
Abstract
Aims/hypothesis
We previously demonstrated that N-glycosylation of plasma proteins and IgGs is different in children with recent-onset type 1 diabetes compared with their healthy siblings. To search for genetic variants contributing to these changes, we undertook a genetic association study of the plasma protein and IgG N-glycome in type 1 diabetes.
Methods
A total of 1105 recent-onset type 1 diabetes patients from the Danish Registry of Childhood and Adolescent Diabetes were genotyped at 183,546 genetic markers, testing these for genetic association with variable levels of 24 IgG and 39 plasma protein N-glycan traits. In the follow-up study, significant associations were validated in 455 samples.
Results
This study confirmed previously known plasma protein and/or IgG N-glycosylation loci (candidate genes MGAT3, MGAT5 and ST6GAL1, encoding beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase, alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase and ST6 beta-galactoside alpha-2,6-sialyltransferase 1 gene, respectively) and identified novel associations that were not previously reported for the general European population. First, novel genetic associations of IgG-bound glycans were found with SNPs on chromosome 22 residing in two genomic intervals close to candidate gene MGAT3; these include core fucosylated digalactosylated disialylated IgG N-glycan with bisecting N-acetylglucosamine (GlcNAc) (pdiscovery=7.65 × 10−12, preplication=8.33 × 10−6 for the top associated SNP rs5757680) and core fucosylated digalactosylated glycan with bisecting GlcNAc (pdiscovery=2.88 × 10−10, preplication=3.03 × 10−3 for the top associated SNP rs137702). The most significant genetic associations of IgG-bound glycans were those with MGAT3. Second, two SNPs in high linkage disequilibrium (missense rs1047286 and synonymous rs2230203) located on chromosome 19 within the protein coding region of the complement C3 gene (C3) showed association with the oligomannose plasma protein N-glycan (pdiscovery=2.43 × 10−11, preplication=8.66 × 10−4 for the top associated SNP rs1047286).
Conclusions/interpretation
This study identified novel genetic associations driving the distinct N-glycosylation of plasma proteins and IgGs identified previously at type 1 diabetes onset. Our results highlight the importance of further exploring the potential role of N-glycosylation and its influence on complement activation and type 1 diabetes susceptibility.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Type 1 diabetes is a chronic disease that is characterised by the autoimmune destruction of insulin-producing pancreatic beta cells [1]. The number of children and adolescents diagnosed with type 1 diabetes has been increasing worldwide at an annual rate of about 3% [2]. Despite the identification of many genetic risk factors [3], the underlying causes of this disease remain unclear, and accumulating evidence suggests that environmental factors play an important role in the development of type 1 diabetes [4].
N-glycosylation is a diverse protein modification process by which complex oligosaccharide structures (glycans) are added to the protein backbone [5]. It is important to stress that glycosylation should not be confused with glycation, since glycosylation is a complex enzymatic process strictly regulated by a network of glycosyltransferases, glycosidases, transcriptional factors, sugar nucleotides and other molecules [6]. Glycation, on the other hand, is a non-enzymatic reaction between reducing sugar and protein, such as the one described for glycated haemoglobin [7]. N-glycosylation changes can influence protein function. For example, addition of sialic acid to the terminal end of N-glycan changes the function of antibodies of the IgG isotype from pro- to anti-inflammatory agents [8], whereas addition of bisecting N‑acetylglucosamine (GlcNAc) is associated with an increased ability of IgGs to destroy target cells through antibody-dependent cellular cytotoxicity [9]. Both the human plasma protein and IgG N-glycomes demonstrate remarkably low intra-individual variance under physiological conditions [10, 11], and at the same time are extremely sensitive to different pathological processes [12], thus supporting their diagnostic and prognostic potential. N-glycosylation changes have been described in various diseases, including type 1 diabetes and other diabetes types [12]. It has been shown that it is possible to distinguish HNF1A-maturity onset diabetes of the young from healthy controls and even from other diabetes types based on proportions of antennary fucose of plasma proteins, and recently that N-glycans with best diagnostic value mostly originate from alpha-1-acid glycoprotein [12, 13]. Remarkably, recent studies have even demonstrated that identification of individuals at an increased risk of future diabetes development is possible based on their N-glycan profiles [12].
Much evidence has been gathered regarding the role of highly branched N-glycans in autoimmunity in general, as well as in type 1 diabetes specifically [12]. Highly branched glycans present on proteins on cell surfaces are involved in interaction with galectins, and thus the formation of glycan–galectin lattices, resulting in increased protein retention time on the cell surface [14]. Defective N-glycosylation of T cells has been implicated in the pathogenesis of type 1 diabetes [14]. N-glycan branching increased the surface retention time of the T cell activation inhibitory glycoprotein CTLA-4 [15] encoded by the CTLA4 gene, which has been identified as one of the causal candidate genes in type 1 diabetes [16].
Genome-wide association studies (GWASs) of the plasma protein and IgG N-glycomes have identified glycosyltransferase loci, as well as loci containing genes that have not previously been shown to be associated with protein glycosylation [17, 18]. Some of these genes (for example IKZF1 and BACH2) have also been shown to be associated with various diseases, including type 1 diabetes [19]. Genetic studies identified a glycosyltransferase gene, FUT2, as one of the causal candidate genes in type 1 diabetes [20], with the possible mechanism involving host resistance to infections [21]. The glycosyltransferase loci MGAT5 (encoding alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase) and MGAT1 have been implicated in the pathogenesis of type 1 diabetes through N-glycan branching and its impact on T cell activation [14, 22]. Decreased expression of ST6GAL1 (encoding ST6 beta-galactoside alpha-2,6-sialyltransferase 1) in B cells has been shown to be associated with type 1 diabetes risk-associated alleles [23].
Protein glycosylation is a complex process that is regulated by a vast network of genes [6], many of which have still not been identified in humans, although they have been identified in a comprehensive study of mouse glycans [24]. We recently showed that plasma protein and IgG N-glycosylation differs between children with recent-onset type 1 diabetes and their healthy siblings [25], and is different from the N-glycan profile of adult type 1 diabetes patients with unregulated blood glucose [26]. In this study, we aimed to obtain knowledge of the genetic impact on the distinct plasma protein and IgG N-glycosylation that has been shown to accompany onset of type 1 diabetes [25], and identify type 1 diabetes risk-associated genes that regulate N-glycosylation. As far as we are aware, this is the first study to correlate genetic and N-glycome data in type 1 diabetes patients.
Methods
Ethics statement
The study was approved by Danish Ethical Committee KA-95139 m and the ethics committee of the University of Zagreb, Faculty of Pharmacy and Biochemistry. The study was performed in accordance with the Declaration of Helsinki. Informed consent was given by all the patients, their parents or guardians.
Study participants
The discovery study comprised 1105 children with new-onset type 1 diabetes (median age 10 years, range 1–18 years) whose plasma samples were collected within three months of type 1 diabetes diagnosis through the Danish Registry of Childhood and Adolescent Diabetes (DanDiabKids) [27]. The follow-up (validation) study comprised 190 children with recent-onset type 1 diabetes and 265 unaffected children from the same family-based DanDiabKids registry. Three of the childhood type 1 diabetes patients have one or more siblings within the control group. Details of the participants in this study are summarised in Table 1.
The inclusion criterion for unaffected children was that a sample from their biological sibling with type 1 diabetes was available in the registry. More than 95% of the sibling samples were collected at the same date as the proband sample, and the sampling dates are quite equally distributed over the year. The year of sampling for unaffected children ranged from 1997 to 2000, and the last registry data extraction and disease status check for unaffected children was performed in January 2019. At the last disease status check, it was established that two individuals had developed type 1 diabetes, one within nearly 6 years and other within 9 years. Some of the unaffected siblings were lost to follow-up if they were subsequently diagnosed at more than 18 years old, at which age they are often referred to adult type 1 diabetes clinics, and therefore their type 1 diabetes status is less certain than for those individuals who were followed for an extended time. However, as the incidence of type 1 diabetes after puberty decreases markedly with increasing age, it is less likely that the older individuals followed for a shorter period developed the disease [28]. Subsets of the cohorts collected through this registry have been used in a number of studies [29,30,31].
Discovery study
In the discovery study, 1105 samples from children with recent-onset type 1 diabetes were genotyped for 183,546 SNPs using Immunochip, a custom-made Infinium array (Illumina, USA), as described previously [32]. A total of 177,022 markers passed the initial sample quality control process, including sample call rate and a concordance check of reported sex vs genotyped sex. Additional quality control was performed by removing SNPs with a genotyping call rate <95% (5% missing) and a minor allele frequency <5%. In total, 108,428 SNPs passed the filtering criteria and were retained in the analysis. The mean genotyping rate in the participants was 99%. All the quality filtering steps were performed using PLINK version 1.07 [33]. To avoid missing true association signals, the SNPs were not filtered for deviations from the Hardy–Weinberg equilibrium, because disease association and population structure can cause deviations from the Hardy–Weinberg equilibrium [34]. The genotype-calling algorithms exported the allele calls aligned to the TOP strand.
After genotype quality control, data were analysed for associations between glycan proportions and individual SNPs genome-wide using the ‘qassoc’ function in PLINK [33], with a p value cut-off of 5 × 10−8. Genome-wide significance thresholds were further adjusted for 21 independent IgG glycan traits [19] (p≤2.4 × 10−9) and 39 plasma protein glycan traits (p≤1.3 × 10−9). Information on linkage disequilibrium (LD) was obtained using SNiPA [35] (SNiPA version 3.4 from November 2020, GRCh37.p13, Ensembl version 87, 1000 genomes phase 3, version 5, European).
Follow-up study
In the follow-up study used for validation, 21 SNPs revealed in the discovery phase were used for genotyping (see electronic supplementary material [ESM] Table 1). Samples from 455 individuals were genotyped using Kompetitive allele-specific PCR genotyping (KASP, LGC Genomics, UK). Of the 21 SNPs, 18 were successfully genotyped (rs137707, rs1047286 and rs137702 failed validation and were not analysed).
The genotype effect on glycan abundance was estimated by mixed modelling, with glycan abundance as the dependent variable, and genotype, disease status and interaction between disease status and genotype as independent variables. Sex and age were included as independent fixed variables, and family identifier was included as a random intercept [36]. The number of independent novel glycan–SNP combinations tested was used to adjust the significance threshold (0.05 for plasma protein glycans and 0.05/2 for IgG glycans).
N-glycome analysis
Briefly, a 10 μl aliquot of plasma was used for plasma protein N-glycome profiling, and 70 μl plasma was used to isolate IgG using a protein G monolithic plate (CIM Protein G 96-monolithic plate, BIA Separations, Slovenia) [37]. N-glycans were then enzymatically released and fluorescently labelled [38]. Hydrophilic interaction ultra-performance liquid chromatography was used to separate N-glycans [37]. Automated integration was applied to separate the chromatograms into 24 peaks for IgG N-glycans (IGP1–IGP24) and 39 peaks for plasma protein N-glycans (GP1–GP39) [39], and all these glycan traits were included in the genetic association analyses. The amount of glycan in each peak was expressed as a percentage of the total integrated area.
N-glycome data for these participants had been obtained previously [25], using 24 and five plates (batches) in the first and second parts of the study, respectively. The first part of the study included N-glycosylation analyses of 1917 children with new-onset type 1 diabetes. The second part of the study included 188 of the 1917 participants involved in the primary study and their 244 unaffected siblings. Within each study part, samples were randomised by age, sex and disease status, and standard and duplicated samples were added to each plate to minimise experimental error. CVs of the measured N-glycans among standard and duplicated samples are presented in ESM Table 2. In order to combine these two parts of the previous study, batch effects were removed using the ComBat method in the R package sva [40]. Data gathered from the samples included in both parts of the study were used for estimation of differences between the study parts. The effect was estimated by mixed modelling using the R package lme4 [36], in which logit-transformed glycan abundance was the dependent variable, while study part was a fixed factor and sample identifier was modelled as a random intercept. The estimated effect, known to originate for technical reasons, was removed from the data.
Results
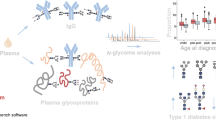
A genetic association study of the plasma protein and IgG N-glycome was performed using data from 1105 recent-onset type 1 diabetes childhood patients from the family-based DanDiabKids registry, who were genotyped at more than 183,000 genetic variants. Data for plasma protein N-glycans as well as those specifically on IgG (which are also represented within plasma protein N-glycans) were used herein [25]. The results are presented in Tables 2 and 3. We identified five genome-wide significant loci associated with plasma protein and/or IgG N-glycans; candidate genes include MGAT3 (encoding beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase), MGAT5, ST6GAL1 and C3 (encoding complement C3) (Fig. 1). Regional association plots are presented in ESM Figures 1–11. The pleiotropy of identified variants in terms of gene expression, protein expression and diseases is summarised in Table 4.

(a) Overview of significant genetic associations with plasma protein and IgG N-glycan proportions in individuals with recent-onset type 1 diabetes. Genes are shown grouped with their associated glycans. Glycans are shown as per GlycoWorkBench standard figures [59]. Glycans that showed novel genetic associations are indicated by a star and presented in colour. The red dotted circles indicate glycan residues that were synthesised by the action of the enzymes encoded by the associated genes. (b) Effect of the C3 missense variant rs1047286. This SNP results in a cyclic to acyclic amino acid substitution, and is associated with altered proportions of the oligomannose plasma protein glycan
Genetic association analysis of plasma protein N-glycans identified a novel locus
Genetic association analyses identified a novel N-glycosylation locus, the C3 gene. Two SNPs in high LD (missense rs1047286 and synonymous rs2230203) located on chromosome 19 within the protein coding region of the C3 gene showed significant genome-wide association with the oligomannose Man9 glycan (GP19) and explained 4% of the variance of the associated glycan. The A alleles of these two SNPs were associated with lower Man9 levels.
This study also confirmed previously reported associations for MGAT3 and MGAT5 [18]. GP6 variation (core fucosylated monogalactosylated glycan with bisecting GlcNAc) was associated with two SNPs located in the intergenic region on chromosome 22 (candidate gene MGAT3): rs5757678 and rs5757680. Also, rs2460382 within an intron of MGAT5 on chromosome 2 showed significant genome-wide association with triantennary trigalactosylated trisialylated plasma protein N-glycan (GP29) (p=3.14 × 10−11).
Novel IgG N-glycan associations uncovered for the N-glycosyltransferase MGAT3
Novel genetic associations of IgG-bound glycans were found with SNPs on chromosome 22; these reside in two genomic intervals defined by their LD (R2>0.5) with the top associated SNP within each interval. Associations within the first genomic interval were the strongest. The complete list of genetic markers that showed significant genome-wide association with IgG N-glycans in the discovery cohort is presented in ESM Table 3.
The most significant novel IgG glycan association was between IGP24 (percentage of core fucosylated biantennary digalactosylated disialylated glycan with bisecting GlcNAc) and two SNPs located in the intergenic region within the first associated genomic interval (candidate gene MGAT3). These SNPs explained 4% of the variance of the associated IGP24. In a previous GWAS on the general European population, IGP24 was exclusively associated with ST6GAL1 on chromosome 3 [17]. In the present study, the most significant genetic associations of IgG-bound glycans were with MGAT3, whereas the most significant genetic associations in the general European population were for ST6GAL1 [17].
Another novel association was found between IGP15 (percentage of core fucosylated biantennary digalactosylated glycan with bisecting GlcNAc) and SNPs within the second genomic interval. The most significantly associated SNP was rs137702 (p=2.88 × 10−10), which resides within an intron of the synaptogyrin 1 gene (SYNGR1, a candidate gene bisecting GlcNAc transferase MGAT3), and explained 4% of the variance of the associated IGP15. IGP15 was associated with MGAT3 in the general European population, but not with this particular genetic interval [17].
This study also confirmed other previously reported IgG N-glycan associations for MGAT3 and ST6GAL1 [17]. In summary, SNPs within the candidate MGAT3 gene were associated with core fucosylated glycans with bisecting GlcNAc in one direction, and core fucosylated digalactosylated disialylated glycan without bisecting GlcNAc in the opposite direction. The most significantly associated SNP was rs5757680 (p=2.12 × 10−26). Two SNPs within an intron of ST6GAL1 showed significant genome-wide association with IGP16, corresponding to the IgG-attached monosialylated N-glycan. The most significant SNP in this region was rs3872724 (p=1.81 × 10−22).
Discussion
This study analysed the genetic impact on distinct plasma protein and IgG N-glycomes accompanying type 1 diabetes onset that we described previously [25]. Within a cohort of 1105 recent-onset type 1 diabetes patients, associations that were not previously reported for the general European population were found. The N-glycosyltransferase gene MGAT3 showed novel association with core fucosylated biantennary digalactosylated disialylated IgG N-glycan with bisecting GlcNAc (IGP24), which was previously exclusively associated with sialyltransferase ST6GAL1 [17], and the new MGAT3 genetic interval was associated with core fucosylated digalactosylated IgG N-glycan with bisecting GlcNAc (IGP15). MGAT3 showed the strongest IgG N-glycan association, which was reported for ST6GAL1 in the general European population [17]. A novel locus influencing plasma protein N-glycosylation was also identified, the C3 gene on chromosome 19. Other previously known associations with plasma protein and IgG N-glycosylation were corroborated (candidate genes MGAT3, MGAT5 and ST6GAL1) [17, 18]. These novel genetic associations were replicated in the follow-up cohort.
C3 encodes the complement component C3, a pivotal protein of all three complement activation pathways that are responsible for host defence against micro-organisms and clearance of self and non-self targets, among other important immune functions [42]. The A alleles of two SNPs in high LD with each other (R2=0.85) within the exons of the C3 were associated with lower proportions of oligomannose Man9 glycan of plasma proteins. These SNPs were a non-synonymous SNP (rs1047286) causing a pPro314Leu substitution and a synonymous SNP (rs2230203). Using the publicly available dataset from GWAS of plasma protein N-glycans in the general European population [43, 44] we found that the same Man9 glycan was associated with rs2230203 (p=1.33 × 10−3), but did not reach genome-wide significance in that cohort, which may mean that it has a bigger effect in type 1 diabetes. Another C3 SNP, rs2230199, was previously shown to be associated with an increased risk of type 1 diabetes among HLA-DR4/4 carriers, one of the highest risk genotypes associated with type 1 diabetes [41,45]. This SNP is in high LD with both C3 SNPs identified in this study (R2=0.82 with rs1047286; R2=0.7 with rs2230203), supporting the significance of our finding.
The association between C3 and plasma protein Man9 may be specifically due to the C3 protein among other plasma proteins, as the Man9 glycan is attached on the C3 surface [46]. The A allele of rs1047286 causes a cyclic to acyclic amino acid substitution within C3 that may increase the accessibility for enzymatic processing of Man9, decreasing its levels. As the Man9 glycan is attached to the domain of C3 that is involved in pathogen binding [47], the alterations may be important for complement activation among carriers of the rs1047286 and rs2230203 A alleles. It has been shown previously that activity of the complement activation alternative pathway was higher among individuals with the rs1047286 A allele [48].
IgG N-glycans with bisecting GlcNAc (IGP24 and IGP15) showed novel associations with SNPs on chromosome 22 located in the intergenic regions or in the introns of the SYNGR1 gene, and close to the N-glycosyltransferase gene MGAT3. As MGAT3 encodes an enzyme that is responsible for addition of bisecting GlcNAc [49], it is the most biologically plausible candidate for these associations. IGP24 was previously shown to be exclusively associated with the sialyltransferase gene ST6GAL1 [17]. MGAT3 showed the strongest IgG N-glycan association in this study, which was reported for ST6GAL1 in the general European population [17]. IGP15 has been previously shown to be associated with MGAT3, but not with this particular MGAT3 genetic interval [17]. Interestingly, the minor alleles of the novel implicated SNPs show pleiotropy with increased expression of soluble glycoprotein 130 (IL-6 receptor subunit β) in plasma [50]. Soluble glycoprotein 130 inhibits IL-6 trans-signalling [51], whereas enhanced T cell responses to IL-6 in type 1 diabetes were shown to be associated with early clinical disease [52].
Increased MGAT3 expression in whole blood and specifically in cell types relevant for IgG biosynthesis (B cells) has been previously shown to be associated with the relevant alleles of SNPs characterised here and associated with increased IGP24 and IGP15 proportions [50, 53]. In our previous intra-family study of these recent-onset type 1 diabetes patients, IgG N-glycans with bisecting GlcNAc were significantly increased in the type 1 diabetes group compared with their healthy siblings, and, of all tested N-glycans, that corresponding to IGP24 differed most significantly between the studied groups [25]. In addition, decreased ST6GAL1 expression in B cells has been associated with type 1 diabetes risk-associated alleles [23]. The altered expression of MGAT3 and ST6GAL1 in tissues relevant for IgG biosynthesis may be the explanation for the observed associations. IgGs with bisecting GlcNAc are involved in increased antibody-dependent cellular cytotoxicity [9], an important process during elimination of viruses, and it has been suggested that one of the autoimmunity triggers in type 1 diabetes may be virus-derived [54].
The sialyltransferase gene ST6GAL1 was associated with monosialylated IgG N-glycan, and the same SNP–glycan association has been identified previously [17]. In our previous family-based study [25], there was no significant difference in the proportion of ST6GAL1-associated IgG glycan between children with type 1 diabetes and their unaffected siblings. However, proportions of disialylated IgGs increased, whereas those of asialylated IgGs decreased, in the participants with recent-onset type 1 diabetes. The increase in disialylated IgGs was mainly driven by IGP24, which was shown here to be regulated by MGAT3 instead of ST6GAL1.
The same sialyltransferase ST6GAL1 SNPs regulated FA2BG2 (IGP15) and FA2BG2S2 (IGP24) glycan proportions in opposite directions within the general European population [17]. FA2BG2 is a core fucosylated digalactosylated glycan with bisecting GlcNAc, and is considered as a substrate for a subsequent addition of sialic acids (thus the synthesis of core fucosylated digalactosylated disialylated glycan with bisecting GlcNAc/FA2BG2S2) in the current standard glycosylation pathway, which implies different subcellular localisation of glycosyltransferases and thus specific order of glycan reactions [55]. Recent in silico and in vitro experiments contradicted previous knowledge and showed that certain glycosyltransferases co-localise across the Golgi and that certain reactions outside the standard pathway may occur [56]. However, disialylated glycans were not measured in these experiments, and thus these reactions could not be predicted [56]. Within the type 1 diabetes population of the present study, the same bisecting GlcNAc-transferase MGAT3 SNPs influenced FA2G2S2 (IGP23; core fucosylated digalactosylated disialylated glycan) and FA2BG2S2 (IGP24) glycan proportions in opposite directions, suggesting that bisecting GlcNAc may be added after the addition of sialic acid.
It has been demonstrated that hyposialylated IgGs activate the endothelial IgG receptor Fcγ receptor IIB (FcγRIIB), resulting in insulin resistance, whereas restored sialylation of IgGs maintained insulin sensitivity [57]. Also, once sialylated, IgG antibodies exert anti-inflammatory properties [8]. The role of these changes in the pathogenesis of type 1 diabetes should be further explored.
There is much evidence in the literature for the role of MGAT5 in autoimmunity, and in type 1 diabetes specifically [12]. MGAT5, which encodes an enzyme responsible for formation of highly branched glycans, was associated with the triantennary GP29 glycan. In previously characterised cohorts, MGAT5 was associated with triantennary glycans in addition to tetra-antennary ones [18]. It has been demonstrated that mammalian N-glycan branching protects against innate immune self-attack in autoimmune pathogenesis [14]. Furthermore, the presence of the highly branched N-glycan produced by the enzyme encoded by MGAT5 restricts T cell activation, and MGAT5-deficient mice exhibit several autoimmune phenotypes [14]. Interestingly, in our previous intra-family study [25], levels of the associated GP29 N-glycan were significantly increased in children with recent-onset type 1 diabetes relative to their unaffected siblings.
Plasma samples used in the study were collected within 3 months of disease diagnosis, and we acknowledge the fact that the temporal order of changes in glycans and diabetes development cannot be inferred. Other studies of N-glycosylation changes before development of autoimmunity or clinical disease are needed to elucidate whether the observed changes are associated with type 1 diabetes development. However, our previous study did identify significant N-glycosylation differences between these recent-onset type 1 diabetes patients and their healthy siblings [25], and novel genetic associations with these markers were found in the present study. Additionally, some of the identified variants were in high LD with another variant previously associated with an increased risk of developing type 1 diabetes [41]. We were not able to standardise the glycan data against medication intake as data on the treatment of study participants were not available. Nevertheless, our previous study also demonstrated that insulin has a low effect on a limited number of glycans [58], which do not include those glycans identified herein as novel associations. Also, this study comprises children at the onset of type 1 diabetes, without the comorbidities seen in the adult population, and glycan changes related to type 1 diabetes could therefore be investigated more precisely. A potential drawback of our study is that we did not have access to a replication cohort from another population. However, we tested SNP–glycan associations from the discovery phase on additional samples from the same registry and validated the novel associations. Other study limitations include the small sample size and use of a genotyping chip that does not cover the whole genome, and thus depends on initial GWASs for variant selection. Future larger-scale studies, identification of potentially causal/functional variants at the identified loci, replication and functional studies are needed to corroborate our findings. It should also be noted that the observed changes may be relevant for other autoimmune disorders rather than being specific to type 1 diabetes, and this should also be addressed in future studies.
In summary, this study on recent-onset type 1 diabetes patients identified associations that were not previously reported for the general European population. Novel associations with IgG N-glycans were uncovered for variants located on chromosome 22. These variants are located near the N-glycosyltransferase gene MGAT3, show pleiotropy with MGAT3 expression in whole blood and specifically in cell types relevant for IgG biosynthesis, and their associated IgG glycans with bisecting GlcNAc were significantly different between the recent-onset type 1 diabetes patients and their healthy siblings. This study also identified a novel genetic locus associated with plasma protein N-glycosylation, the C3 gene locus. C3 variants identified in this study are located in the coding region, and the associated Man9 glycan is attached on a domain that is involved in pathogen binding of the complement component C3 [47], thus the influence of this alteration on complement activation in type 1 diabetes presents an interesting target for future studies. Additionally, the identified C3 variants were in high LD with another type 1 diabetes risk-associated variant. These findings suggest the need for further studies of N-glycosylation mechanisms regulating type 1 diabetes susceptibility. We would like to highlight the importance of further exploring gene-specific polymorphisms and their associated N-glycosylation changes, as such study may reveal underlying molecular mechanisms, which are still unknown for many identified type 1 diabetes risk-associated SNPs.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- C3 :
-
Complement C3 gene
- GlcNAc:
-
N-acetylglucosamine
- GP:
-
Plasma protein glycan peak
- GWAS:
-
Genome-wide association study
- IGP:
-
IgG glycan peak
- LD:
-
Linkage disequilibrium
References
Mathis D, Vence L, Benoist C (2001) β-Cell death during progression to diabetes. Nature 414(6865):792–798. https://doi.org/10.1038/414792a
Tuomilehto J, Ogle GD, Lund-Blix NA, Stene LC (2020) Update on worldwide trends in occurrence of childhood type 1 diabetes in 2020. Pediatr Endocrinol Rev 17(Suppl 1):198–209. https://doi.org/10.17458/per.vol17.2020.tol.epidemiologychildtype1diabetes
Morahan G (2012) Insights into type 1 diabetes provided by genetic analyses. Curr Opin Endocrinol Diabetes Obes 19(4):263–270. https://doi.org/10.1097/MED.0b013e328355b7fe
Rewers M, Ludvigsson J (2016) Environmental risk factors for type 1 diabetes. Lancet 387(10035):2340–2348. https://doi.org/10.1016/S0140-6736(16)30507-4
Varki A, Cummings RD, Esko JD et al (eds) (2009) Essentials of glycobiology. Cold Spring Harbor Laboratory Press. Available from https://www.ncbi.nlm.nih.gov/books/NBK1908/
Lauc G, Rudan I, Campbell H, Rudd PM (2010) Complex genetic regulation of protein glycosylation. Mol Biosyst 6(2):329–335. https://doi.org/10.1039/B910377E
Rahbar S (2005) The discovery of glycated hemoglobin: a major event in the study of nonenzymatic chemistry in biological systems. Ann NY Acad Sci 1043:9–19. https://doi.org/10.1196/annals.1333.002
Anthony RM, Ravetch JV (2010) A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol 30(Suppl 1):S9–S14. https://doi.org/10.1007/s10875-010-9405-6
Davies J, Jiang L, Pan LZ, LaBarre MJ, Anderson D, Reff M (2001) Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FCγRIII. Biotechnol Bioeng 74(4):288–294. https://doi.org/10.1002/bit.1119
Gornik O, Wagner J, Pucić M, Knezević A, Redzic I, Lauc G (2009) Stability of N-glycan profiles in human plasma. Glycobiology 19(12):1547–1553. https://doi.org/10.1093/glycob/cwp134
Novokmet M, Lukić E, Vučković F et al (2014) Changes in IgG and total plasma protein glycomes in acute systemic inflammation. Sci Rep 4:4347. https://doi.org/10.1038/srep04347
Rudman N, Gornik O, Lauc G (2019) Altered N-glycosylation profiles as potential biomarkers and drug targets in diabetes. FEBS Lett 593(13):1598–1615. https://doi.org/10.1002/1873-3468.13495
Tijardović M, Štambuk T, Juszczak A et al (2022) Fucosylated AGP glycopeptides as biomarkers of HNF1A-Maturity onset diabetes of the young. Diabetes Res Clin Pract 185:109226. https://doi.org/10.1016/j.diabres.2022.109226
Demetriou M, Granovsky M, Quaggin S, Dennis JW (2001) Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409(6821):733–739. https://doi.org/10.1038/35055582
Lau KS, Partridge EA, Grigorian A et al (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129(1):123–134. https://doi.org/10.1016/j.cell.2007.01.049
Nisticò L, Buzzetti R, Pritchard LE et al (1996) The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Hum Mol Genet 5(7):1075–1080. https://doi.org/10.1093/hmg/5.7.1075
Klarić L, Tsepilov YA, Stanton CM et al (2020) Glycosylation of immunoglobulin G is regulated by a large network of genes pleiotropic with inflammatory diseases. Sci Adv 6(8):eaax0301. https://doi.org/10.1126/sciadv.aax0301
Sharapov SZ, Shadrina AS, Tsepilov YA et al (2021) Replication of 15 loci involved in human plasma protein N-glycosylation in 4802 samples from four cohorts. Glycobiology 31(2):82–88. https://doi.org/10.1093/glycob/cwaa053
Lauc G, Huffman JE, Pučić M et al (2013) Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLOS Genet 9(1):e1003225. https://doi.org/10.1371/journal.pgen.1003225
Onengut-Gumuscu S, Chen WM, Burren O et al (2015) Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 47(4):381–386. https://doi.org/10.1038/ng.3245
Smyth DJ, Cooper JD, Howson JMM et al (2011) FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes 60(11):3081–3084. https://doi.org/10.2337/db11-0638
Yu Z, Li CF, Mkhikian H, Zhou RW, Newton BL, Demetriou M (2014) Family studies of type 1 diabetes reveal additive and epistatic effects between MGAT1 and three other polymorphisms. Genes Immun 15(4):218–223. https://doi.org/10.1038/gene.2014.7
Ram R, Mehta M, Nguyen QT et al (2016) Systematic evaluation of genes and genetic variants associated with type 1 diabetes susceptibility. J Immunol 196(7):3043–3053. https://doi.org/10.4049/jimmunol.1502056
Krištić J, Zaytseva OO, Ram R et al (2018) Profiling and genetic control of the murine immunoglobulin G glycome. Nat Chem Biol 14(5):516–524. https://doi.org/10.1038/s41589-018-0034-3
Rudman N, Kifer D, Kaur S et al (2022) Children at onset of type 1 diabetes show altered N-glycosylation of plasma proteins and IgG. Diabetologia 65(8):1315–1327. https://doi.org/10.1007/s00125-022-05703-8
Bermingham ML, Colombo M, McGurnaghan SJ et al (2018) N-glycan profile and kidney disease in type 1 diabetes. Diabetes Care 41(1):79–87. https://doi.org/10.2337/dc17-1042
Svensson J, Cerqueira C, Kjærsgaard P et al (2016) Danish registry of childhood and adolescent diabetes. Clin Epidemiol 8:679–683. https://doi.org/10.2147/CLEP.S99469
Ostman J, Lönnberg G, Arnqvist HJ et al (2008) Gender differences and temporal variation in the incidence of type 1 diabetes: results of 8012 cases in the nationwide diabetes incidence study in Sweden 1983-2002. J Intern Med 263(4):386–394. https://doi.org/10.1111/j.1365-2796.2007.01896.x
Sorensen JS, Birkebaek NH, Bjerre M et al (2015) Residual β-cell function and the insulin-like growth factor system in Danish children and adolescents with type 1 diabetes. J Clin Endocrinol Metab 100(3):1053–1061. https://doi.org/10.1210/jc.2014-3521
Thorsen SU, Pipper CB, Mortensen HB et al (2017) Levels of soluble TREM-1 in children with newly diagnosed type 1 diabetes and their siblings without type 1 diabetes: a Danish case-control study. Pediatr Diabetes 18(8):749–754. https://doi.org/10.1111/pedi.12464
Thorsen SU, Pipper CB, Mortensen HB, Pociot F, Johannesen J, Svensson J (2016) No Contribution of GAD-65 and IA-2 Autoantibodies around Time of Diagnosis to the Increasing Incidence of Juvenile Type 1 Diabetes: A 9-Year Nationwide Danish Study. Int J Endocrinol 2016:8350158. https://doi.org/10.1155/2016/8350158
Brorsson CA, Onengut S, Chen WM et al (2015) Novel association between immune-mediated susceptibility loci and persistent autoantibody positivity in type 1 diabetes. Diabetes 64(8):3017–3027. https://doi.org/10.2337/db14-1730
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. https://doi.org/10.1086/519795
Wittke-Thompson JK, Pluzhnikov A, Cox NJ (2005) Rational inferences about departures from Hardy–Weinberg equilibrium. Am J Hum Genet 76(6):967–986. https://doi.org/10.1086/430507
Arnold M, Raffler J, Pfeufer A, Suhre K, Kastenmüller G (2015) SNiPA: an interactive, genetic variant-centered annotation browser. Bioinforma 31(8):1334–1336. https://doi.org/10.1093/bioinformatics/btu779
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
Pučić M, Knezević A, Vidic J et al (2011) High throughput isolation and glycosylation analysis of IgG-variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics 10(10):M111.010090. https://doi.org/10.1074/mcp.M111.010090
Akmačić IT, Ugrina I, Štambuk J et al (2015) High-throughput glycomics: optimization of sample preparation. Biochemistry (Mosc.) 80(7):934–942. https://doi.org/10.1134/S0006297915070123
Agakova A, Vučković F, Klarić L, Lauc G, Agakov F (2017) Automated integration of a UPLC glycomic profile. Methods Mol Biol 1503:217–233. https://doi.org/10.1007/978-1-4939-6493-2_17
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6):882–883. https://doi.org/10.1093/bioinformatics/bts034
Törn C, Liu X, Hagopian W et al (2016) Complement gene variants in relation to autoantibodies to beta cell specific antigens and type 1 diabetes in the TEDDY Study. Sci Rep 6:27887. https://doi.org/10.1038/srep27887
Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11(9):785–797. https://doi.org/10.1038/ni.1923
Huffman J, Hayward C (2017) Dataset pertaining to the publication ‘Polymorphisms in B3GAT1, SLC9A9 and MGAT5 are associated with variation within the human plasma N-glycome of 3533 European adults’. Available from https://datashare.is.ed.ac.uk/handle/10283/2759. Accessed 13 Jun 2018
Huffman JE, Knezevic A, Vitart V et al (2011) Polymorphisms in B3GAT1, SLC9A9 and MGAT5 are associated with variation within the human plasma N-glycome of 3533 European adults. Hum Mol Genet 20(24):5000–5011. https://doi.org/10.1093/hmg/ddr414
Erlich H, Valdes AM, Noble J et al (2008) HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57(4):1084–1092. https://doi.org/10.2337/db07-1331
Ritchie GE, Moffatt BE, Sim RB, Morgan BP, Dwek RA, Rudd PM (2002) Glycosylation and the complement system. Chem Rev 102(2):305–319. https://doi.org/10.1021/cr990294a
Janssen BJC, Huizinga EG, Raaijmakers HCA et al (2005) Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 437(7058):505–511. https://doi.org/10.1038/nature04005
Abrera-Abeleda MA, Nishimura C, Frees K et al (2011) Allelic variants of complement genes associated with dense deposit disease. J Am Soc Nephrol 22(8):1551–1559. https://doi.org/10.1681/ASN.2010080795
Taniguchi N, Kizuka Y (2015) Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res 126:11–51. https://doi.org/10.1016/bs.acr.2014.11.001
Pietzner M, Wheeler E, Carrasco-Zanini J et al (2020) Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat Commun 11(1):6397. https://doi.org/10.1038/s41467-020-19996-z
Jostock T, Müllberg J, Ozbek S et al (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268(1):160–167. https://doi.org/10.1046/j.1432-1327.2001.01867.x
Hundhausen C, Roth A, Whalen E et al (2016) Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci Transl Med 8(356):356ra119. https://doi.org/10.1126/scitranslmed.aad9943
Fairfax BP, Makino S, Radhakrishnan J et al (2012) Genetics of gene expression in primary immune cells identifies cell-specific master regulators and roles of HLA alleles. Nat Genet 44(5):502–510. https://doi.org/10.1038/ng.2205
Knip M, Simell O (2012) Environmental triggers of type 1 diabetes. Cold Spring Harb Perspect Med 2(7):a007690. https://doi.org/10.1101/cshperspect.a007690
Berger EG, Thurnher M, Müller U (1987) Galactosyltransferase and sialyltransferase are located in different subcellular compartments in HeLa cells. Exp Cell Res 173(1):267–273. https://doi.org/10.1016/0014-4827(87)90352-1
Benedetti E, Pučić-Baković M, Keser T et al (2017) Network inference from glycoproteomics data reveals new reactions in the IgG glycosylation pathway. Nat Commun 8(1):1483. https://doi.org/10.1038/s41467-017-01525-0
Tanigaki K, Sacharidou A, Peng J et al (2018) Hyposialylated IgG activates endothelial IgG receptor FcγRIIB to promote obesity-induced insulin resistance. J Clin Invest 128(1):309–322. https://doi.org/10.1172/JCI89333
Knezević A, Polasek O, Gornik O et al (2009) Variability, heritability and environmental determinants of human plasma N-glycome. J Proteome Res 8(2):694–701. https://doi.org/10.1021/pr800737u
Ceroni A, Maass K, Geyer H et al (2008) GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res 7(4):1650–1659. https://doi.org/10.1021/pr7008252
Acknowledgements
The authors are grateful for and would like to acknowledge the invaluable contributions of all the participants, research nurses, local investigators, administrative teams and other clinical staff. Some of the data were presented as an abstract and/or oral and poster presentation at the Virtual Congress of Croatian Pharmacy and Medical Biochemistry Students’ Association (CPSA) and Portuguese Pharmacy Students’ Association (APEF) ‘Understanding genetics – the key to a healthier GENEration’ in 2022, at the meeting of PhD candidates organised by the Croatian Science Foundation (PhD Caffe #14 meeting) in 2022, at the Congress of the Croatian Society of Biochemistry and Molecular Biology (HDMBM) HDBMB22: From Science to Knowledge in 2022, at the 55th EASD Annual Meeting in 2019, in Glycoconjugate Journal preceding the GLYCO-25 International Symposium on Glycoconjugates in 2019, at the Infoday of Postgraduate Doctoral Study ‘Pharmaceutical-Biochemical Sciences’ of University of Zagreb, Faculty of Pharmacy and Biochemistry in 2019, and at the 29th Joint Glycobiology meeting in 2018, the 2nd GlycoCom in 2018, the 1st Human Glycome Project Meeting in 2018 and the 28th Joint Glycobiology Meeting in 2017.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
OG and GM conceived, designed and supervised the study. NR, SK, VS, DK, DŠ, TK, TŠ, LK and FP contributed to the data collection, acquisition or analysis, and interpretation of data. NR, GM and OG wrote the manuscript. All authors reviewed the manuscript and approved the final version of the manuscript. OG and GM are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The study was supported by the Croatian National Science Foundation, grant agreement number UIP-2014-09-7769 (to OG). The DanDiabKids biobank is supported by a grant from the Danish Diabetes Association. GM’s laboratory is supported by the Western Australia Diabetes Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 1.01 MB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rudman, N., Kaur, S., Simunović, V. et al. Integrated glycomics and genetics analyses reveal a potential role for N-glycosylation of plasma proteins and IgGs, as well as the complement system, in the development of type 1 diabetes. Diabetologia 66, 1071–1083 (2023). https://doi.org/10.1007/s00125-023-05881-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-05881-z