption on and desorption from clay minerals: application to formation and mining of ion-adsorption REE deposits. Ore Geol Rev 157:105446. https://doi.org/10.1016/j.oregeorev.2023.105446
Xia F, Brugger J, Chen G et al (2009) Mechanism and kinetics of pseudomorphic mineral replacement reactions: a case study of the replacement of pentlandite by violarite. Geochim Cosmochim Acta 73:1945–1969. https://doi.org/10.1016/j.gca.2009.01.007
Xu N, Gao Y (2008) Characterization of hematite dissolution affected by oxalate coating, kinetics and pH. Appl Geochem 23:783–793. https://doi.org/10.1016/j.apgeochem.2007.12.026
Xu C, Taylor RN, Li W et al (2012) Comparison of fluorite geochemistry from REE deposits in the Panxi region and Bayan Obo, China. J Asian Earth Sci 57:76–89. https://doi.org/10.1016/j.jseaes.2012.06.007
Yang M, Liang X, Ma L et al (2019) Adsorption of REEs on kaolinite and halloysite: a link to the REE distribution on clays in the weathering crust of granite. Chem Geol 525:210–217. https://doi.org/10.1016/j.chemgeo.2019.07.024
Yao X, Xia F, Brugger J et al (2021) Rapid marcasite to pyrite transformation in acidic low-temperature hydrothermal fluids and saturation index control on FeS2Precipitation dynamics and phase selection. ACS Earth Space Chem 5:2453–2465. https://doi.org/10.1021/acsearthspacechem.1c00137
Yaxley GM, Anenburg M, Tappe S et al (2022) Carbonatites: classification, sources, evolution, and emplacement. Annu Rev Earth Planet Sci 50:261–293. https://doi.org/10.1146/annurev-earth-032320-104243
Zhu M, Frandsen C, Wallace AF et al (2016) Precipitation pathways for ferrihydrite formation in acidic solutions. Geochim Cosmochim Acta 172:247–264. https://doi.org/10.1016/j.gca.2015.09.015
Zhukova IA, Stepanov AS, Jiang SY et al (2021) Complex REE systematics of carbonatites and weathering products from uniquely rich Mount Weld REE deposit, Western Australia. Ore Geol Rev 139:104539. https://doi.org/10.1016/j.oregeorev.2021.104539
Zolotov MY, Shock EL (2005) Formation of jarosite-bearing deposits through aqueous oxidation of pyrite at Meridiani Planum, Mars. Geophys Res Lett 32. https://doi.org/10.1029/2005GL024253
Acknowledgements
The authors thank Leon Faulkner (Environmental Copper Ltd.) and Eric Whittaker (formerly of Terramin Australia Ltd.) for access to the Kapunda mine site and unpublished geochemical datasets. We thank Malcolm Roberts and Alexadra Suvorova at CMCA for their support in collecting EPMA and SEM data, respectively, as well as David J Paterson and Barbara Etschmann for their support in collecting XFM data. This work benefited from discussions with Allan Pring (Flinders University) and Sudeshna Baku (University College London). We recognise the Ngadjuri people as the traditional owners of the land from which the KV00X sample originated. Part of this work was undertaken by T. G. Bamforth as a dissertation component of a University College London/University of South Australia Global Management of Natural Resources Masters program. T.G.B acknowledges subsequent financial support from an ARC-funded scholarship, an International Tuition Fee Scholarship from Murdoch University, and a CSIRO top-up scholarship to support his PhD studies. Research activities were partly funded by an Australian Research Council Discovery Grant (DP200102284) and Australian Synchrotron beamtime award. ChatGPT was used sparingly to improve the readability of the Abstract. Lastly, we thank Karen Kelley for handing our manuscript and providing valuable feedback, as well as Michael Anenburg, one anonymous reviewer and one anonymous associate editor whose comments largely improved the quality of the manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
Tobias Bamforth: Conceptualisation, Investigation, Formal Analysis, Writing - Original Draft, Writing - Review & Editing, Visualization; Fang Xia: Conceptualisation, Resources, Validation, Writing – Review and Editing, Funding Acquisition, Supervision; Caroline Tiddy: Conceptualisation, Resources, Writing – Review and Editing, Supervision; Ignacio Gonzalez-Alvarez: Conceptualisation, Writing – Review and Editing, Supervision; Joel Brugger: Investigation, Formal Analysis, Writing - Review & Editing; Siyu Hu: Investigation, Writing - Review & Editing; Louise Shoneveld: Investigation, Data Curation, Writing - Review & Editing; Mark Pearce: Writing - Review & Editing, Supervision; Andrew Putnis: Funding Acquisition, Writing – Review and Editing, Supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Editorial handing: F. Melcher
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bamforth, T.G., Xia, F., Tiddy, C.J. et al. High-Grade REE accumulation in regolith: Insights from supergene alteration of an apatite-rich vein at the Kapunda Cu mine, South Australia. Miner Deposita (2024). https://doi.org/10.1007/s00126-024-01283-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00126-024-01283-2