
Abstract
Aims/hypothesis
The in vivo role of mechanistic target of rapamycin (mTOR) in the development and function of adipose tissue, especially brown adipose tissue (BAT), is not well understood. Here, we aimed to assess the effect of mTOR (also known as Mtor) knockout on adipose tissues and systemic energy metabolism.
Methods
We generated adipocyte-specific mTOR-knockout mice (Adipoq-mTOR) by crossing adiponectin-Cre (Adipoq-Cre) mice with mTOR flox/flox mice. The mice were then subjected to morphological, physiological (indirect calorimetry, glucose and insulin tolerance tests) and gene expression analyses to determine the role of mTOR in adipose tissues.
Results
We provide in vivo evidence that mTOR is essential for adipose tissue development and growth. Deletion of mTOR decreased the mass of both BAT and white adipose tissues (WAT) and induced browning of WAT. In addition, ablation of mTOR in adipose tissues caused insulin resistance and fatty liver in the Adipoq-mTOR mice. Furthermore, mTOR was required for adipocyte differentiation in vivo and activation of PPARγ ameliorated the differentiation deficiency of the mTOR-null adipocytes.
Conclusions/interpretation
Our findings demonstrate that mTOR is a critical regulator of adipogenesis and systemic energy metabolism. Our study provides key insights into the role of mTOR in adipose tissues; such knowledge may facilitate the development of novel strategies with which to treat obesity and related metabolic diseases.
Similar content being viewed by others


Introduction
Adipose tissue plays a critical role in regulating energy balance and metabolism and its dysfunction is closely associated with metabolic diseases such as obesity and insulin resistance. In mammals, there are two main types of adipose: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is the major energy storage site that stores excess energy, whereas BAT contains numerous mitochondria with high levels of uncoupling protein 1 (UCP1) that dissipates energy as heat [1]. Adipocytes in the WAT and BAT are called white and brown adipocytes, respectively. Recently, a third type of adipocyte, called beige or brite [2], has been found in WAT. Beige adipocytes can be induced by chemical or hormonal stimulation, cold exposure or genetic manipulation [3–7]. Similar to classical brown adipocytes, beige adipocytes have high levels of UCP1 [3, 7, 8]. Both brown and beige adipocytes can increase energy expenditure and counteract obesity [7–9]. Thus, understanding the development and function of brown and beige adipocytes may provide novel strategies with which to counteract obesity.
Mammalian target of rapamycin (mTOR) is a conserved serine/threonine protein kinase involved in regulating protein synthesis, cell growth and energy metabolism [10]. mTOR functions in two distinct complexes: mTOR complex 1 (mTORC1) and mTORC2 [11]. mTORC1, consists of mTOR, raptor and deptor, is sensitive to rapamycin [10]. mTORC2, containing mTOR, rictor and deptor, is insensitive to rapamycin [10]. Recent studies demonstrate that both complexes play a crucial role in adipogenesis [12, 13], lipid homeostasis [10, 14, 15], glucose metabolism and insulin actions [16, 17]. Inhibition of mTORC1 signalling by rapamycin or small interfering RNA against Raptor (also known as Rptor) decreases adipocyte proliferation and differentiation [18–21] and protects mice against high-fat diet (HFD)-induced obesity [22]. Adipose-specific ablation of Raptor also enhances energy expenditure and protects the mutant mice against HFD-induced obesity [23, 24]. Mice lacking ribosomal S6 kinase 1 (S6K1), a key downstream target of mTORC1, have relatively lower fat mass and higher energy expenditure [13, 25]. Moreover, deletion or overexpression of Tsc1/Tsc2, upstream regulators of mTORC1, further confirms a key role for mTORC1 in adipocyte differentiation and energy metabolism [26]. Compared with mTORC1, the function of mTORC2 in adipocytes is not well understood. Rictor-null mice die at around embryonic day 10.5 [27, 28]. In Caenorhabditis elegans, Rictor-null worms have comparatively more body fat [14, 29], indicating that mTORC2 may also play a role in regulating adipogenesis. In mice, adipose-specific deletion of Rictor decreases glucose uptake and glucose tolerance in vivo without affecting adipocyte size or fat mass [16, 17, 30]. These reports indicate that mTORC1 and mTORC2 play distinct roles in adipogenesis and lipid metabolism. However, most of the earlier studies involved perturbations of either mTORC1 or mTORC1. The direct effect of genetic deletion of mTOR (also known as Mtor), which has the effect of simultaneously suppressing mTORC1 and mTORC2, in adipose tissues has not been reported. Moreover, previous studies used FABP4-Cre to drive the adipose-specific deletion of Raptor or Rictor and this may have led to off-target deletion in non-adipose tissues [31, 32] and confound the interpretation of results. In this study, we directly and specifically deleted mTOR in adipocytes by crossing adipocyte-restricted adiponectin-Cre (Adipoq-Cre) mice with mTOR flox/flox mice.
Methods
Animals
All procedures involving mice were guided by Purdue University Animal Care and Use Committee. Additionally, all experimental protocols were approved by Purdue University Animal Care and Use Committee. Adipoq-Cre (stock no. 010803) and mTOR flox/flox (stock no. 011009) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were of a C57BL/6 J background and housed in an animal facility with free access to water and standard rodent chow food or HFD (TD.06414; Harlan, Indianapolis, IN, USA). PCR genotyping (Sigma-Aldrich, St Louis, MO, USA) was carried out as described by the supplier.
Indirect calorimetry study
Oxygen consumption (\( \overset{.}{V}{\mathrm{O}}_2 \)), carbon dioxide production (\( \overset{.}{V}{\mathrm{CO}}_2 \)), respiratory exchange ratio (RER) and heat production were measured by using an indirect calorimetry system (Oxymax, Columbus Instruments, Columbus, OH, USA) [7].
GTTs and insulin tolerance tests
GTTs and insulin tolerance tests (ITT) were performed as described previously [7].
Haematoxylin–eosin staining
For adipose tissues, section and haematoxylin and eosin (H&E) staining was carried out as described previously [7]. For liver tissues, frozen livers were cut into 10 μm thick cross sections using a Leica CM1850 cryostat (Leica Microsystems, Wetzlar, Germany) for H&E staining. Whole-slide digital images were collected at magnification × 20 with an Aperio Scan Scope slide scanner (Aperio, Vista, CA, USA). Scanned images were analysed by Photoshop CS3 (San Jose, CA, USA) to calculate nuclei numbers.
Primary adipocyte isolation and culture
The BAT and WAT stromal vascular fraction (SVF) cells were isolated using collagenase digestion [7]. For adipogenic differentiation, cells were induced to differentiate when they reached 90% confluence [7].
Oil Red O staining
Cultured cells or liver sections were stained using Oil Red O work solutions containing 6 ml stock solution (5 g/l in isopropanol) and 4 ml double-distilled H2O for 30 min. After staining, the cells or liver sections were washed and pictured.
Triacylglycerol and NEFA assay
The triacylglycerol (TG) levels in liver, muscle and serum were determined using enzymatic methods (Cayman kit, no. 10010303; Ann Arbor, MI, USA). The levels of NEFA in serum were measured by fluorimetric assay kit (Cayman, no. 700310).
Total RNA extraction, cDNA synthesis and real-time PCR
Total RNA was extracted from cells or tissues using Trizol Reagent (Sigma-Aldrich). The purity and concentration of total RNA were measured and then 5 μg of total RNA were reversed transcribed. Real-time PCR was carried out and the \( {2}^{-\Delta \Delta {\mathrm{C}}_{\mathrm{t}}} \) method was used to analyse the relative changes in gene expression normalised against 18S rRNA as internal control [7].
Protein extraction and western blot analysis
Total protein was extracted from cells or tissues using RIPA buffer [7]. The UCP1 antibody and the mitochondrial antibody cocktail were from Abcam (Abcam, Cambridge, MA, USA); the p-S6, S6, p-4EBP1, 4EBP1, p-AKT, AKT antibodies were from Cell Signalling Technology (Danvers, MA, USA); all other antibodies were from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Dallas, TX, USA). Protein separation and western blot analysis were conducted as described previously [33].
Data analysis
No data were excluded and all experimental data are presented as means ± SEM. Comparisons were made by unpaired two-tailed Student’s t tests. Effects were considered significant at p < 0.05. Samples were randomised. Experimenters were not completely blinded to group assignment and outcome assessment.
Results
Generation of adipocyte-specific mTOR-knockout mice
To directly investigate the role of mTOR in adipogenesis, we used the Cre–loxP recombination system. mTOR flox/flox mice were crossed with Adipoq-Cre mice that express Cre recombinase under control of the adiponectin gene promoter. The adipocyte-specific expression pattern of Adipoq-Cre has been verified by previous studies [31, 34, 35]. Thus, in the Adipoq-Cre/mTOR flox/flox mice (abbreviated as Adipoq-mTOR) the exons 1–5 of mTOR should be deleted in adipocytes, leading to loss of expression of mTOR protein (Fig. 1a). Real-time PCR and western blotting analysis confirmed the efficient and specific deletion of mTOR in BAT as well as in inguinal WAT (iWAT), anterior subcutaneous WAT (asWAT) and epididymal WAT (eWAT) (Fig. 1b and electronic supplementary material [ESM] Fig. 1a, b) but not in non-adipose tissues including muscle, kidney, liver, heart and lung (Fig. 1c and ESM Fig. 1c, d).

Adipoq-Cre mediated deletion of mTOR decreases fat mass. (a) Targeting strategy for deletion of mTOR. (b, c) mRNA levels of mTOR in adipose (b, n = 4) and non-adipose tissues (c, n = 3). (d) The body weight of WT (mTOR flox/flox) and KO (Adipoq-mTOR) mice (n = 6). (e) Representative images of BAT and WAT depots. Scale bar, 3 mm. (f, g) The weights of adipose (f) and non-adipose tissues (g) (n = 6). Mice 8–10 weeks of age were used. White bars, WT mice; black bars, KO mice. Error bars represent SEM; *p < 0.05, **p < 0.01 and ***p < 0.001 for KO vs WT. EDL, extensor digitorum longus muscle; Gas, gastrocnemius muscle; Hrt, heart; Kid, kidney; Liv, liver; Sol, soleus muscle; Spl, spleen; TA, tibialis anterior muscle
Adipocyte-specific deletion of mTOR causes reduction of fat mass
Adipoq-mTOR mice were born at expected Mendelian ratios and were indistinguishable from their wild-type (WT) littermates. On the normal chow diet, the Adipoq-mTOR mice showed a similar growth curve and food intake to the WT littermates (data not shown) and the body weight was similar for WT and knockout (KO) mice at 8 weeks of age (Fig. 1d). Notably, the adipose tissues from Adipoq-mTOR mice were smaller in size than those from WT mice (Fig. 1e). The mass of BAT, iWAT, asWAT and eWAT taken from Adipoq-mTOR mice was 55%, 69%, 86% and 43% lighter, respectively, than the mass of tissue taken from their WT littermates (Fig. 1f). The muscle mass in Adipoq-mTOR mice was unchanged (Fig. 1g). Strikingly, the liver, spleen and heart of the Adipoq-mTOR mice were 58%, 42% and 32% heavier, respectively, than those of WT mice (Fig. 1g). The lipid and TG content was much higher in liver (ESM Fig. 2a–c) and skeletal muscles (ESM Fig. 2d) of Adipoq-mTOR mice compared with WT mice. In addition, we found lower TG levels and higher NEFA levels in serum of Adipoq-mTOR mice (ESM Fig. 2e, f). Taken together, adipocyte-specific deletion of mTOR caused reduction of adipose tissues but expansion of non-adipose tissues, especially the liver.
Ablation of mTOR affects adipogenesis of brown adipocytes
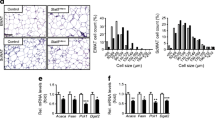
We next examined whether the decreased BAT mass in Adipoq-mTOR mice is due to reduction in adipocyte cell number, cell size or both. Results from H&E staining and nuclei density analysis (number of nuclei per unit area) showed there to be a higher density of nuclei in the KO mice (335 ± 21/area) than in the WT mice (171 ± 14/area), suggesting smaller BAT adipocyte size in the KO mice (Fig. 2a). Consistently, the genomic DNA content per BAT depot was lower in the Adipoq-mTOR mice than in the WT mice (Fig. 2b). Taken together, the reduced BAT mass in Adipoq-mTOR mice is a consequence of a decrease in cell size as well as cell numbers.

mTOR deficiency inhibits BAT development. (a) H&E staining of BAT sections. Scale bar, 100 μm. (b) Genomic DNA content per BAT depot (n = 4). (c) Levels of UCP1, FABP4 and mitochondrial proteins in BAT. (d, e) mRNA levels of mature adipocyte marker genes and BAT-selective genes (d, n = 4 or 5) and lipolysis-related genes (e, n = 3) in BAT from WT and KO mice. White bars, WT mice; black bars, KO mice. Error bars represent SEM; *p < 0.05, **p < 0.01 and ***p < 0.001 for KO vs WT. ATP5A, ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1; GAPDH, glyceraldehyde-3-phosphate; dehydrogenase; MTCO1, mitochondrially encoded cytochrome c oxidase I; NDUFB8, NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8; SDHB, succinate dehydrogenase complex, subunit B, iron sulfur (Ip); UQCRC2, ubiquinol cytochrome c reductase core protein 2
We next analysed the molecular phenotype of BAT from WT and Adipoq-mTOR mice. Mitochondrial biogenesis and brown adipocyte marker proteins and their genes, such as Ucp1, Prdm16, Cidea and Ppara, were expressed at similar levels in WT and Adipoq-mTOR mice (Fig. 2c, d). However, the BAT from Adipoq-mTOR mice contained lower levels of fatty acid binding protein 4 (FABP4), a mature adipocyte marker (Fig. 2c). Consistently, the mRNA levels of mature adipocyte markers (Fabp4, Adipoq, Lep) were lower in the BAT of Adipoq-mTOR mice compared with WT mice (Fig. 2d). The expression levels of lipolysis-related genes Pnpla2 and Lipe were unchanged (Fig. 2e) by mTOR deletion. These data strongly suggest that deletion of mTOR inhibits brown adipocyte adipogenesis in vivo.
To verify whether deletion of mTOR affects brown pre-adipocyte differentiation in culture, we isolated BAT SVF cells from the WT and Adipoq-mTOR mice. We observed that mTOR depletion had a more dramatic effect on BAT in vitro than in vivo (ESM Fig. 3a). mTOR deletion inhibited brown adipocyte differentiation and TG accumulation (ESM Fig. 3a). Consistent with the in vivo data, mTOR-deficient brown adipocytes expressed lower levels of FABP4 and Adipoq in culture (ESM Fig. 3b, c). In addition, the expression of Ucp1 and Cidea was also decreased by mTOR knockout (ESM Fig. 3c). Together, these results indicate that deletion of mTOR inhibits brown adipocyte differentiation and adipogenesis in vivo and in vitro.
Deletion of mTOR induces browning of iWAT
To investigate the effect of mTOR deficiency on WAT, we examined the morphology of iWAT. Interestingly, H&E staining revealed that the iWAT of Adipoq-mTOR mice contained numerous brown adipocyte-like cells filled with multilocular lipid droplets (Fig. 3a). The iWAT of KO mice had a higher number of nuclei per area than iWAT of WT mice (WT 24 ± 3 vs KO 110 ± 11) but had a similar genomic DNA content per depot (Fig. 3a, b). We also observed more robust integrin αM (Cd11b) labelling in the iWAT of KO compared with WT mice (data not shown), indicative of increased inflammatory infiltration. In addition, deletion of mTOR decreased the expression of FABP4 (Fig. 3c). Interestingly, higher levels of UCP1 protein were detected in the iWAT of Adipoq-mTOR mice compared with WT mice (Fig. 3c). Consistently, Adipoq-mTOR iWAT expressed higher levels of BAT signature markers Ucp1, Cidea and Ppara (Fig. 3d). Also, mTOR deletion upregulated the expression of Lipe but downregulated the expression of Adipoq, Lep and Pnpla2 in iWAT (Fig. 3d, e). These data suggest that lack of mTOR inhibits the development of iWAT but induces its browning.

mTOR deficiency inhibits WAT development and induces browning of WAT. (a) H&E staining of iWAT sections. Scale bar, 100 μm. (b) Genomic DNA content per iWAT depot of WT and KO mice (n = 4). (c) Protein levels of UCP1 and FABP4 in iWAT. (d, e) mRNA levels of mature adipocyte marker genes and BAT-selective genes (d) and lipolysis-related genes (e) in iWAT from WT and KO mice (n = 5). White bars, WT mice; black bars, KO mice. Error bars represent SEM; *p < 0.05, **p < 0.01 and ***p < 0.001 for KO vs WT. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
To examine whether the effects of mTOR deletion on browning of iWAT is cell autonomous, we isolated SVF cells from iWAT of WT and Adipoq-mTOR mice. Oil Red O staining indicated that deletion of mTOR inhibits adipocyte differentiation and TG accumulation (ESM Fig. 4a). Notably, the expression level of Ucp1 and mature adipocyte markers was significantly lower in mTOR KO white adipocytes (ESM Fig. 4b, c). These results indicated that deletion of mTOR induces browning of WAT in a non-cell autonomous manner.
mTOR deletion affects insulin sensitivity but not energy metabolism
Adipose tissues regulate systemic glucose metabolism and insulin sensitivity [7, 36]. To determine whether reduction of BAT and WAT mass in Adipoq-mTOR mice affects systemic glucose metabolism and insulin sensitivity, we conducted GTTs and ITTs. Compared with WT littermates, Adipoq-mTOR mice had higher blood glucose levels after glucose injection (Fig. 4a) and a slower rate of insulin-stimulated glucose clearance (Fig. 4b). However, compared with WT mice, the Adipoq-mTOR mice had similar values for \( \overset{.}{V}{\mathrm{O}}_2 \), \( \overset{.}{V}{\mathrm{CO}}_2 \), heat production and RER (Fig. 4c–f). Taken together, these results suggest that adipocyte-specific deletion of mTOR does not affect energy metabolism but results in insulin resistance.

Induced insulin resistance in Adioq-mTOR mice. (a, b) Blood glucose concentrations during IPGTT (a, n = 8) and ITT (b, n = 7) performed on WT and KO mice. (c–f) Average oxygen consumption (c), CO2 production (d), heat production (e) and RER (f) of WT and KO mice (n = 6). White circles and bars, WT mice; black circles and bars, KO mice. Error bars represent SEM; *p < 0.05 and ***p < 0.001 for KO vs WT
To further confirm the effects of mTOR deficiency on energy metabolism and insulin sensitivity, we fed the WT and KO mice with HFD. Although the body weight of Adipoq-mTOR mice and WT mice was similar before and after HFD feeding, the fat mass of the KO mice was consistently less than that of the WT mice (ESM Fig. 5a, b). The liver mass in the Adipoq-mTOR mice was dramatically increased, whereas other tissues, including kidney, heart, lung, tibialis anterior muscle and gastrocnemius muscle, were unchanged (ESM Fig. 5c, d). Notably, the Adipoq-mTOR mice retained insulin resistance after HFD feeding (ESM Fig. 5e, f). These results together suggest that adipocyte-specific deletion of mTOR causes insulin resistance regardless of dietary conditions.
mTOR affects adipogenesis through PPARγ
To understand the molecular mechanism through which mTOR affects fat development and adipogenesis, we examined peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer binding protein α (C/EBPα), two key transcription factors that regulate adipogenesis [37]. Western blotting and real-time PCR results showed that mTOR deficiency dramatically decreased the expression of PPARγ and C/EBPα in BAT (Fig. 5a, b), as well as in brown adipocytes (Fig. 5c). Similar results were obtained using iWAT tissue and differentiated iWAT adipocytes from the KO and WT mice (ESM Fig. 6a–c). Rosiglitazone is a PPARγ agonist that induces PPARγ expression and increases its activity [6, 38]. Consistently, rosiglitazone treatment significantly enhanced expression of Pparg, Cebpa, Fabp4, Adipoq and Ucp1 in brown adipocytes (Fig. 5d).

mTOR deletion affects brown pre-adipocyte differentiation through PPARγ. (a) Protein levels of PPARγ and C/EBPα in BAT of Adipoq-mTOR (KO) and WT mice. (b) mRNA levels of Pparg, Cebpa, Fasn, Lpl and Srebf1c in BAT (n = 4 or 5). White bars, WT mice; black bars, KO mice. (c) Protein levels of FABP4, PPARγ and C/EBPα in brown adipocytes. (d) mRNA levels of Pparg, Cebpa, Fabp4, Adipoq and Ucp1 in brown adipocytes (n = 6). White bars, vehicle control group; black bars, rosiglitazone (Ros) group. (e) Oil Red O staining of brown adipocytes treated with or without Ros. (f) mRNA levels of Fabp4, Adipoq and Ucp1 in brown adipocytes treated with or without Ros (n = 6). White bars, WT cells treated with vehicle; light-grey bars, WT cells treated with Ros; dark-grey bars, KO cells treated with vehicle; black bars, KO cells treated with Ros. Error bars represent SEM; *p < 0.05, **p < 0.01 and ***p < 0.001 for KO vs WT, Ros vs control or indicated comparisons. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
To confirm whether PPARγ mediates the effects of mTOR deficiency on adipogenesis, we treated the WT and KO brown adipocytes with rosiglitazone. Remarkably, rosiglitazone treatment rescued the differentiation of the mTOR-deficient brown pre-adipocytes (Fig. 5e). Consistently, the expression of Fabp4, Adipoq and Ucp1 in brown adipocytes was significantly increased in both WT and KO brown adipocytes after rosiglitazone treatment (Fig. 5f). Taken together, we conclude that mTOR deletion affects adipogenesis, at least in part, through the PPARγ signalling pathway.
Discussion
Here, we generated the Adipoq-mTOR mouse model and directly identified a role for mTOR in BAT and WAT. We provided evidence that deletion of mTOR inhibits fat development and leads to insulin resistance. Consistently, we demonstrated that mTOR deletion decreases lipid accumulation and differentiation of brown and white adipocytes. We further elucidated the PPARγ signalling through which mTOR regulates the differentiation of pre-adipocytes. Our study reveals the critical role of the mTOR signalling pathway in regulating adipose tissues development, whole-body energy metabolism and insulin sensitivity.
The roles of mTORC1 and mTORC2 in adipose tissues have been studied separately by genetic perturbation of key components involved [15, 17, 24, 30], but the overall role of mTOR signalling has not been reported. Here, we used a highly adipocyte-specific Adipoq-Cre [31, 34, 35] to delete mTOR in mice and demonstrated that mTOR deletion inhibits BAT and WAT development. We found that adipose-specific deletion of mTOR dramatically decreased the levels of mTOR and phosphorylated S6 and 4EBP1 proteins but, surprisingly, increased phosphorylated AKT (S473) in BAT and iWAT. The increased levels of phosphorylated AKT might be due to the feedback activation of AKT signalling induced by mTORC1 deletion or a kinase that phosphorylates AKT, such as DNA-protein kinase [39] and integrin-linked kinase [40]. In this regard, the increased phosphorylated AKT level cannot improve insulin sensitivity due to blockage of downstream mTOR.
We found that both the number and size of adipocytes were decreased in KO mice. Consistent with our findings, adipose-specific deletion of Raptor also results in leaner mice with lighter fat pads due to smaller and fewer WAT cells [24]. Inhibition of mTORC1 with rapamycin or by knockout of S6k1 (also known as Rps6kb1) also reduced fat mass and cell size [25, 41]. These results together suggest that mTORC1 plays a positive role in regulating the development of adipose. The role of mTORC2 in adipose tissue has been studied recently [15, 17, 30], but the results are controversial. Two studies using FABP4-Cre to drive the deletion of Rictor reported no effect on fat mass or individual adipocyte size [17, 30]. However, these results are inconsistent with the phenotypes observed in Myf5-Cre driven Rictor-knockout mice, in which the mass of Myf5-lineage adipocytes was decreased [15]. The different phenotypes from these studies may be due to the Cre mice that were used. Although BAT and some white adipocytes are from Myf5 lineage, the Myf5-Cre also drives gene deletion in skeletal muscles [42, 43], which can secrete myokines that regulate adipose development and function [3, 33]. Moreover, leaky expression of FABP4-Cre was found in adipocyte progenitors and non-adipose tissues [31, 32, 44]. Therefore, the leaky expression of FABP4-Cre or Myf5-Cre makes it difficult to interpret the data from these studies. Our Adipoq-mTOR mouse model drives specific deletion of mTOR in adipocytes and provides an excellent model with which to examine the role of mTOR in adipocytes and adipose tissues.
We found that adipocyte-specific deletion of mTOR drives browning of WAT. The KO iWAT has numerous small adipocytes with higher levels of Ucp1 and Lipe expression. Likewise, the browning of WAT is also commonly seen in lipodystrophic models, such as Bscl2 knockout mice [45], in which fatty acids produced from unbridled lipolysis induce UCP1 and browning of WAT [46]. Moreover, previous studies have also reported that adipose-specific deletion of Raptor upregulated the expression of genes related to mitochondrial biogenesis in WAT [24]. S6k1 deletion also induces formation of multilocular adipocytes and increases expression of Ucp1 and Pgc1a [25]. In contrast, deletion of Tsc1 induced the brown-to-white adipocyte phenotypic switch [26]. These results indicate that inhibition of mTORC1 signalling contributes to the white-to-brown adipocyte switching. In addition, a recent study reported that adipocytes lacking Rictor have stronger UCP1 staining and brown-adipocyte-like characteristics [15], suggesting that mTORC2 may also regulate white-to-brown switching.
Adipose tissues participate in regulating whole-body energy metabolism, glucose homeostasis and insulin sensitivity [17]. Notably, we found that adipocyte-specific deletion of mTOR has no effect on whole-body energy metabolism but causes insulin resistance. It has been reported that mTORC1 is a regulator of adipose metabolism and controls whole-body energy homeostasis [24]. Adipose-specific deletion of Raptor improves energy expenditure and insulin sensitivity and protects mice against HFD-induced obesity [24]. Consistently, S6k1-deficient mice have enhanced β-oxidation, faster glucose clearance and insulin sensitivity and are resistant to HFD-induced obesity [25]. However, administration of rapamycin prevents HFD-induced obesity and exacerbates glucose intolerance [41]. Distinct from mTORC1, loss of Rictor reduces glucose transport in fat cells [16, 17] and adipose-specific knockout of Rictor in mice leads to mild glucose intolerance and severe insulin resistance [17]. These reports suggest that the insulin resistance in Adipoq-mTOR mice might mainly result from the absence of mTOC2 in adipose tissues. However, we cannot exclude the effects of other factors, such as adipose tissue inflammation (data not shown), on insulin resistance in the KO mice.
Numerous studies have shown that hepatic steatosis is strongly associated with insulin resistance [47]. Notably, higher serum NEFA and increased hepatic steatosis were found in the Adipoq-mTOR mice. Consistent with our results, adipose-specific deletion of Rictor increases hepatic steatosis [17, 30]. However, the weight of liver was not affected in the S6k1-deletion mice [25] and even less lipid accumulation was found after rapamycin treatment [41]. Taken together, the insulin resistance in Adipoq-mTOR mice may be mainly due to hepatic steatosis induced by mTOC2 deficiency.
We found that deletion of mTOR inhibits pre-adipocyte differentiation through PPARγ, a master regulator of adipogenesis [37]. Treatment with the PPAR agonist rosiglitazone rescued the differentiation and lipid accumulation in mTOR-knockout adipocytes. Previous studies have demonstrated that mTORC1 affects adipocyte differentiation and lipid accumulation by regulating lipogenesis and lipolysis [48]. In 3 T3-L1 adipocytes, activation of mTORC1 signalling suppresses lipolysis and promotes lipid accumulation [48]. Basal rates of lipolysis were increased in S6k1-knockout mice [25]. Inhibition of mTORC1 signalling by knockdown of raptor or by rapamycin stimulates lipolysis by activation of Pnpla2 [48]. In addition, S6K1 depletion or rapamycin treatment inhibits adipocyte differentiation [13, 19, 20]. mTOR regulates pre-adipocyte differentiation and PPARγ activity probably not through the production of a ligand but rather through its direct impact on PPARγ transactivation activity [49] or through upregulation of AKT, a regulator of PPAR [12]. However, absence of Raptor in Myf5-lineage cells promotes osteogenesis and inhibits adipogenesis, while deletion of Rictor promotes adipogenesis and inhibits osteogenesis [50]. Therefore, mTORC1 and mTORC2 appear to counteract each other during adipocyte differentiation.
In conclusion, we demonstrate that mTOR plays several important roles in adipose development and adipocyte differentiation. These results provide a novel insight into the important role of mTOR in systemic metabolism. Such knowledge may be useful for developing strategies for treating obesity and related metabolic diseases.
Abbreviations
- asWAT:
-
Anterior subcutaneous WAT
- BAT:
-
Brown adipose tissue
- C/EBPα:
-
CCAAT/enhancer binding protein α
- eWAT:
-
Epididymal WAT
- FABP4:
-
Fatty acid binding protein 4
- H&E:
-
Haematoxylin and eosin
- HFD:
-
High-fat diet
- ITT:
-
Insulin tolerance test
- iWAT:
-
Inguinal WAT
- KO:
-
Knockout
- mTOR:
-
Mechanistic target of rapamycin
- mTORC:
-
mTOR complex
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RER:
-
Respiratory exchange ratio
- S6K1:
-
Ribosomal S6 kinase 1
- SVF:
-
Stromal vascular fraction
- TG:
-
Triacylglycerol
- UCP1:
-
Uncoupling protein 1
- WAT:
-
White adipose tissue
- WT:
-
Wild-type
References
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Walden TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J (2012) Recruited vs. nonrecruited molecular signatures of brown, ʻbriteʼ, and white adipose tissues. Am J Physiol Endocrinol Metab 302:E19–E31
Bostrom P, Wu J, Jedrychowski MP et al (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468
Cao L, Choi EY, Liu XL et al (2011) White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab 14:324–338
Fisher FM, Kleiner S, Douris N et al (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26:271–281
Ohno H, Shinoda K, Spiegelman BM, Kajimura S (2012) PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab 15:395–404
Bi P, Shan T, Liu W et al (2014) Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med 20:911–918
Crane JD, Palanivel R, Mottillo EP et al (2015) Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med 21:166–172
Rosen ED, Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156:20–44
Lamming DW, Sabatini DM (2013) A central role for mTOR in lipid homeostasis. Cell Metab 18:465–469
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Yoon MS, Zhang C, Sun Y, Schoenherr CJ, Chen J (2013) Mechanistic target of rapamycin controls homeostasis of adipogenesis. J Lipid Res 54:2166–2173
Carnevalli LS, Masuda K, Frigerio F et al (2010) S6K1 plays a critical role in early adipocyte differentiation. Dev Cell 18:763–774
Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G (2009) Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 23:496–511
Hung CM, Calejman CM, Sanchez-Gurmaches J et al (2014) Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep 8:256–271
Olsen JM, Sato M, Dallner OS et al (2014) Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation. J Cell Biol 207:365–374
Kumar A, Lawrence JC, Jung DY et al (2010) Fat cell-specific ablation of Rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 59:1397–1406
El-Chaar D, Gagnon A, Sorisky A (2004) Inhibition of insulin signaling and adipogenesis by rapamycin: effect on phosphorylation of p70 S6 kinase vs eIF4E-BP1. Int J Obes 28:191–198
Bell A, Grunder L, Sorisky A (2000) Rapamycin inhibits human adipocyte differentiation in primary culture. Obes Res 8:249–254
Yeh WC, Bierer BE, Mcknight SL (1995) Rapamycin inhibits clonal expansion and adipogenic differentiation of 3t3-L1 cells. Proc Natl Acad Sci U S A 92:11086–11090
Vila-Bedmar R, Lorenzo M, Fernandez-Veledo S (2010) Adenosine 5′-monophosphate-activated protein kinase-mammalian target of rapamycin cross talk regulates brown adipocyte differentiation. Endocrinology 151:980–992
Chang GR, Chiu YS, Wu YY et al (2009) Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci 109:496–503
Polak P, Hall MN (2009) mTOR and the control of whole body metabolism. Curr Opin Cell Biol 21:209–218
Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN (2008) Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8:399–410
Um SH, Frigerio F, Watanabe M et al (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205
Xiang X, Lan H, Tang H et al (2015) Tuberous sclerosis complex 1-mechanistic target of rapamycin complex 1 signaling determines brown-to-white adipocyte phenotypic switch. Diabetes 64:519–528
Guertin DA, Stevens DM, Thoreen CC et al (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11:859–871
Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA (2006) Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 11:583–589
Jones KT, Greer ER, Pearce D, Ashrafi K (2009) Rictor/TORC2 regulates Caenorhabditis elegans fat storage, body size, and development through sgk-1. PLoS Biol 7:e60
Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN (2009) mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A 106:9902–9907
Lee KY, Russell SJ, Ussar S et al (2013) Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62:864–874
Wang JQ, Liu RX, Wang F et al (2013) Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat Cell Biol 15:1455–1463
Shan TZ, Liang XR, Bi PP, Kuang SH (2013) Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1 alpha-Fndc5 pathway in muscle. FASEB J 27:1981–1989
Jeffery E, Berry R, Church CD et al (2014) Characterization of Cre recombinase models for the study of adipose tissue. Adipocytes 3:206–211
Wang ZV, Deng Y, Wang QA, Sun K, Scherer PE (2010) Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151:2933–2939
Stanford KI, Middelbeek RJW, Townsend KL et al (2013) Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest 123:215–223
Rosen ED, MacDougald OA (2006) Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7:885–896
Barquissau V, Beuzelin D, Giroud M et al (2014) PPAR gamma and PPAR alpha agonists induce white-to-brown conversion of human white adipocytes along with a metabolic shift from glucose to fatty acid oxidation. Diabetologia 57:S283
Davidson D, Amrein L, Panasci L, Aloyz R (2013) Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol 4:5
Zhu MY, Chen D, Li DY et al (2013) Luteolin inhibits angiotensin II-induced human umbilical vein endothelial cell proliferation and migration through downregulation of Src and Akt phosphorylation. Circ J 77:772–779
Chang GR, Wu YY, Chiu YS et al (2009) Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol 105:188–198
Kuang S, Kuroda K, Le Grand F, Rudnicki MA (2007) Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129:999–1010
Dong F, Sun X, Liu W et al (2006) Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development 133:4891–4899
Shan TZ, Liu WY, Kuang SH (2013) Fatty acid binding protein 4 expression marks a population of adipocyte progenitors in white and brown adipose tissues. FASEB J 27:277–287
Chen W, Zhou H, Liu S et al (2013) Altered lipid metabolism in residual white adipose tissues of Bscl2 deficient mice. Plos One 8:e82526
Fedorenko A, Lishko PV, Kirichok Y (2012) Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151:400–413
Monetti M, Levin MC, Watt MJ et al (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6:69–78
Chakrabarti P, English T, Shi J, Smas CM, Kandror KV (2010) Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 59:775–781
Kim JE, Chen J (2004) regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 53:2748–2756
Martin SK, Fitter S, Dutta AK et al (2015) Brief report: the differential roles of mTORC1 and mTORC2 in mesenchymal stem cell differentiation. Stem Cells 33:1359–1365
Acknowledgements
We thank J. Wu (Department of Animal Sciences, Purdue University, West Lafayette, IN, USA) for mouse colony maintenance and technical support, and members of the Kuang Laboratory for comments.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
The project was partially supported by funding from the National Institutes of Health (NIH, R01AR060652) and an incentive grant from Purdue University Office of Vice President for Research (OVPR) to SHK, ‘Hundred Talents Program’ funding from Zhejiang University to TZS and Nanhu Scholars Program for Young Scholars of Xinyang Normal University (XYNU) to PPZ.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
TZS, PPZ and SHK designed and conceived the research. TZS, PPZ, QYJ and YX performed the experiments, analysed and interpreted the data and revised the manuscript. TZS and SHK wrote the manuscript. YZW assisted with data analysis and interpretation and with revision of the article. All authors approved the final version of the manuscript. TZS and SHK are responsible for the integrity of the work as a whole.
Additional information
Tizhong Shan, Pengpeng Zhang, Qinyang Jiang and Yan Xiong contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 93 kb)
ESM Fig. 2
(PDF 157 kb)
ESM Fig. 3
(PDF 114 kb)
ESM Fig. 4
(PDF 152 kb)
ESM Fig. 5
(PDF 17 kb)
ESM Fig. 6
(PDF 93 kb)
Rights and permissions
About this article

Cite this article
Shan, T., Zhang, P., Jiang, Q. et al. Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia 59, 1995–2004 (2016). https://doi.org/10.1007/s00125-016-4006-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4006-4