
Abstract
Aims/hypothesis
The aim of this study was to determine the mechanism(s) for hypoglycaemia occurring late following oral glucose loading in patients with cystic fibrosis (CF).
Methods
A 3 h 75 g OGTT was performed in 27 non-diabetic adults with CF who were classified based on this test as experiencing hypoglycaemia (glucose <3.3 mmol/l with or without symptoms or glucose <3.9 mmol/l with symptoms, n = 14) or not (n = 13). Beta cell function, incretin (glucagon-like peptide-1 [GLP-1] and glucose-dependent insulinotropic peptide [GIP]) and counterregulatory hormone responses (glucagon, catecholamines, growth hormone and cortisol) were assessed.
Results
The two groups did not differ in age, weight or BMI. There were more male participants and individuals with pancreatic exocrine insufficiency in the hypoglycaemia group. Fasting plasma glucose did not differ between the two groups (5.3 ± 0.16 vs 5.3 ± 0.10 mmol/l). Both fasting insulin (20.7 ± 2.9 vs 36.5 ± 4.8 pmol/l; p = 0.009) and C-peptide (0.38 ± 0.03 vs 0.56 ± 0.05 nmol/l; p = 0.002) were lower in those who experienced hypoglycaemia. Following glucose ingestion, glucose concentrations were significantly lower in the hypoglycaemia group from 135 min onwards, with a nadir of 3.2 ± 0.2 vs 4.8 ± 0.3 mmol/l at 180 min (p < 0.001). The test was terminated early in three participants because of a glucose level <2.5 mmol/l. Insulin and C-peptide concentrations were also lower in the hypoglycaemia group, while incretin hormone responses were not different. Modelling demonstrated that those experiencing hypoglycaemia were more insulin sensitive (439 ± 17.3 vs 398 ± 13.1 ml min−1 m−2, p = 0.074 based on values until 120 min [n = 14]; 512 ± 18.9 vs 438 ± 15.5 ml min−1 m−2, p = 0.006 based on values until 180 min [n = 11]). In line with their better insulin sensitivity, those experiencing hypoglycaemia had lower insulin secretion rates (ISRfasting: 50.8 ± 3.2 vs 74.0 ± 5.9 pmol min−1 m−2, p = 0.002; ISROGTT: 44.9 ± 5.0 vs 63.4 ± 5.2 nmol/m2, p = 0.018) and beta cell glucose sensitivity (47.4 ± 4.5 vs 79.2 ± 7.5 pmol min−1 m−2 [mmol/l]−1, p = 0.001). Despite the difference in glucose concentrations, there were no significant increases in glucagon, noradrenaline, cortisol or growth hormone levels. Adrenaline increased by only 66% and 61% above baseline at 165 and 180 min when glucose concentrations were 3.8 ± 0.2 and 3.2 ± 0.2 mmol/l, respectively.
Conclusions/interpretation
Hypoglycaemia occurring late during an OGTT in people with CF was not associated with the expected counterregulatory hormone response, which may be a consequence of more advanced pancreatic dysfunction/destruction.
Similar content being viewed by others


Introduction
Improved survival of individuals with cystic fibrosis (CF) has resulted in the emergence of additional consequences and complications of the disease [1]. One of these is disturbances of glucose metabolism, which can result in age-dependent elevations in glucose concentrations. The hyperglycaemia associated with CF manifests as impaired glucose tolerance (IGT) in 10–20% of patients dependent on age, and CF-related diabetes (CFRD) in nearly half of patients aged 40 to 50 [1].
A different disturbance of glucose metabolism has also become apparent in people with CF. Both youth and adults have been noted to experience hypoglycaemia following oral glucose loading, the prevalence varying from 7% to 30% depending in part on the definition of low glucose and the duration of the test [2,3,4,5]. The development of hypoglycaemia during an OGTT is not predictable and can contribute to anxiety and fear and discourage patients from undergoing the recommended screening for diabetes. In line with this latter possibility, we recently reported in an unselected cohort of individuals attending the CF clinic at the University of Washington who underwent a 3 h OGTT that hypoglycaemia occurred in more than 60% of adults without CFRD who had a routine OGTT as part of their clinical care [6].
On the basis on these observations, we performed an exploratory, cross-sectional study seeking a mechanistic explanation for this hypoglycaemia. We hypothesised that the post-glucose load hypoglycaemia observed in individuals with CF results from an insulin response that is exaggerated and delayed and/or a deficient counterregulatory hormone response. We further postulated that any alterations in islet function that underlie the development of hypoglycaemia late during an OGTT could be due to or accentuated by alterations in incretin hormone release. To test this hypothesis with adequate sampling and duration to detect differences, we performed a frequently sampled, 3 h OGTT in adult patients with CF without CFRD and compared beta cell function, counterregulatory responses (comprised of glucagon, catecholamines, growth hormone and cortisol) and responses of the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) in those who did or did not experience hypoglycaemia.
Methods
Participants
Study participants were recruited from the Cystic Fibrosis Clinic at the University of Washington Medical Center, Seattle, WA, USA. Individuals were identified by review of the electronic medical record and offered the opportunity to participate. To be eligible, they needed to be >18 years of age, clinically stable with no history of CFRD, i.e. have either normal glucose tolerance or IGT at 2 h on an OGTT (see Procedures below) [7, 8], and not have taken antibiotics for pulmonary exacerbation for at least 2 weeks prior to being studied. Individuals who were pregnant, using intravenous antibiotics or systemic corticosteroids, had known liver or chronic kidney disease, hematocrit <30% in the prior 12 months or a history of Burkholderia cepacia genomovar III infection in the prior 24 months were not eligible. The study was approved by the University of Washington Human Subjects Review Committee and all participants gave written informed consent prior to being studied.
Hypoglycaemia was defined as either: (1) a glucose concentration <3.3 mmol/l at any time during the OGTT with or without symptoms of hypoglycaemia; or (2) symptoms of hypoglycaemia with a glucose concentration <3.9 mmol/l and an unwillingness to continue the test. Autonomic and neuroglycopenic symptoms were considered indicative of hypoglycaemia. Participants rated these symptoms on an in-house questionnaire using a seven-point Likert scale. The glucose criteria for hypoglycaemia were similar to those defined by the International Hypoglycaemia Study Group [9]; however they were not identical as the current study was commenced in July 2015, prior to publication of these recommendations.
Procedures
After a minimum 10 h overnight fast, eligible participants were admitted to the Clinical Research Center where they underwent a 3 h, 75 g OGTT. An indwelling catheter for repeated blood sampling was placed in a forearm vein and kept patent by a slow infusion of normal saline (154 mmol/l NaCl). A minimum of 15 min transpired between placing the catheter and drawing of the first sample. Samples for subsequent assay were drawn at −10 and −5 min prior to commencing glucose ingestion. Thereafter, additional blood samples were drawn 10, 20, 30, 60, 90, 105, 120, 135, 150, 165 and 180 min after beginning consumption of the glucose. Capillary blood glucose monitoring was performed if participants developed symptoms of hypoglycaemia. Sampling was discontinued if the glucose level fell below 2.5 mmol/l or if the glucose fell below 3.9 mmol/l with the development of symptoms of hypoglycaemia incompatible with further testing.
Assays
All blood samples, except for those used for capillary blood glucose monitoring, were drawn on EDTA. For the catecholamine samples, glutathione was included as a preservative, while for glucagon a protease inhibitor was added to the collection tubes. All samples were immediately placed on ice and then separated and frozen at −80°C prior to shipment to the respective laboratories for analyses.
Plasma samples for glucose, insulin, C-peptide, glucagon, GLP-1 and GIP were assayed at the University of Washington Northwest Lipid and Diabetes Research Laboratories. Glucose was measured by the glucose hexokinase method using Roche reagent on a Roche c501 autoanalyser (Roche Diagnostics, Indianapolis, IN, USA). Insulin and C-peptide were measured by a two site immuno-enzymometric assay performed on the Tosoh 2000 autoanalyser (Tosoh Biosciences, South San Francisco, CA, USA). A radioimmunoassay was used to measure glucagon (Millipore Sigma, Burlington, MA, USA; GL-32K). ELISAs were used to measure total GLP-1 (ALPCO, Salem, NH, USA; 43-GPTHU-E-01) and total GIP (Millipore Sigma; EZHGIP-54K).
Cortisol and growth hormone were assayed in serum using one-step competitive and one-step immunoenzymatic assays, respectively (Beckman Coulter, Brea, CA, USA). Catecholamines (adrenaline [epinephrine] and noradrenaline [norepinephrine]) were measured by HPLC (Bio-Rad Laboratories, Hercules, CA, USA). The minimum detection limit of both the adrenaline and noradrenaline assays was 110 pmol/l. All these assays were performed at the Clinical and Translational Research Centers (CTRC) Laboratory at the University of Colorado, Denver, CO, USA. A number of samples for adrenaline were reported to have interfering substances that rendered the results unreliable. Thus, where residual samples were available, adrenaline was re-measured at the Mayo Clinic Laboratories (Rochester, MN, USA) by HPLC following chromatographic purification [10]. Additional information regarding these samples and the results are provided in electronic supplementary material (ESM) Fig. 1.
Calculations
The two samples drawn prior to glucose ingestion were averaged. Insulin sensitivity was estimated as the inverse of fasting insulin [11]. The early insulin response (insulinogenic index) was calculated as the ratio of the incremental insulin and glucose responses from 0 to 30 min of the OGTT (∆I0–30/∆G0–30). To account for the modulating effect of insulin sensitivity on this response and thereby provide a measure of beta cell function, we calculated the oral disposition index (DIo) [12]. The incremental AUC (iAUC) above baseline was calculated using the trapezoidal method. In instances where there were decrements below baseline in the response, the iAUC represents the net of the increment plus the decrement.
Modelling
Glucose and C-peptide data were further analysed by deconvolution using the Mari model to quantify parameters of beta cell function dependent on the insulin secretion rate (ISR) [13]. The first model-derived beta cell component represents the dependence of ISR on glucose concentration through a dose–response function relating the two variables. From this dose–response relationship, the slope is calculated and represents beta cell glucose sensitivity. The dose–response is modulated by a time-varying potentiation factor to explain observed insulin secretion that cannot be accounted for as a function of glucose alone and provides a quantification of relative potentiation/inhibition of ISR over the course of the OGTT. This factor was calculated as the ratio between the ISR values from 100–120 min and 0–20 min. The second beta cell component quantified by the model is known as rate sensitivity and represents the dependence of ISR on the rate of change of glucose concentration.
Insulin sensitivity was also quantified by modelling of OGTT glucose and insulin data [14].
Data management and analyses
Three participants in the hypoglycaemia group and one in the no hypoglycaemia group were found to have interfering substances in the adrenaline assay. Thus, their adrenaline data were excluded from the analyses.
As the study was specifically designed to employ 3 h OGTTs, all available values are presented except for those in which data were missing for the three individuals in the hypoglycaemia group who had a truncated OGTT.
All statistical analyses were performed using Stata (vs 12.1, College Station, TX, USA). Data are presented as mean±SE. Comparisons between the two groups were computed using Students t tests or χ2 tests. Multiple regression analysis was used to examine change in peptide concentrations over time while accounting for multiple measurements per participant using generalised estimating equation modelling and a robust variance estimator. Differences in slope of these changes by development of hypoglycaemia was assessed by interaction terms between time × substrate inserted into regression models. Nominal p values are presented. Except where noted, a p < 0.05 was considered statistically significant, with no adjustments made for multiple comparisons.
Results
Demographic, physical and glucose tolerance characteristics
Of the 205 individuals whose electronic medical records were screened, 101 were ineligible as they had known CFRD. An additional 68 individuals either did not meet study inclusion criteria or declined to participate. Thus, 36 individuals with CF agreed to participate and underwent the 3 h OGTT. As we had predefined that we would only study individuals known not to have CFRD, those nine individuals whose 2 h glucose level on the OGTT was compatible with a diagnosis of diabetes were not included in subsequent analyses. Table 1 shows the characteristics of the remaining 27 individuals, 14 of whom became hypoglycaemic and 13 of whom did not.
The two groups were similar with regard to age, weight and BMI. The number of male and female participants was similar, but more men had hypoglycaemia. The hypoglycaemia group comprised more individuals who were homozygous for the F508 deletion and all individuals who experienced hypoglycaemia had pancreatic exocrine insufficiency, compared with 62% who did not experience hypoglycaemia. There was no difference in pulmonary function between the two groups as assessed by predicted forced expiratory volume in the first second (FEV1). The prevalence of Pseudomonas aeruginosa positivity also did not differ between the two groups.
With regard to glucose metabolism, the two groups had similar fasting plasma glucose concentrations. Both fasting plasma insulin and C-peptide concentrations were significantly lower in those with hypoglycaemia.
Glucose, insulin and C-peptide responses during the OGTT
In response to oral glucose loading, there was a prompt increase in glucose in both groups (Fig. 1a). Until 90 min, glucose concentrations were generally lower in the group that did not experience hypoglycaemia, after which they were higher in this group. At 135 min, glucose was significantly lower in the hypoglycaemia group (5.7 ± 0.5 vs 7.4 ± 0.1 mmol/l, p = 0.019). Three individuals did not complete the OGTT, investigators stopping two at 135 min and another at 165 min owing to a capillary glucose level below 2.5 mmol/l. Five individuals experienced symptoms of hypoglycaemia, all of whom completed the OGTT. One participant asked to discontinue the test because of symptoms of hypoglycaemia just prior to the final sample being drawn when their capillary glucose was 3.4 mmol/l. At the final 180 min time point, the glucose level in the 11 participants who experienced hypoglycaemia and completed the test (with or without symptoms) was 3.2 ± 0.2 vs 4.8 ± 0.3 mmol/l (p < 0.001) in those without hypoglycaemia. In the 11 participants with hypoglycaemia who completed the 3 h OGTT and the 13 without hypoglycaemia, the iAUC glucose was not significantly different (Table 2).

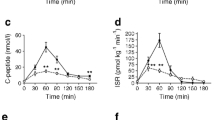
Plasma glucose (a), insulin (b), C-peptide (c) and glucagon (d) concentrations during the 3 h OGTT in 14 participants who experienced hypoglycaemia late in the test (blue circles and solid line) and 13 participants who did not (red squares and dashed line). *p < 0.05 for concentrations that differed between the two groups at the indicated time points. Data are presented as mean ± SE
Following glucose ingestion there was a rapid increase in the concentrations of both insulin and C-peptide, which remained greater in the non-hypoglycaemia group for most of the duration of the test (Fig. 1b, c). The early insulin response (∆I0–30/∆G0–30) was lower in the hypoglycaemia vs the non-hypoglycaemia group (26.9 ± 5.7 vs 65.0 ± 14.6 pmol/mmol, p = 0.019). Similarly, the iAUCs over the 3 h for insulin and C-peptide were lower in those with hypoglycaemia (Table 2). When this insulin response was adjusted for the glucose stimulus as the ratio of iAUCinsulin/iAUCglucose, there was no difference between those with hypoglycaemia (57.4 ± 10.5 pmol/mmol) and those without (85.6 ± 12.8 pmol/mmol, p = 0.109 between groups). Similarly, the ratio of iAUCC-peptide and iAUCglucose was not different in those who did and did not experience hypoglycaemia (0.55 ± 0.08 vs 0.66 ± 0.08 nmol/mmol, p = 0.328).
Insulin sensitivity and beta cell function
Model-based insulin sensitivity was numerically higher in those with hypoglycaemia when data up to 120 min were modelled, but this did not reach statistical significance (439 ± 17.3 vs 398 ± 13.1 ml min−1 m−2, p = 0.074). When the additional time points to 180 min were included in those who completed the OGTT, those with hypoglycaemia were more insulin sensitive than those without (512 ± 18.9 vs 438 ± 15.5 ml min−1 m−2, p = 0.006).
Using output from the model, a number of beta cell parameters were compared. The ISR at fasting and during the OGTT were both lower in the hypoglycaemia group (ISRfasting 50.8 ± 3.2 vs 74.0 ± 5.9 pmol min−1 m−2, p = 0.002; ISROGTT 44.9 ± 5.0 vs 63.4 ± 5.2 nmol/m2, p = 0.018). These values are in keeping with the pattern of lower calculated ISRs throughout the OGTT in the hypoglycaemia group (Fig. 2a). The dose–response of the relationship of ISR and plasma glucose concentration in the two groups is illustrated in Fig. 2b. This dose–response relationship, also known as beta cell glucose sensitivity, was lower in the hypoglycaemia group (47.4 ± 4.5 vs 79.2 ± 7.5 pmol min−1 m−2 [mmol/l]−1, p = 0.001). While the ISR was significantly lower in the hypoglycaemia group when the glucose during the OGTT was 8.0 mmol/l or higher (p < 0.05), it was also significantly higher at a glucose concentration of 4.6 mmol/l down to the lowest glucose level (3.8 mmol/l) achieved by the non-hypoglycaemia group (p < 0.05). Rate sensitivity was numerically lower in those with hypoglycaemia compared with those without, but this did not reach statistical significance (263 ± 85 vs 573 ± 133 pmol m−2 [mmol/l]−1, p = 0.059). The potentiation factor differed between the two groups being greater in the hypoglycaemia group (2.46 ± 0.24 vs 1.43 ± 0.20, p = 0.003).

Model-derived ISR during the OGTT (a) and the dose–response relationship between plasma glucose concentration during the OGTT and the model-derived ISR across the whole glucose range (b). The hypoglycaemia group is illustrated in blue and the non-hypoglycaemia group in red. In the hypoglycaemia group, compared with the non-hypoglycaemia group, the ISR was significantly lower at 8.0 mmol/l glucose and above and significantly higher at 4.6 mmol/l and lower (all p < 0.05). The slope of the line describing this relationship differed between those who did and did not develop hypoglycaemia (p = 0.001). Data are presented as mean ± SE
As beta cell function is represented by insulin responses adjusted for the prevailing insulin sensitivity, using the known hyperbolic relationship between 1/fasting insulin (as a surrogate of insulin sensitivity) and the early insulin response over 30 min in the OGTT [12], we calculated beta cell function as DIo. This measure was not different in those with and without hypoglycaemia (1.31 ± 0.21 vs 1.75 ± 0.23 (mmol/l)−1, p = 0.159). We also explored the role of insulin sensitivity to modulate the five model-derived beta cell measures by adjusting these measures for model-based insulin sensitivity (excluding the three participants in the hypoglycaemia group with a truncated OGTT). In these analyses, only beta cell glucose sensitivity remained significantly different between the groups after this adjustment (p = 0.032).
Incretin hormone responses
We hypothesised that hypoglycaemia late in the OGTT may be due to an enhanced incretin response that would increase the beta cell response and thereby glucose disposal. However, despite the insulin and C-peptide responses being lower in those with hypoglycaemia, the profiles of the GLP-1 and GIP responses during the OGTT did not differ between the two groups (Fig. 3). Neither fasting concentrations of the two incretins nor iAUCs for GLP-1 and GIP differed between the two groups (Table 2). The GLP-1 data exclude one participant in the non-hypoglycaemia group who had GLP-1 concentrations that were more than three SDs greater than the mean. Inclusion of these data did not change the findings, i.e. GLP-1 responses were not different between those who did and did not experience hypoglycaemia.

Plasma GLP-1 (a) and GIP (b) concentrations during the 3 h OGTT in 14 participants who experienced hypoglycaemia late in the test (blue circles) and 13 participants who did not (red circles). Data are presented as mean ± SE
Counterregulatory hormone responses
We assessed the responses of glucagon, adrenaline, noradrenaline, cortisol and growth hormone in the two groups to determine whether hypoglycaemia was associated with the expected counterregulation. Glucagon was sampled at all time points during the OGTT, while the other counterregulatory hormones were sampled at baseline and from 60 min onwards.
For the alpha cell peptide glucagon, fasting concentrations were not different between the two groups (Table 2). The glucagon profiles following glucose ingestion are illustrated in Fig. 1d. With the increase in glucose, plasma glucagon concentrations significantly declined in both groups over time (p < 0.001), not differing by occurrence of hypoglycaemia (p = 0.486). At 180 min, the glucagon concentrations in the hypoglycaemia and non-hypoglycaemia groups were 68.7 ± 5.0 and 69.1 ± 4.3 ng/l, respectively (p = 0.958). No difference in glucagon concentration was observed in the hypoglycaemia and non-hypoglycaemia groups after adjustment for 180 min glucose concentration by multiple regression analysis (β = 1.51, p = 0.867). In the hypoglycaemia group, the glucagon concentration at 180 min was 82% of that prior to glucose ingestion. In further support of a lack of a glucagon response in the hypoglycaemia group, the iAUC was not different between the two groups (Table 2).
The adrenaline and noradrenaline responses are illustrated in Fig. 4 a and b, respectively. The adrenaline data exclude the four participants considered to have substances interfering in the assay (three from the hypoglycaemia and one from the non-hypoglycaemia group; the individual data for all participants are illustrated in ESM Fig. 1). The adrenaline profile highlights that the concentrations in those with hypoglycaemia were on average numerically higher, with only values at 165 and 180 min being significantly higher (p = 0.019 and p = 0.012, respectively). The values at these two times were 66% and 61% greater than baseline in those with hypoglycaemia, at a time when their glucose levels were 3.8 ± 0.2 and 3.2 ± 0.2 mmol/l, respectively. In the participants without hypoglycaemia, the adrenaline responses were 22% and 7% lower than baseline at these same two time points. Noradrenaline concentrations did not differ between the two groups, with no evidence of an increase in response to hypoglycaemia and similar iAUCs (Table 2).

Plasma adrenaline (a), noradrenaline (b), cortisol (c) and growth hormone (d) concentrations during the 3 h OGTT in 14 participants who experienced hypoglycaemia late in the test (blue circles and solid line) and 13 participants who did not (red squares and dashed line). Owing to interfering substances in the adrenaline assay, data are presented for 11 participants who experienced hypoglycaemia and 12 participants who did not. *p < 0.05 for concentrations that differed between the two groups at the indicated time points. Data are presented as mean ± SE
There were no differences in the responses of either cortisol or growth hormone between the groups (Fig. 4c, d). With glucose administration, plasma growth hormone concentrations declined significantly in both groups over time (p = 0.008), not differing between the groups (p = 0.469). However, there was a suggestion in five participants with hypoglycaemia that the growth hormone response was increasing, resulting in a small, but not significant, increase in the mean growth hormone concentrations in the hypoglycaemia group during the last 15 min of the test. When considering the integrated concentrations over the full duration of the OGTT, neither cortisol nor growth hormone differed between the two groups (Table 2).
Discussion
Advances in the treatment of CF have increased longevity, resulting in the unveiling of abnormalities in glucose tolerance [1]. In addition to CFRD, postprandial hypoglycaemia is being recognised as a relatively common occurrence, particularly in those who do not have CFRD [2, 3]. This particular abnormality in glucose metabolism in CF has not been well studied and why it occurs is unclear. A recent editorial suggested ‘There is clear need for more investigation into the underlying pathophysiology of hypoglycaemia and low glucose in cystic fibrosis.’ [15]. Here we demonstrate for the first time to our knowledge that these individuals lack an adequate counterregulatory response to hypoglycaemia and that this failure appears to affect all components, with the exception of adrenaline. Further, we found no evidence that hypoglycaemia occurred because of alterations in beta cell function that would result in increased insulin release shortly after glucose was ingested or as a delayed insulin response.
The counterregulatory response to hypoglycaemia has been well studied under experimental clamp conditions with insulin-induced hypoglycaemia [16, 17]. Using this approach, it has been shown that the increase in glucose production that occurs with insulin-induced hypoglycaemia is dependent on intact glucagon secretion, but not that of growth hormone, and that adrenergic mechanisms responsible for the sympathoadrenal response are critical for recovery when the glucagon response is impaired. It is also well recognised that in diabetes recurrent episodes of hypoglycaemia are associated with a reduced counterregulatory response to hypoglycaemia, a condition commonly known as hypoglycaemia-associated autonomic failure (HAAF) [18]. This abnormality in counterregulation occurs when there is a lack of glucagon response, due to an islet abnormality, and an attenuated sympathoadrenal response, frequently the result of recent, antecedent hypoglycaemia. The lack of the sympathoadrenal response, which appears to result from changes within the central nervous system or its afferent or efferent connections, is also felt to be responsible for the lack of hypoglycaemic symptoms. In our participants, nine did not experience symptoms despite glucose reaching concentrations below 3.3 mmol/l. Further, given the minimal increase in adrenaline and lack of a noradrenaline response, it is quite possible that recurrent hypoglycaemia with unawareness occurs in individuals such as those we studied. Clearly, this area needs further exploration.
We can hypothesise some potential mechanisms at the islet level that could explain our observation of a lack of the critical glucagon response to hypoglycaemia. First, pancreatic insufficiency in CF is associated with functional abnormalities of the beta, alpha and pancreatic polypeptide cells; however, this has typically been examined in those with abnormal glucose tolerance [19,20,21]. In one of these studies, in a cohort of CF patients of whom 66% had abnormal glucose tolerance (either IGT or diabetes), exocrine insufficiency was associated with an impaired glucagon response to insulin-induced hypoglycaemia but a normal recovery from hypoglycaemia [20]. More recently, in those with normal glucose tolerance, pancreatic exocrine insufficiency was associated with both reduced insulin secretion and abnormal regulation of alpha cell function, which included reduced glucagon secretion in response to arginine injection [22]. In our cohort, all who developed hypoglycaemia had pancreatic exocrine insufficiency, compared with 62% who did not experience hypoglycaemia late in the OGTT. When we compared only those with pancreatic insufficiency, our findings were not different (data not shown). Second, it is well recognised that CF is associated with morphological abnormalities in the pancreas, particularly at the islet level. In fact, in a recent study using human autopsy samples, distinct abnormalities were already present in individuals without known diabetes [23]. These changes included increased islet glucagon and IL-1β immunoreactivity, both of which were also present in those with diabetes. Thus, it is of interest that, despite presumed adequate glucagon stores, we failed to observe a glucagon response in our participants. As the glucagon response to hypoglycaemia appears to also be critically dependent on islet innervation and the resultant release of noradrenaline [24], it is possible that the morphological changes occurring in CF include destruction of the sympathetic nerve endings. Third, it has been demonstrated that the CF transmembrane conductance regulator (CFTR) is present in glucagon-secreting alpha cells and that loss-of-function mutations in CFTR contribute to dysregulated glucagon secretion in CFRD [25]. Whether the dysregulation in glucagon release we observed is also impacted by altered CFTR function needs to be determined. Interestingly, we observed a preponderance of individuals homozygous for the F508 deletion amongst the hypoglycaemia group. Collectively, these observations all provide an islet-related basis for the lack of glucagon response and deserve further exploration.
When designing our study, we hypothesised that one reason for the hypoglycaemia late during an OGTT could be a more vigorous early and/or late insulin response that could be related to enhanced release of the incretin peptides GLP-1 and/or GIP. We were, however, surprised to find that, rather than an increased beta cell response, those with late hypoglycaemia in fact had a reduced response. As the C-peptide response was also lower, the reduced insulin response does not appear to be due to alterations in hepatic extraction of insulin, but rather the result of a primary change in the islet. We also examined the role of glucose in modulating beta cell insulin release by accounting for the glucose excursions during the test using the ratios of the areas of the two beta cell peptides to glucose. When doing so, we again found no evidence in those who experienced hypoglycaemia that the beta cell’s response was more vigorous. Further, as the total glucose excursions were not different and the glucose profiles demonstrated that early in the OGTT those with hypoglycaemia in fact had higher glucose concentrations, the hypoglycaemia cannot be attributed to a robust beta cell response in the absence of adequate glucose absorption. Modelling of the OGTT, in keeping with the lower fasting C-peptide and insulin levels, showed the hypoglycaemia group to be more insulin sensitive. Thus, the lower insulin and C-peptide responses were appropriate in this group as they did not require as much insulin to maintain glucose homeostasis [26]. In addition, modelling raised another thought-provoking point regarding beta cell function. When glucose levels during the OGTT were below the normal range, insulin secretion in the hypoglycaemia group did not appear to decrease appropriately, possibly driving glucose concentrations to a lower level. An inappropriate insulin response in the presence of low glucose has also been suggested in a recent study of patients with CF experiencing hypoglycaemia [27]. The mechanism for this apparent dysfunction cannot be discerned from the current data, but it is credible this may result from insufficient local noradrenaline release so that insulin secretion is not suppressed normally [28].
We did not observe differences in either GLP-1 or GIP concentrations, suggesting that beta cell function is not altered because of changes in incretin release. It is interesting that GLP-1 also regulates alpha cell function, suppressing glucagon secretion at elevated glucose concentrations but not affecting responses at low glucose levels. Thus, we cannot discern from the current data whether the fact that GLP-1 concentrations are not lower in the face of hypoglycaemia represents abnormal release of this peptide or whether the normal ability for hypoglycaemia to overcome any inhibitory effect of GLP-1 on the alpha cell is lost [29]. Nevertheless, these observations underscore the likelihood that the incomplete counterregulatory response, rather than beta cell abnormality, is the critical component of the late hypoglycaemia observed in individuals with CF.
While we did not study individuals with reactive hypoglycaemia, which may be considered a control group, we believe our observations in these cases of late hypoglycaemia in CF distinguish it from reactive hypoglycaemia. The latter also occurs late after nutrient ingestion and may be associated with symptoms of neuroglycopenia and sympathoadrenal activation [30, 31]. In some instances of reactive hypoglycaemia, symptoms attributable to hypoglycaemia develop but there is no evidence of hypoglycaemia, while in others they are unrelated to the rate of fall in plasma glucose or the timing of the glucose nadir. When symptoms are associated with glucose fall, this condition is thought to result from: (1) an exaggerated insulin response; (2) increased insulin sensitivity in which the insulin response is not appropriately reduced as a compensatory mechanism; (3) abnormalities in glucagon release; or (4) renal glycosuria.
The magnitude of the problem of late hypoglycaemia during an OGTT and its clinical implications are not entirely clear. The reported prevalence rate varies, being as great as 30% at 2 h using a glucose threshold of 3.9 mmol/l. Importantly, the OGTT represents a diagnostic test and may not reflect glucose metabolism in everyday life. In a recent report of 169 individuals with CF who had an OGTT, 25 were reported as having hypoglycaemia during the test [5]. In this same report, 14 participants self-reported symptoms suggestive of hypoglycaemia in their medical record, but only one had hypoglycaemia during an OGTT. Further studies are thus needed that could include continuous glucose monitoring to ascertain the time and frequency of hypoglycaemia in daily living. It is also possible that the magnitude of the problem will vary depending on the nature of the nutrient ingested, with oral glucose clearly different from daily dietary consumption. While the long-term consequences of hypoglycaemia are not fully defined, follow-up for up to 10 years does suggest that the risk of CFRD may be lower in those experiencing hypoglycaemia [3, 4].
There are a number of additional questions that could not be answered by the design and limitations of our study. While our study was small and is thus somewhat exploratory in nature, we believe it is the first to provide a comprehensive assessment of potential mechanisms for the late hypoglycaemia observed in CF. More detailed studies of counterregulation such as insulin-induced hypoglycaemic clamps should be performed to confirm our observations of an inappropriately low counterregulatory response, noting that the counterregulatory response could vary depending on prandial status [32]. We did not use continuous glucose monitoring to ascertain how frequently participants were experiencing hypoglycaemia in daily life and thus whether a component of our findings was related to HAAF. We also did not collect data that allowed us to examine the degree of pancreatic insufficiency, and whether this was related to beta and alpha cell function as has been found by others [20, 22]. Finally, given the large number of tests performed, there is the possibility of an inflated type 1 error and an increased chance of finding a spurious result. Future studies will help delineate this possibility.
In conclusion, by performing a 3 h OGTT on a cohort of adults with CF, we found that individuals who experience hypoglycaemia late in the test do so because of an insufficient counterregulatory response rather than either increased or delayed insulin release. This abnormality involves both deficient glucagon and sympathoadrenal responses, the aetiology of which is not clear and warrants further study.
Data availability
The datasets generated during and/or analysed during the current study are not publicly available due to information security but are available from the corresponding author on reasonable request.
Abbreviations
- CF:
-
Cystic fibrosis
- CFRD:
-
CF-related diabetes
- CFTR:
-
CF transmembrane conductance regulator
- CTRC:
-
Clinical and Translational Research Centers
- DIo:
-
Oral disposition index
- FEV1 :
-
Forced expiratory volume in the first second
- GIP:
-
Glucose-dependent insulinotropic peptide
- GLP-1:
-
Glucagon-like peptide-1
- HAAF:
-
Hypoglycaemia-associated autonomic failure
- iAUC:
-
Incremental AUC
- IGT:
-
Impaired glucose tolerance
- ISR:
-
Insulin secretion rate
References
Cystic Fibrosis Foundation (2017) Cystic Fibrosis Foundation patient registry: 2016 annual data report. Cystic Fibrosis Foundation, Bethesda, MD
Battezzati A, Battezzati PM, Costantini D et al (2007) Spontaneous hypoglycemia in patients with cystic fibrosis. Eur J Endocrinol 156(3):369–376. https://doi.org/10.1530/eje.1.02344
Radike K, Molz K, Holl RW, Poeter B, Hebestreit H, Ballmann M (2011) Prognostic relevance of hypoglycemia following an oral glucose challenge for cystic fibrosis-related diabetes. Diabetes Care 34(4):e43. https://doi.org/10.2337/dc10-2286
Mannik LA, Chang KA, Annoh PQK et al (2018) Prevalence of hypoglycemia during oral glucose tolerance testing in adults with cystic fibrosis and risk of developing cystic fibrosis-related diabetes. J Cyst Fibros 17(4):536–541. https://doi.org/10.1016/j.jcf.2018.03.009
Armaghanian N, Markovic TP, Brand-Miller JC, Bye PTP, Moriarty CP, Steinbeck KS (2018) Hypoglycaemia in cystic fibrosis: an analysis of a single centre adult cystic fibrosis clinic. J Cyst Fibros 17(4):542–547. https://doi.org/10.1016/j.jcf.2017.11.015
Hirsch IB, Janci MM, Goss CH, Aitken ML (2013) Hypoglycemia in adults with cystic fibrosis during oral glucose tolerance testing. Diabetes Care 36(8):e121–e122. https://doi.org/10.2337/dc12-1859
American Diabetes Association (1997) Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 20:1183–1197
Moran A, Brunzell C, Cohen RC et al (2010) Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 33(12):2697–2708. https://doi.org/10.2337/dc10-1768
International Hypoglycaemia Study Group (2017) Glucose concentrations of less than 3.0 mmol/l (54 mg/dl) should be reported in clinical trials: a joint position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 60(1):3–6. https://doi.org/10.1007/s00125-016-4146-6
Jiang NS, Machacek D, Wadel OP (1976) Further study on the two-column plasma catecholamine assay. Mayo Clin Proc 51(2):112–116
Kahn SE, Prigeon RL, McCulloch DK et al (1993) Quantification of the relationship between insulin sensitivity and β-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42:1663–1672
Utzschneider KM, Prigeon RL, Faulenbach MV et al (2009) Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 32(2):335–341. https://doi.org/10.2337/dc08-1478
Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E (2002) Meal and oral glucose tests for assessment of β-cell function: modeling analysis in normal subjects. Am J Physiol Endocrinol Metab 283:E1159–E1166
Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ (2001) A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care 24(3):539–548
Moheet A, Ode KL (2018) Hypoglycaemia in patients with cystic fibrosis- harbinger of poor outcomes or innocent bystander? J Cyst Fibros 17(4):428–429. https://doi.org/10.1016/j.jcf.2018.05.012
Sacca L, Sherwin R, Hendler R, Felig P (1979) Influence of continuous physiologic hyperinsulinemia on glucose kinetics and counterregulatory hormones in normal and diabetic humans. J Clin Invest 63(5):849–857. https://doi.org/10.1172/JCI109384
Rizza RA, Cryer PE, Gerich JE (1979) Role of glucagon, catecholamines, and growth hormone in human glucose counterregulation. Effects of somatostatin and combined alpha- and beta-adrenergic blockade on plasma glucose recovery and glucose flux rates after insulin-induced hypoglycemia. J Clin Invest 64(1):62–71. https://doi.org/10.1172/JCI109464
Cryer PE (2013) Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369(4):362–372. https://doi.org/10.1056/NEJMra1215228
Mohan V, Alagappan V, Snehalatha C, Ramachandran A, Thiruvengadam KV, Viswanathan M (1985) Insulin and C-peptide responses to glucose load in cystic fibrosis. Diabete Metab 11(6):376–379
Moran A, Diem P, Klein DJ, Levitt MD, Robertson RP (1991) Pancreatic endocrine function in cystic fibrosis. J Pediatr 118(5):715–723. https://doi.org/10.1016/s0022-3476(05)80032-0
Lanng S, Thorsteinsson B, Roder ME et al (1993) Pancreas and gut hormone responses to oral glucose and intravenous glucagon in cystic fibrosis patients with normal, impaired, and diabetic glucose tolerance. Acta Endocrinol 128(3):207–214. https://doi.org/10.1530/acta.0.1280207
Sheikh S, Gudipaty L, De Leon DD et al (2017) Reduced β-cell secretory capacity in pancreatic-insufficient, but not pancreatic-sufficient, cystic fibrosis despite normal glucose tolerance. Diabetes 66(1):134–144. https://doi.org/10.2337/db16-0394
Hull RL, Gibson RL, McNamara S et al (2018) Islet interleukin-1β immunoreactivity is an early feature of cystic fibrosis that may contribute to β-cell failure. Diabetes Care 41(4):823–830. https://doi.org/10.2337/dc17-1387
Havel PJ, Mundinger TO, Taborsky GJ Jr (1996) Pancreatic sympathetic nerves contribute to increased glucagon secretion during severe hypoglycemia in dogs. Am J Phys 270(1 Pt 1):E20–E26. https://doi.org/10.1152/ajpendo.1996.270.1.E20
Edlund A, Pedersen MG, Lindqvist A, Wierup N, Flodstrom-Tullberg M, Eliasson L (2017) CFTR is involved in the regulation of glucagon secretion in human and rodent alpha cells. Sci Rep 7(1):90. https://doi.org/10.1038/s41598-017-00098-8
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444(7121):840–846. https://doi.org/10.1038/nature05482
Kilberg MJ, Sheikh S, Stefanovski D et al (2019) Dysregulated insulin in pancreatic insufficient cystic fibrosis with post-prandial hypoglycemia. J Cyst Fibros. https://doi.org/10.1016/j.jcf.2019.07.006
Campfield LA, Smith FJ (1983) Neural control of insulin secretion: interaction of norepinephrine and acetylcholine. Am J Phys 244(5):R629–R634. https://doi.org/10.1152/ajpregu.1983.244.5.R629
Nauck MA, Heimesaat MM, Behle K et al (2002) Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab 87(3):1239–1246. https://doi.org/10.1210/jcem.87.3.8355
Johnson DD, Dorr KE, Swenson WM, Service FJ (1980) Reactive hypoglycemia. JAMA 243(11):1151–1155
Brun JF, Fedou C, Mercier J (2000) Postprandial reactive hypoglycemia. Diabetes Metab 26(5):337–351
Porcellati F, Pampanelli S, Rossetti P et al (2003) Counterregulatory hormone and symptom responses to insulin-induced hypoglycemia in the postprandial state in humans. Diabetes 52(11):2774–2783. https://doi.org/10.2337/diabetes.52.11.2774
Acknowledgements
The authors wish to thank the participants for their time and effort in addressing this important clinical problem. We appreciate the advice of A. Mari (Institute of Neuroscience, National Research Council, Padova, Italy) regarding the mathematical modelling and discussions with R. Singh (Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, USA) and K. Morgenthaler (Colorado Clinical and Translational Sciences Institute, University of Colorado Denver, Aurora, CO, USA) regarding the catecholamine assays. The support of B. Ramsey, S. Heltshe and S. McNamara (all at Seattle Children’s Research Institute, University of Washington, Seattle, WA, USA) during the performance of the study is greatly appreciated.
Funding
This study was supported in part by a Pilot and Feasibility Award to SEK from the University of Washington Cystic Fibrosis Research and Translation Center (NIH grant P30 DK089507) and the Diabetes Research Center at the University of Washington (NIH grant P30 DK017047). Additional support to SEK, KMU and EJB was provided by the Department of Veterans Affairs. MAS was supported by NIH grant T32 DK007247. The study sponsors were not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication.
Author information
Authors and Affiliations
Contributions
SEK designed the study and all authors acquired data. SEK, EJB and KMU analysed the data and all authors participated in interpreting it. SEK drafted and all other authors reviewed/edited the manuscript and gave final approval for its publication. SEK and EJB had access to all the data and are guarantors of the work.
Corresponding author
Ethics declarations
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM Figure
(PDF 317 kb)
Rights and permissions
About this article

Cite this article
Aitken, M.L., Szkudlinska, M.A., Boyko, E.J. et al. Impaired counterregulatory responses to hypoglycaemia following oral glucose in adults with cystic fibrosis. Diabetologia 63, 1055–1065 (2020). https://doi.org/10.1007/s00125-020-05096-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05096-6