
Abstract
Aims/hypothesis
The mechanisms underlying pancreatic islet mass expansion have attracted considerable interest as potential therapeutic targets to prevent or delay the onset of type 2 diabetes. While several factors promoting beta cell proliferation have been identified, in the context of nutrient excess the roles of glucose or NEFA in relation to insulin resistance remain unclear. Here we tested the hypothesis that glucose and NEFA synergistically and reversibly promote beta cell proliferation in the context of nutrient-induced insulin resistance.
Methods
Using 72 h infusions of glucose (GLU) or the oleate-enriched lipid emulsion ClinOleic (CLI), singly or in combination, we assessed beta cell proliferation, islet mass and insulin sensitivity in male Lewis rats. The effects of nutrients and endogenous circulating factors were examined in isolated and transplanted islets. Reversibility was studied 3 and 6 days after the end of the infusion.
Results
GLU infusions modestly stimulated beta cell proliferation, CLI alone had no effect and GLU+CLI infusions markedly stimulated beta cell proliferation. Insulin sensitivity was equally decreased in GLU and GLU+CLI infusions. GLU+CLI infusions also stimulated beta cell proliferation in islets transplanted under the kidney capsule, albeit to a lesser extent compared with endogenous islets. Ex vivo, the combination of glucose and NEFA enhanced beta cell proliferation in rat and human islets independently from secreted insulin, and serum from GLU+CLI-infused rats potentiated the effect of glucose. Glucose tolerance, beta cell proliferation and islet mass were all restored to normal levels 6 days after termination of the infusion.
Conclusions/interpretation
Glucose and NEFA synergistically and reversibly promote beta cell proliferation in part via direct action on the beta cell and independently from secreted insulin.
Similar content being viewed by others

Introduction
The pancreatic beta cell has a remarkable capacity to adjust to a changing metabolic environment by increasing its functional mass [1, 2]. In rodents, and possibly humans, the increase in beta cell mass is due, in part, to replication. Despite the recent identification of several candidates [3–5], the nature of the signals that trigger beta cell proliferation in response to a given metabolic situation remains elusive [6–8]. While glucose is an established beta cell mitogen [9–12], NEFA have been reported to stimulate [9] or inhibit [10] beta cell proliferation in vivo. Several factors can explain these discrepancies, including the confounding effect of insulin resistance and whether or not glucose and NEFA levels are simultaneously elevated.
We previously developed a rat model of nutrient excess in vivo in which combined infusion of glucose and NEFA for 72 h leads, in an age-dependent manner, to impaired beta cell function, despite a marked increase in beta cell proliferation and mass [13–15]. This model provides a unique opportunity to examine the individual and combined effects of elevated glucose and NEFA levels in vivo in relation to insulin sensitivity. Using this model, the present study was designed to test the hypothesis that combined glucose and NEFA infusion reversibly promotes beta cell proliferation in part via circulating factors released in response to insulin resistance. Specifically, we sought the following information: (1) What are the effects of glucose and NEFA, singly or in combination, on beta-cell proliferation? (2) Does nutrient-induced beta cell proliferation correlate with the level of insulin sensitivity? (3) What is the relative contribution of circulating factors to beta cell proliferation? (4) Is nutrient-induced beta cell proliferation reversible?
Methods
Animal surgeries
All procedures were approved by the Institutional Committee for the Protection of Animals at the Centre Hospitalier de l’Université de Montréal. Male Lewis rats weighing 250–350 g (∼2 months old) and 450–550 g (∼6 months old) (Charles River, Saint-Constant, QC, Canada) were individually housed under controlled temperature on a 12 h light–dark cycle with free access to water and standard laboratory chow. The rats underwent catheterisation of the jugular vein for nutrient infusions and the carotid artery for sampling as described [15] and were allowed to recover for 6 days. For islet transplantation studies, 200 age-matched Lewis rat islets were injected via a cannula under the left kidney capsule during the catheterisation surgery.
Infusions
Rats were randomly assigned to four groups receiving the following intravenous infusions: saline (SAL) (0.9% wt/vol. NaCl; Baxter, Mississauga, ON, Canada); 70% (wt/vol.) glucose (GLU) (McKesson, Montreal, QC, Canada); 20% (wt/vol.) ClinOleic (CLI) (Fresenius Kabi, Uppsala, Sweden; an olive/soybean triacylglycerol emulsion composed of 65% monounsaturated/20% polyunsaturated/15% saturated fatty acids) co-infused with 20 U/ml heparin (Sandoz, Boucherville, QC, Canada) or GLU plus 20% (wt/vol.) ClinOleic plus heparin (GLU+CLI). The glucose infusion rate (GIR) was adjusted to maintain plasma glucose at 13.9–19.4 mmol/l throughout the 72 h infusion. See ESM Methods.
Hyperinsulinaemic–euglycaemic clamps
Hyperinsulinaemic–euglycaemic clamps (HIEC) were performed as described previously (in mice) [16] with minor modifications. See ESM Methods.
IVGTT
Glucose (0.5 g/kg) was injected into the jugular catheter of 5 h-fasted rats. Blood samples were taken at 0, 3, 6, 9, 15, 30, 45 and 60 min.
Analysis of islet mass and beta cell proliferation, size and apoptosis
Islet mass was measured on paraffin sections prepared as described [13] using an anti-chromogranin A antibody (Ab85554; Abcam, Toronto, ON, Canada) to label endocrine cells. Beta cell proliferation was measured by immunohistochemical staining of pancreas cryosections for Ki67 (Ab66155; Abcam) and insulin as described [13] or by intravenous injection of BrdU (B5002; Sigma-Aldrich, Oakville, ON, Canada) 50 mg/kg at 12, 36 and 60 h during the infusion followed by immunostaining for BrdU (347580; Becton Dickinson, San Jose, CA, USA) and insulin. Beta cell proliferation was expressed as a percentage of double-positive Ki67+ insulin+ (or BrdU+ insulin+) cells to total insulin+ cells. Apoptosis was measured using the FragEL DNA Fragmentation Detection Kit (Millipore, Etobicoke, ON, Canada) according to the manufacturer’s instructions (see ESM Methods). The experimenter was blind to group assignments.
Rat and human islet culture
Islets were isolated from male Lewis rat as described [17] and cultured in RPMI-1640 with 10% (vol./vol.) FBS (Invitrogen, Burlington, VT, USA) for 72 h in the presence of glucose, a NEFA mixture mimicking the composition of CLI (65% oleate, 20% linoleate and 15% palmitate) or insulin (Humulin; Lilly, Toronto, ON, Canada) as indicated in figure legends. To assess the role of circulating factors, FBS was replaced by 10% (vol./vol.) serum (de-complemented for 30 min at 56°C) from infused rats. The final glucose concentration in the culture medium was adjusted to take into account the glucose present in the serum from GLU- and GLU+CLI-infused rats. See ESM Methods.
Islets from non-diabetic human donors were provided by the Clinical Islet Laboratory at the University of Alberta, the Alberta Diabetes Institute Islet Core and the National Institute of Diabetes and Digestive and Kidney Diseases - sponsored Integrated Islet Distribution Program. The use of human islets was approved by the Institutional Ethics Committee of the Centre Hospitalier de l’Université de Montréal (protocol no. ND-05-035). Human islets were cultured in cGMP Prodo Islet Media (Standard; Prodo Laboratories, Aliso Viejo, CA, USA) with 5% (vol./vol.) human AB serum and 1% (vol./vol.) glutamine/glutathione mixture. Human islet proliferation was determined as for rat islets.
Analytical measurements
NEFA, C-peptide and insulin measurements are described briefly in ESM Methods.
Statistical analysis
Data are expressed as means ± SEM. Statistical analyses were performed using Student’s t test or ANOVA followed by two-by-two comparisons using Tukey or Sidak post hoc test (GraphPad Prism 6 version 6.0; La Jolla, CA, USA); p < 0.05 was considered significant.
Results
Characterisation of the 72 h-infused Lewis rat model
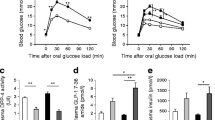
The 72 h infusions had no effect on body weight in either age group (ESM Fig. 1a, b). Total energy intake was decreased in GLU- and GLU+CLI-infused 2-month-old rats but not 6-month-old rats (ESM Fig. 1c, d). In 2-month-old rats, blood glucose was increased in the target range (∼15 mmol/l) in GLU- and GLU+CLI-infused rats (Fig. 1a). Plasma insulin was increased in GLU- and GLU+CLI- but not CLI-infused rats (Fig. 1b). Plasma NEFA was increased in CLI- and GLU+CLI-infused rats (Fig. 1c). The GIR required to maintain target blood glucose levels during the infusion was lower in GLU+CLI-infused rats than in GLU-infused rats (Fig. 1d). Similar differences between infusion groups were observed in 6-month-old rats (Fig. 1e–h).

Characterisation of the nutrient-infused rat model. (a–d) Two-month-old and (e–h) 6-month-old rats were infused with SAL, GLU, CLI or GLU+CLI for 72 h. (a, e) Blood glucose, (b, f) plasma insulin and (c, g) plasma NEFA were measured during the infusion. Data are means ± SEM (n = 9–27). (d, h) Average GIR during the infusion. Data represent individual values and mean (n = 21–27). For (a–c) and (e–g), ***p < 0.001 vs SAL, ANOVA. For (d) and (h), ††† p < 0.001 vs GLU+CLI, unpaired Student’s t test
Islet mass and beta cell proliferation increase in response to GLU and GLU+CLI infusion in 2- and 6-mo-old rats
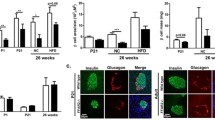
Because of the weakness of insulin staining in GLU- and GLU+CLI-infused rats (likely related to a reduction in insulin biosynthesis following nutrient infusion [14]), we used chromogranin A as a marker of endocrine cells to measure islet mass. In 2-month-old rats, the islet area (as a percentage of total pancreas area) was significantly increased in GLU- and GLU+CLI-infused groups compared with SAL (Fig. 2a). A similar trend was observed for islet mass, although the differences did not reach statistical significance (Fig. 2b) because of lower pancreas weight (ESM Fig. 2a). The increase in islet mass was mostly accounted for by a greater number of large islets (ESM Fig. 2b). The proportion of beta to alpha cells was greater following GLU and GLU+CLI infusions (ESM Fig. 2c). The size of beta cells was greatest in the GLU+CLI group (ESM Fig. 2d). Using Ki67 staining, we observed a 12-fold increase in beta cell proliferation in GLU+CLI-infused rats compared with SAL and a threefold increase compared with GLU (Fig. 2c, d). Using BrdU staining, only the GLU+CLI infusion induced a significant increase in beta cell proliferation (Fig. 2e). We could not detect apoptosis in islets from any of the infusion groups (ESM Fig. 2e).

Measurement of beta cell expansion. (a–e) Two-month-old and (f–j) 6-month-old rats were infused with nutrients as indicated. (a, f) Islet area (as a percentage of total pancreas area). (b, g) Islet mass. (c, h) Representative immunostaining for insulin (green), Ki67 (red) and nuclei (blue) in pancreatic sections is shown. (d, i) The percentage of Ki67+ insulin+ cells over insulin+ cells. (e, j) Percentage of BrdU+ insulin+ cells over insulin+ cells was determined. Data are means ± SEM (n = 3–6). Scale bars, 50 μm. *p < 0.05, **p < 0.01 and ***p < 0.001 vs SAL, ANOVA
In 6-month-old rats, islet area and islet mass were slightly but not significantly increased in GLU- and GLU+CLI-infused rats compared with controls (Fig. 2f, g). This was mainly due to the presence of larger islets and greater beta cell size in GLU- and GLU+CLI-infused groups (ESM Fig. 2f, h). Pancreas weight was reduced in GLU+CLI-infused rats (ESM Fig. 2g). The number of Ki67-positive beta cells was increased in GLU-infused rats and even more so in GLU+CLI-infused rats (Fig. 2h, i). The profile observed with BrdU staining was similar (Fig. 2j).
Taken together, these data indicate that glucose and NEFA synergistically stimulate beta cell proliferation in vivo and that this effect is more pronounced in older animals.
Infusions of GLU or GLU+CLI induce insulin resistance
To investigate whether the increase in beta cell proliferation correlates with insulin resistance, we performed HIEC at the end of the infusions in a second cohort of rats. In 2-month-old rats, blood glucose levels (Fig. 3a) and GIR (Fig. 3b) during the clamp were essentially identical in all four groups. In contrast, the M/I index (ratio between GIR (μmol kg−1 min−1) and insulin levels (pmol/l) during the clamp; Fig. 3c) and insulin sensitivity index (ESM Fig. 3a) were significantly lower in GLU- and GLU+CLI-infused rats but remained unchanged by the CLI infusion. Despite the constant insulin infusion rate and co-infusion of somatostatin, the levels of circulating insulin were significantly increased in GLU- and GLU+CLI-infused rats (Fig. 3d). This was not due to residual endogenous (rat) insulin, which was negligible in all groups (ESM Fig. 3b). Rather, levels of human insulin were different (ESM Fig. 3c), which can be explained by changes in insulin clearance rates in GLU- and GLU+CLI-infused rats (ESM Fig. 3d).

Assessment of insulin sensitivity. HIEC were performed following nutrient infusion in (a–d) 2-month-old and (e–h) 6-month-old rats. (a, e) Average blood glucose, (b, f) GIR, (c, g) M/I index and (d, h) plasma insulin during the clamp are shown. Data are means ± SEM (n = 5 or 6). *p < 0.05 and ***p < 0.001 vs SAL, ANOVA
Similar changes were observed in 6-month-old rats (Fig. 3e–h and ESM Fig. 3e–h). Regardless of the infusion regimen, the GIR was significantly lower in 6-month-old rats than in 2-month-old rats, reflecting age-dependent insulin resistance (Fig. 3f vs Fig. 3b).
Thus, GLU and GLU+CLI infusions induce a similar degree of insulin resistance, when compared with SAL infusion, in both 2-month-old and 6-month-old rats. In contrast, the amplitude of beta cell proliferation is much greater in GLU+CLI-infused rats than in GLU-infused rats (Fig. 2), suggesting that insulin resistance is not the only driver of beta cell replication in this context.
GLU+CLI induces beta cell proliferation in islet grafts
We transplanted 200 islets from 2-month-old rats (insufficient to alter the recipient’s circulating glucose, insulin or NEFA levels [data not shown]) under the kidney capsule of age-matched Lewis rats 6 days prior to infusion of SAL or GLU+CLI. The GLU+CLI infusion increased beta cell proliferation in transplanted islets (Fig. 4a, b) albeit to a lesser extent than in endogenous islets (Fig. 4c).

Measurement of beta cell proliferation in transplanted vs endogenous islets. Two-month-old rats were infused with nutrients. (a) Representative images of insulin (green), Ki67 (red) and nuclei (blue) staining in transplanted islets are shown. Arrows show positive nuclei for Ki67. The percentage of Ki67+ insulin+ cells over insulin+ cells in (b) transplanted and (c) endogenous islets was determined. Data represent individual values and mean (n = 3). Scale bars, 50 μm. For (b), **p < 0.01 vs SAL, paired Student’s t test. For (c), *p < 0.05, unpaired Student’s t test
Glucose and NEFA stimulate beta cell proliferation in isolated islets ex vivo
To examine the direct effect of nutrients, we subjected rat isolated islets to conditions mimicking those found in vivo. High glucose alone did not significantly increase beta cell proliferation in islets from either 2-month-old or 6-month-old rats (Fig. 5a–c). Surprisingly, a mixture of NEFA enriched in oleate strongly induced beta cell proliferation at 5.5 mmol/l glucose and the addition of 16.7 mmol/l glucose did not further enhance this response. Thus, the synergistic effect between glucose and NEFA observed in vivo (Fig. 2) was not replicated ex vivo. Apoptosis was detected in islets but was not further enhanced by either glucose or NEFA (Fig. 5d, ESM Fig. 4a). We then performed similar experiments in islets isolated from humans (ESM Table 1). Although addition of NEFA to the low-glucose condition did not increase beta cell proliferation (Fig. 5e), a significant increase was detected in the presence of high glucose (Fig. 5f).

Measurement of beta cell proliferation in rat and human isolated islets. Islets from (a, b, g, h) 2-month-old and (c) 6-month-old rats or from (e, f) humans were treated with: (a–f) glucose and NEFA mixture (0.5 mmol/l); (g) glucose and NEFA mixture (0.5 mmol/l) and 10% serum from SAL-infused (white bars), GLU-infused (grey bars) or GLU+CLI-infused (black bars) rats: or (h) glucose and NEFA mixture (0.5 mmol/l) with/without 1500 nmol/l insulin. (a) Representative images of insulin (green), Ki67 (red) and nuclei (blue) staining are shown, with glucose (G) concentrations shown in mmol/l. Arrows show positive nuclei for Ki67. (b, c, e–h) The percentage of Ki67+ insulin+ cells over insulin+ cells. (d) Percentage of beta cell apoptosis. Data are means ± SEM (n = 4–6). Scale bars, 50 μm. For (b–d), (g) and (h), *p < 0.05, **p < 0.01 and ***p < 0.001 vs 5.5 mmol/l glucose, ANOVA. For (e) and (f), *p < 0.05 vs 5.5 mmol/l glucose, paired Student’s t test
Serum from GLU+CLI-infused rats potentiates glucose-induced beta cell proliferation ex vivo
To find out if circulating factors other than nutrients themselves present in nutrient-infused rats might account for the differences observed between in vivo and ex vivo situations, we treated rat isolated islets with nutrients plus 10% serum from infused rats. Serum from GLU- or GLU+CLI-infused rats did not stimulate beta cell proliferation at 5.5 mmol/l glucose but serum from GLU+CLI-infused rats increased beta cell proliferation at 16.7 mmol/l glucose (Fig. 5g). The addition of NEFA did not further increase this response, probably because the maximal proliferative capacity had been attained.
These data indicate that glucose and NEFA can directly stimulate beta cell proliferation and that endogenous factors present in the serum from GLU+CLI-infused rats potentiate glucose-induced beta cell proliferation, consistent with our in vivo data.
Insulin secretion in response to glucose and NEFA does not contribute to beta cell proliferation
We then asked whether insulin released in response to nutrients might act in an autocrine/paracrine fashion to promote beta cell proliferation. Insulin levels in the medium were significantly higher when islets from 2-month-old or 6-month-old rats were cultured in the presence of 16.7 mmol/l glucose with 0.5 mmol/l NEFA (ESM Fig. 4b, c). Addition of 0.5 mmol/l NEFA alone only modestly increased insulin levels in islets from 6-month-old but not 2-month-old rats. As beta cell proliferation was not increased by 16.7 mmol/l glucose but was strongly stimulated by the addition of 0.5 mmol/l NEFA (Fig. 5b, c), these data reveal a lack of correlation between insulin levels and beta cell proliferation.
Finally, we exposed rat islets to a concentration of insulin (1500 nmol/l) similar to the insulin levels in the supernatant fraction of islets exposed to 16.7 mmol/l glucose for 72 h (ESM Fig. 4b, c). Insulin did not increase beta cell proliferation at 5.5 mmol/l glucose either in the absence or presence of 0.5 mmol/l NEFA (Fig. 5h). Similar results were obtained when islets were treated with a lower insulin concentration of 10 nmol/l (data not shown). These data indicate that secreted insulin does not mediate nutrient-induced beta cell proliferation.
Nutrient-induced glucose intolerance and beta cell proliferation is reversible
To ascertain whether the changes brought about by nutrient excess are reversible, we performed another series of GLU+CLI vs SAL infusions in 2-month-old rats and followed them up at 3 and 6 days after the end of the infusions. Circulating glucose, insulin, NEFA and C-peptide levels in GLU+CLI-infused rats returned to basal levels after 6 days (ESM Fig. 5a–d). Immediately after termination of the infusion, basal blood glucose was significantly lower in GLU+CLI-infused rats (Fig. 6a) but the rats were glucose intolerant (Fig. 6b) despite elevated insulin levels during the IVGTT (Fig. 6c), consistent with insulin resistance (Fig. 3). Basal blood glucose and glucose and insulin levels during the IVGTT gradually returned to normal after 3 and 6 days of recovery (Fig. 6d–i). Islet area, proliferating beta cells and the number of large islets remained elevated 3 days after the end of the infusion but returned to basal level at 6 days (Fig. 7) as did pancreas weight (data not shown), whereas beta cell size remained slightly increased (Fig. 7). No beta cell apoptosis was detected on either day 3 or day 6 following infusion (ESM Fig. 5e). Thus, the major functional and morphological changes brought about by nutrient infusion are fully reversible and do not lead to permanent beta cell damage.

Measurement of glucose tolerance during recovery. IVGTTs (0.5 g/kg body weight) were performed at (a–c) 0, (d–f) 3 and (g–i) 6 days after nutrient infusion in 2-month-old rats. (a, d, g) Basal blood glucose and (b, e, h) glucose levels during the IVGTT expressed as a percentage of basal glucose were measured. AUC for glucose are shown inset. (c, f, i) Plasma insulin levels during the IVGTT. AUC for insulin are shown inset. White circles, SAL; black circles, GLU+CLI. Data are means ± SEM (n = 3–6). **p < 0.01 and ***p < 0.001 vs SAL, unpaired Student’s t test

Measurement of beta cell mass expansion during recovery. Two-month-old rats were infused with nutrients and beta cell expansion was assessed at the end of the infusion (D0) and at day 3 (D3) and 6 (D6) after infusion. (a) The percentage of islet area to total pancreas area and (b) percentage of Ki67+ insulin+ cells over total insulin+ cells were determined. Islet size distribution was determined on (c) D3 and (e) D6. Beta cell size was also determined on (d) D3 and (f) D6. White bars, SAL; black bars, GLU+CLI. Data are means ± SEM (n = 3–6). Data presented at D0 for (a, b) are the same as in Fig. 2a, d. For (a), (b), (d) and (f), *p < 0.05, **p < 0.01 and ***p < 0.001 vs SAL, unpaired Student’s t test. For (c) and (e), **p < 0.01 vs SAL, ANOVA
Discussion
In this study, we investigated the effects of glucose and NEFA, singly or in combination, on beta cell proliferation in vivo and ex vivo. First, in 72 h-infused rats we showed that while glucose alone modestly induced beta cell proliferation and NEFA alone did not, the effect of both nutrients was synergistic with a marked increase in beta cell proliferation in GLU+CLI-infused animals. Second, islets transplanted into GLU+CLI-infused rats had a proliferative response, albeit of lower magnitude than that of the endogenous islets. Third, in contrast to our in vivo observations, a mixture of NEFA strongly induced rat and human beta cell proliferation ex vivo even at low glucose levels, independently from secreted insulin, and circulating factors present in the serum of GLU+CLI-infused rats potentiated glucose-induced beta cell proliferation. Finally, the changes in glucose homeostasis and beta cell proliferation and mass brought about by the GLU+CLI infusion were fully reversed 6 days after termination of the infusion.
Ageing is associated with insulin resistance in rodents and humans [18, 19]. Accordingly, the M/I index of insulin sensitivity during HIEC was lower in 6-month-old rats than in 2-month-old rats across all infusion groups. Further, infusion of glucose with or without NEFA induced insulin resistance as described previously in rats [9–11, 20]. The magnitude of beta cell proliferation in response to GLU and GLU+CLI infusion was higher in 6-month-old rats than in 2-month-old rats. This suggests that in this context the proliferative effect of nutrients is enhanced in a background of pre-existing insulin resistance, despite the well-known age-dependent loss of beta cell replicative capacity [21, 22], which was also observed in our ex vivo experiments (Fig. 5c vs Fig. 5b). A number of studies using pharmacological [23, 24] or genetic [25–27] tools have demonstrated that insulin resistance leads to beta cell hyperplasia in rodents, and the protease inhibitor serpin B1 was recently identified as a liver-derived circulating factor mediating this response in insulin-resistant mice [5]. In the context of nutrient-induced insulin resistance, however, the respective contributions of nutrients themselves vs their indirect effect through insulin resistance on beta cell proliferation remain unclear, and in this regard our study provides important insights. Infusion of GLU and GLU+CLI induced a similar degree of insulin resistance, yet the rate of beta cell proliferation was much greater in GLU+CLI-infused rats than in GLU-infused rats, in both age groups, suggesting the involvement of additional factors. However, we cannot exclude the possibility that changes in insulin sensitivity in the liver and adipose tissue were masked due to the predominant role of muscle in insulin-dependent glucose disposal in HIEC. Metabolic labelling studies will be required to test this possibility.
Consistent with previous findings [26, 28, 29], we observed significant beta cell proliferation in transplanted islets following GLU+CLI infusions. This may involve factors generated locally, direct effects of nutrients themselves or other circulating factors. In support of the latter, serum from GLU+CLI-infused rats potentiated glucose-induced beta cell proliferation in islets ex vivo. As discussed above, levels of insulin and insulin resistance were similar in rats infused with GLU and GLU+CLI yet only serum from the latter group was found to stimulate beta cell proliferation, suggesting the contribution of circulating factors distinct from those arising from insulin resistance alone.
Our observation that glucose promotes beta cell proliferation in vivo and ex vivo is consistent with the results of previous infusion studies in rodents [9, 10, 20] and with the notion that intracellular glucose metabolism controls beta cell replication [29]. In contrast, variable responses have been described following NEFA infusions. Steil et al [9] detected an increase in beta cell proliferation in response to Intralipid infusions in rats, whereas Pascoe et al [10] observed that Liposyn II inhibited glucose-induced beta cell proliferation in mice. The opposite was seen in our model in which CLI markedly enhanced the proliferative effect of glucose. Several differences can explain these discrepancies, including the species (mice vs rats), the strain [13] and the nature of the lipid emulsion. Liposyn II is highly enriched in polyunsaturated fatty acids, whereas monounsaturated fatty acids are predominant in CLI used in this study. Consistent with this, we found that a combination of NEFA mimicking CLI effectively stimulated beta cell proliferation ex vivo. Similarly, Vernier et al [30] showed that a mixture of oleate and palmitate increases beta cell proliferation in rat islets, whereas Pascoe et al [10] found that a lipid emulsion mimicking Liposyn II blocked the stimulatory effect of glucose. Further studies will be required to identify the mechanism whereby oleate promotes beta cell proliferation.
We observed interesting differences in the effects of NEFA in vivo and ex vivo, whereby NEFA were able to stimulate beta cell proliferation in rat isolated islets even at low glucose levels, while CLI infusions had no effect if not combined with GLU. Thus, in vivo, NEFA potentiate the effects of glucose but cannot trigger beta cell proliferation by themselves, similar to their action on insulin secretion. However, we cannot exclude the possibility that differences in the composition and concentration of the NEFA to which the islets were exposed may be responsible. Importantly, the potentiating effect of NEFA on glucose-induced proliferation was confirmed in human islets ex vivo in the presence of high glucose.
We demonstrated that insulin levels do not correlate with nutrient-induced beta cell proliferation and that insulin does not promote beta cell proliferation in islets ex vivo. These results are consistent with recent findings in mouse islets [31] but contrast with previous finding in insulin-secreting cell lines [32]. Although our data do not exclude a role for insulin signalling in the regulation of beta cell mass, as suggested from beta cell-specific insulin-receptor-knockout mouse studies [33, 34], they do indicate that the increase in beta cell proliferation in response to glucose and NEFA is independent of the autocrine/paracrine action of insulin.
Glucose tolerance and insulin secretion returned to normal in GLU+CLI-infused rats on day 6 post infusion. Furthermore, beta cell proliferation progressively returned to basal levels following termination of the infusion. Interestingly, islet area was restored after 6 days despite elevated beta cell proliferation in GLU+CLI-infused rats up to 3 days post infusion and no detectable increase in apoptosis. These data imply that mechanisms restoring beta cell numbers are rapidly recruited after removal of the stimulus and confirm the plasticity of the beta cell response to metabolic stress [35] and at the end of pregnancy [36].
In conclusion, using a rat model of nutrient excess we showed that excessive glucose and NEFA levels synergistically and reversibly promote beta cell proliferation in vivo through a combination of mechanisms including a direct effect of nutrients that is not mediated by secreted insulin.
Abbreviations
- CLI:
-
ClinOleic
- GIR:
-
Glucose infusion rate
- GLU:
-
Glucose, administered by infusion
- HIEC:
-
Hyperinsulinaemic–euglycaemic clamps
- SAL:
-
Saline
References
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116:1802–1812
Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383:1068–1083
Wang P, Alvarez-Perez JC, Felsenfeld DP et al (2015) A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 21:383–388
Yi P, Park JS, Melton DA (2013) Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 153:747–758
El Ouaamari A, Dirice E, Gedeon N et al (2015) SerpinB1 promotes pancreatic β cell proliferation. Cell Metab 23:194–205
Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF (2012) Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 61:2205–2213
Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A (2014) Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 63:819–831
Stewart AF, Hussain MA, Garcia-Ocana A et al (2015) Human β-cell proliferation and intracellular signaling: part 3. Diabetes 64:1872–1885
Steil GM, Trivedi N, Jonas JC et al (2001) Adaptation of β-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Phys Endocrinol Metab 280:E788–E796
Pascoe J, Hollern D, Stamateris R et al (2012) Free fatty acids block glucose-induced β-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes 61:632–641
Bernard C, Thibault C, Berthault MF et al (1998) Pancreatic β-cell regeneration after 48-h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes 47:1058–1065
Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A (2003) Specific and combined effects of insulin and glucose on functional pancreatic β-cell mass in vivo in adult rats. Endocrinology 144:2717–2727
Zarrouki B, Benterki I, Fontes G et al (2014) Epidermal growth factor receptor signaling promotes pancreatic β-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes 63:982–993
Fontes G, Zarrouki B, Hagman DK et al (2010) Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 53:2369–2379
Hagman DK, Latour MG, Chakrabarti SK et al (2008) Cyclical and alternating infusions of glucose and intralipid in rats inhibit insulin gene expression and Pdx-1 binding in islets. Diabetes 57:424–431
Fergusson G, Ethier M, Guevremont M et al (2014) Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol Metab 3:848–854
Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V (2000) Inhibition of insulin gene expression by long-term exposure of pancreatic β cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metab Clin Exp 49:532–536
Escriva F, Agote M, Rubio E et al (1997) In vivo insulin-dependent glucose uptake of specific tissues is decreased during aging of mature Wistar rats. Endocrinology 138:49–54
Defronzo RA (1979) Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes 28:1095–1101
Bonner-Weir S, Deery D, Leahy JL, Weir GC (1989) Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes 38:49–53
Rankin MM, Kushner JA (2009) Adaptive β-cell proliferation is severely restricted with advanced age. Diabetes 58:1365–1372
Tschen SI, Dhawan S, Gurlo T, Bhushan A (2009) Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes 58:1312–1320
Shirakawa J, Okuyama T, Yoshida E et al (2014) Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, β-cell functions, and β-cell proliferation in male mice. Endocrinology 155:2102–2111
Rafacho A, Abrantes JL, Ribeiro DL et al (2011) Morphofunctional alterations in endocrine pancreas of short- and long-term dexamethasone-treated rats. Horm Metab Res 43:275–281
Michael MD, Kulkarni RN, Postic C et al (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6:87–97
El Ouaamari A, Kawamori D, Dirice E et al (2013) Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep 3:401–410
Flier SN, Kulkarni RN, Kahn CR (2001) Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci U S A 98:7475–7480
Levitt HE, Cyphert TJ, Pascoe JL et al (2011) Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 54:572–582
Porat S, Weinberg-Corem N, Tornovsky-Babaey S et al (2011) Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 13:440–449
Vernier S, Chiu A, Schober J et al (2012) β-cell metabolic alterations under chronic nutrient overload in rat and human islets. Islets 4:379–392
Stamateris RE, Sharma RB, Kong Y et al (2016) Glucose induces mouse β-cell proliferation via IRS2, MTOR, and cyclin D2 but not the insulin receptor. Diabetes 65:981–995
Assmann A, Ueki K, Winnay JN, Kadowaki T, Kulkarni RN (2009) Glucose effects on β-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol 29:3219–3228
Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999) Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339
Okada T, Liew CW, Hu J et al (2007) Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A 104:8977–8982
Alarcon C, Boland BB, Uchizono Y et al (2016) Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes 65:438–450
Scaglia L, Smith FE, Bonner-Weir S (1995) Apoptosis contributes to the involution of β cell mass in the post partum rat pancreas. Endocrinology 136:5461–5468
Acknowledgements
We are grateful to M. Éthier and G. Fergusson (CRCHUM) for valuable technical assistance with in vivo experiments, to G. Dodier (CRCHUM) for technical assistance with immunohistochemistry, to M. Guévremont, J. Morin and the Cellular Physiology core facility of CRCHUM for beta cell mass analyses and to T. Alquier, M. Madiraju and M. Prentki (CRCHUM) for their valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request. All data generated or analysed during this study are included in this published article (and its supplementary information files).
Funding
This study was supported by the National Institutes of Health (grant R01-DK-58096 to VP) and the Canadian Institutes of Health Research (grant MOP 77686 to VP). KV and VSM are supported by Postdoctoral Fellowships from the Fonds de recherche Québec – Santé and the CRCHUM, respectively. VP holds the Canada Research Chair in Diabetes and Pancreatic Beta Cell Function.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
VSM designed the experiments, acquired the data, analysed the results and wrote the manuscript. KV and CT designed the experiments and acquired the data. VSM, BZ and VP conceived and designed the project. JG and VP analysed the results and wrote the manuscript. All authors revised the manuscript and approved the final version. VSM and VP are the guarantors of this work and take full responsibility for the content of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 1714 kb)
Rights and permissions
About this article

Cite this article
Moullé, V.S., Vivot, K., Tremblay, C. et al. Glucose and fatty acids synergistically and reversibly promote beta cell proliferation in rats. Diabetologia 60, 879–888 (2017). https://doi.org/10.1007/s00125-016-4197-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4197-8