
Abstract
Islet inflammation contributes to beta cell demise in both type 1 and type 2 diabetes. 12-Lipoxygenase (12-LO, gene expressed as ALOX12 in humans and 12-Lo in rodents in this manuscript) produces proinflammatory metabolites such as 12(S)-hydroxyeicosatetraenoic acids through dioxygenation of polyunsaturated fatty acids. 12-LO was first implicated in diabetes when the increase in 12-Lo expression and 12(S)-hydroxyeicosatetraenoic acid was noted in rodent models of diabetes. Subsequently, germline 12-Lo −/− was shown to prevent the development of hyperglycemia in mouse models of type 1 diabetes and in high-fat fed mice. More recently, beta cell-specific 12-Lo −/− was shown to protect mice against hyperglycaemia after streptozotocin and a high-fat diet. In humans, 12-LO expression is increased in pancreatic islets of autoantibody-positive, type 1 diabetic and type 2 diabetic organ donors. Interestingly, the high expression of ALOX12 is associated with the alteration in first-phase glucose-stimulated insulin secretion in human type 2 diabetic islets. To further clarify the role of islet 12-LO in diabetes and to validate 12-LO as a therapeutic target of diabetes, we have studied selective pharmacological inhibitors for 12-LO. The compounds we have identified show promise: they protect beta cell lines and human islets from apoptosis and preserve insulin secretion when challenged by proinflammatory cytokine mixture. Currently studies are underway to test the compounds in mouse models of diabetes. This review summarises a presentation given at the ‘Islet inflammation in type 2 diabetes’ symposium at the 2015 annual meeting of the EASD. It is accompanied two other mini-reviews on topics from this symposium (by Simone Baltrusch, DOI: 10.1007/s00125-016-3891-x and Marc Donath, DOI: 10.1007/s00125-016-3873-z) and a commentary by the Session Chair, Piero Marchetti (DOI: 10.1007/s00125-016-3875-x).
Similar content being viewed by others


Introduction
Activation of inflammatory pathways in pancreatic islets has long been appreciated to be central to the pathology of type 1 diabetes, and has more recently been considered as a major component of type 2 diabetes, contributing to pancreatic beta and alpha cell failure [1]. Proinflammatory mediators, such as cytokines and chemokines, produced by cells of the adaptive or innate immune system that infiltrate the islet [2, 3], islet intrinsic molecules, such as amyloid polypeptide deposits [3] and excess fatty acids [4], all contribute to the inflammatory milieu. Collectively, these molecules are considered to activate proinflammatory cascades including inflammasome formation [5], Toll-like receptor activation [4, 5] and endoplasmic reticulum stress, which in turn promote the progressive functional demise of the beta cells.
Proinflammatory lipid mediators resulting from increased processing of fatty acids are another potentially significant source of inflammation in islets under diabetogenic stress. In support, the activation of the lipoxygenase pathway is noted in other chronic inflammatory conditions such as obese adipose tissue, atherosclerosis and the brain affected by Alzheimer’s disease [6]. Lipoxygenases convert polyunsaturated fatty acids into eicosanoids, including leukotrienes, lipoxins, hepoxilins, eoxins, resolvins and protectins [7]. Of five lipoxygenase isoforms identified in humans, 12-lipoxygenase (12-LO; encoded by the ALOX12 gene) is the major lipoxygenase isoform in human islets and produces predominantly 12(S)-hydroxyeicosatetraenoic acid (12-S-HETE) from arachidonic acid (Fig. 1) [8]. In mice, the enzyme primarily responsible for the production of 12-S-HETE in islets is leucocyte 12-LO (also known as 12/15-LO, encoded by Alox15. Throughout the manuscript, 12-Lo refers to Alox15 and 12-LO refers to leucocyte 12-LO protein in mice/rats) (Fig. 1) [6]. Increased production of 12-S-HETE in islets and adipose tissue has been reported in rodent models of type 1 and type 2 diabetes and in diabetic humans [8, 9]. This metabolite exerts pathogenic inflammatory effects by activating downstream cytokines such as IL-12 [10] or by activating the second messengers c-Jun N-terminal kinase and p38 mitogen-activated protein kinase (MAPK) [11]. Also, mitochondrial and oxidative stress, conditions to which beta cells are highly sensitive, have been reported to be increased following 12-LO activation in the heart as a result of an increase in mitochondrial nitric oxide and intramitochondrial calcium [12].

Generation of lipid metabolites from arachidonic acid. Arachidonic acid [n-6 polyunsaturated fatty acid (20:4)] is generated from membrane phospholipids by phospholipase A2. 12-Lipoxygenase (12-LO) converts arachidonic acid to 12(S)-hydroperoxyeicosatetraenoic acid [12(S)- H(p)ETE] through oxidation at carbon-12 (9th carbon from the ω tail). 12(S)-H(p)ETE is an unstable intermediate metabolite and is quickly converted to 12-S-HETE. The human enzyme most involved in generating 12-S-HETE in islets is 12-LO and is encoded by the ALOX12 gene. In mice, the products of both Alox12 and Alox15 (platelet 12-LO and leucocyte 12-LO, respectively) generate 12-S-HETE [9]. The enzymes 5-lipoxygenase (5-LO) and cyclo-oxygenase also metabolise arachidonic acid; the former produces leukotrienes, the latter produces prostaglandins. 12-S-HETE increases proinflammatory mediators including IL-12 [10], the phosphorylated form of p38 MAPK [23] and NOX1 [24], thus causing endoplasmic reticulum (ER) stress, mitochondrial stress and oxidative stress, cumulatively contributing to the impairment in insulin secretion and beta cell death. Thus, the inhibitors of 12-LO, such as ML127 and ML355, hold therapeutic promise in the prevention and recovery of beta cell function in type 1 and type 2 diabetes
This mini-review will focus on evidence substantiating a significant role of the 12-LO pathway in islet dysfunction and beta cell demise in both type 1 and type 2 diabetes and will describe pre-clinical studies using selective and potent 12-LO inhibitors, which represent promising future therapeutics.
The contribution of 12-LO in the development of diabetes: lessons learned from mouse models
Mouse models of diabetes have proven useful in determining a role for 12-LO in the pathogenesis of diabetes. We believe that activation of proinflammatory cytokine pathways by the 12-S-HETE produced as a result of the metabolism of arachidonic acid by 12-LO is the key to the development of both forms of diabetes.
With respect to type 2 diabetes, the deletion of mouse 12-Lo prevents high-fat diet (HFD)-induced beta cell dysfunction, insulin resistance and adipose tissue inflammation. Indeed, global 12-Lo deletion allows HFD-fed mice to maintain glucose tolerance levels equivalent to wild-type C57BL/6 mice fed a normal chow diet [13]. Further investigation into the role of 12-LO in type 2 diabetes suggests that these effects are highly dependent upon adipose tissue 12-Lo expression. Inflammation in fat has detrimental effects on islet health as shown by adipose tissue-specific 12-Lo −/− mice [14]. However, islet expression of 12-Lo also plays a role in beta cell health in models of type 2 diabetes. In an in vivo study, islet health was improved following HFD feeding of 12-Lo −/− mice compared with littermate controls [13]. In addition, in vitro testing of islets with inflammatory cytokines over a 24 h period resulted in diminished insulin secretion by islets from wild-type mice, while islets from pancreas-specific 12-Lo −/− animals (pLOKO mice) maintained insulin levels similar to those seen after a 4 h cytokine treatment [15]. While islets from wild-type mice show significant decreases in insulin production following stimulation with proinflammatory cytokines over a 24 h period, islets from pLOKO mice are relatively protected from the detrimental effects of cytokines [15]. Under these conditions, they produce about 30% less insulin compared with non-cytokine treated islets. However, islets from wild-type littermates, exhibit an almost 70% reduction in insulin production under the same culture conditions [15].
Type 1-like diabetes is prevented following the global deletion of 12-Lo in a multiple low-dose streptozotocin (STZ) model of diabetes on a C57BL/6 background [16]. Since the multiple low-dose STZ model approximates human type 1 diabetes, an NOD mouse line congenic for the 12-Lo −/− was generated. The resulting NOD mice, which are globally deficient for 12-Lo, are protected against developing spontaneous type 1-like diabetes [17]. The disease incidence in female mice is reduced to about 2%, compared with the normal range of 60–80% for spontaneous disease development. Indeed, these mice show reduced levels of proinflammatory cytokines in both islets and macrophages compared with NOD mice with normal levels of 12-LO [18].
Beta cell mass is higher in global 12-Lo −/− mice, even prior to the initiation of overt inflammation and true insulitis in the NOD model. This suggests that 12-LO contributes to the inflammatory process very early in disease pathogenesis. Indeed, quantification of islet-infiltrating macrophages is significantly reduced in 12-Lo −/− mice as early as 4 weeks of age [17].
Mouse macrophages are known to express high levels of 12-Lo, while B and T cells express virtually undetectable levels of the enzyme [18]. In 12-Lo −/− NOD mice, thioglycollate-elicited macrophages contain significantly decreased levels of proinflammatory cytokines, including IFNγ and IL-12, which are critical for the stimulation of autoimmune T cell responses. Macrophages have been detected very early in the development of type 1 diabetes, prior to T cell-mediated insulitis. Therefore, effects of 12-LO on T cells are likely mediated by upstream immune responses. On the basis of these data, we are generating a mouse with macrophage-specific 12-Lo −/− on the NOD background using the Lyz2 promoter to drive Cre recombinase.
Furthermore, pLOKO mice on the C57BL6 background are protected against low-dose STZ-induced diabetes [15]. Beta cell mass is maintained in the pLOKO mice, and the mice fair better in both random-fed glucose tests and classical glucose tolerance tests, compared with control littermates (12-Lo fl/fl without Cre). A mouse with islet-specific 12-Lo −/− on the NOD background is currently being developed.
The evidence suggests that 12-LO is a potential therapeutic target expressed early in both disease processes. We are hopeful that 12-LO inhibitors (see below) used in these mouse models will provide a rationale for the continuing development of pharmacological inhibitors for clinical development.
Evidence supporting the role of 12-LO in beta cell demise in human diabetes
To determine the contribution of 12-LO in the development of beta cell loss in human diabetes, we examined pancreatic sections of organ donors provided by the Network for Pancreatic Organ Donors with Diabetes (nPOD, www.jdrfnpod.org/). When tested using an antibody against 12-LO, the expression of 12-LO was negligible in pancreatic islets of non-diabetic donors. In contrast, the 12-LO signal was significantly increased in pancreatic islets of autoantibody-positive, type 1 diabetic and type 2 diabetic donors [19]. This study provided several important insights regarding the role of islet 12-LO in both human type 1 and type 2 diabetes. First, the fact that 12-LO staining was increased in pancreatic islets of autoantibody-positive donors—a group of individuals that includes subclinical type 1 diabetes at a high level—suggests that 12-LO activation may occur at an early stage during the development of type 1 diabetes. Among the donors with type 1 diabetes, 12-LO staining was limited to pancreases with residual beta cells. This observation again implicates that 12-LO activation may be associated with ongoing loss of beta cells and that blocking 12-LO activation may rescue residual beta cells. Interestingly, islet cells expressing 12-LO were mostly negative for insulin staining but showed weak staining for pancreatic polypeptide (PP). Although further study is required to define the nature of these 12-LO/PP double-positive cells, the loss of insulin production in beta cells (de-differentiation) during the development of diabetes has been demonstrated in mouse models and implicated in human diabetes as well [20, 21]. These 12-LO/PP double-positive cells may potentially indicate de-differentiated beta cells. Finally, the presence of 12-LO-positive islets in type 2 diabetic donors who do not have autoantibodies strongly supports the notion that 12-LO is activated in both type 1 and type 2 diabetes in humans and can potentially serve as a therapeutic target for both forms of diabetes [1].
The upregulation of ALOX12 expression was demonstrated in human islets isolated from type 2 diabetic donors [22]. Interestingly, the expression of ALOX12 was not correlated with that of CCL2 or TNFα—two other proinflammatory genes shown to be increased in type 2 diabetic human islets. ALOX12 expression was particularly high in type 2 diabetic islets with blunted but preserved glucose-stimulated insulin secretion. On the other hand, the expression of CCL2 and TNFa was increased in type 2 diabetic islets that showed little response to glucose in terms of insulin secretion. Collectively, 12-LO may serve as a target to prevent the loss of functional beta cells in both type 1 and type 2 diabetes, most likely at the stage when ongoing beta cell loss is underway.
Once 12-LO is activated, the concentration of the proinflammatory product, 12-S-HETE, will increase locally within pancreatic islets. The rise in 12-S-HETE can impair beta cell viability and function. We have shown that treatment of human islets in culture with 12-S-HETE reduces glucose-stimulated insulin secretion and induces beta cell apoptosis in association with an increase in phosphorylation of p38 MAPK [23] and NADPH oxidase-1 (NOX1) [24], implicating proinflammatory and oxidative stress pathways as downstream events of 12-LO activation. Importantly, the expression of ALOX12 and the production of 12-S-HETE are both upregulated by proinflammatory cytokines [23, 24], and oxidative stress itself could provoke an inflammatory response in human islets [1]. Once activated, 12-LO could create feed-forward amplification of a detrimental proinflammatory response within the pancreatic islet environment. Given that 12-LO is expressed in pancreatic islets at an early stage of diabetes development in humans, the blockade of 12-LO activation may serve as a powerful tool to prevent the progression of beta cell loss in both forms of diabetes.
Small molecule inhibitors of 12-LO: therapeutic potential
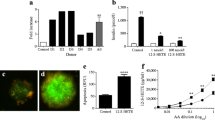
Clarification of the role of 12-LO activity in cell pathophysiology would be greatly aided by the identification of potent and selective inhibitors of 12-LO. Motivated by the interest in 12-LO inhibition as a therapeutic, active efforts have been made based on medicinal chemistry, computational chemistry and natural product isolation. These studies identified agents with micromolar potency for 12-LO inhibition and moderate selectivity, including, for example, nordihydroguaiaretic acid (NDGA) [25], brominated aryl phenols [26] and hinokitiol [27]. However, these compounds are not selective inhibitors of 12-LO. In addition, baicalein, which is marketed as a selective inhibitor of 12-LO, has been reported to be equipotent against both 15-LO-1 and 12-LO [28]. With the need for selective inhibitors of 12-LO, driven by the potential therapeutic benefit for such compounds, quantitative high-throughput screening (qHTS) against large libraries of small molecules has been conducted. Two chemical scaffolds resulting from the qHTS define new chemical series of potent inhibitors of 12-LO. A subsequent structure–activity relationship (SAR) study of the chemical scaffolds, 8-hydroxyquinoline and 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide, identified lead compounds ML127 and ML355, respectively [29–31]. Both ML127 and ML355 exhibit submicromolar potency for 12-LO inhibition and a greater than fiftyfold selectivity relative to other lipoxygenase enzymes. For in vitro/ex vivo validation studies, ML127 and ML355 are equally efficacious. In terms of drug development, ML355 exhibits a more expansive and descriptive SAR, along with favourable in vitro absorption, distribution, metabolism and elimination (ADME) and in vivo pharmacokinetic profile. In whole cell studies, both ML127 and ML355 proved to be potent inhibitors of 12-LO. The IC50 for ML127/ML355 for inhibition of 12-S-HETE production in the whole cell studies was analogous to the efficacy for inhibition of the 12-LO enzyme in cell-free models. These two compounds have facilitated the dissection of 12-LO activity in beta cells as it relates to beta cell dysfunction induced by inflammatory cytokines. Following acute exposure to inflammatory cytokines (TNFα, IL-1β, and IFNγ), beta cell function and survival is compromised. Many of the effects of inflammatory cytokines can be reproduced by the direct addition of 12-S-HETE; implying a role for 12-LO. ML127 and ML355, bioavailable selective inhibitors of 12-LO, have helped to identify 12-LO as a mediator of inflammatory cytokine-induced beta cell dysfunction (Fig. 1). Initial studies performed in homogeneous beta cell lines revealed that exposure of the cells to inflammatory cytokines resulted in defective glucose-stimulated insulin secretion and an elevation in cell apoptosis. With the addition of either ML127 or ML355, both these markers of beta cell function and survival are preserved in the presence of inflammatory cytokines [32]. Similar studies have also been performed in human islets. Encouragingly, ML127 and ML355 are efficacious in human islets, which bodes well for the development of future therapeutics to preserve and protect beta cells in diabetes. As in human islets from type 2 diabetic individuals, the first phase of insulin secretion following a glucose challenge is blunted in human islets from non-diabetic donors exposed to inflammatory cytokines. Our preliminary data indicate that compound ML355 may improve first-phase insulin secretion in non-diabetic human islets exposed to inflammatory cytokines.
Inflammation arises from access to and intake of excess nutrients and is considered to be a key driver of type 2 diabetes in genetically predisposed individuals [1]. Thus, a clinically relevant strategy to preserve functional beta cell mass under conditions of ongoing inflammation is needed. Given the substantial evidence for 12-LO activation in islets affected by diabetes in humans and mouse models, inhibition of 12-LO is a candidate strategy. The current selective inhibitors of 12-LO will serve to aid future proof-of-concept studies. The selective 12-LO inhibitors also offer a useful new tool for pathway dissection and the identification of new potential targets for intervention with the goal of preserving beta cell function. Gene changes related to cell stress have been identified as being modulated by the compounds ML127 and ML355. The discovery, characterisation and validation of selective 12-LO inhibitors have been an important first step in progressing a new therapeutic approach for diabetes. Consequently, 12-LO inhibition is now on the radar of major pharmaceutical industries.
Closing remarks
Considerable progress has been made in recent years towards understanding the modulation of the lipoxygenase pathway in beta cells and pancreatic islets in type 1 and type 2 diabetes. Robust evidence from animal models of diabetes indicates that germline 12-Lo deletion significantly reduces the frequency of development of type 1 diabetes and improves insulin sensitivity and glucose tolerance in obesity. Importantly, evidence from islets of type 2 diabetic patients suggests that ALOX12 is the major isoform that increases expression and that 12-S-HETE is the principal metabolite produced in dysfunctional islets. Also, some of the protective mechanisms resulting from the genetic deletion or pharmacological inhibition of 12-LO, such as decreased oxidative stress, endoplasmic reticulum stress and apoptosis, following a proinflammatory cytokine challenge have been described in rodent and human islets in vitro. Significant progress in dissecting the pathogenic components of the lipoxygenase pathway has been made possible by the recent discovery of selective and potent small molecule inhibitors of different lipoxygenase isoforms. However, it is critical to further our understanding of the biological mechanisms involved, particularly those directly responsible for enzyme activation in diabetes and the downstream molecular effectors. Investigating candidate activators of the different lipoxygenase isoforms may include local oxygen availability, redox cellular potential, glucose concentration, substrate availability and abundance, and amyloid deposits. We also need further insight into the mechanisms by which lipoxygenase products, such as 12-S-HETE, can propagate inflammation and endoplasmic reticulum or oxidative stress. These mechanisms can be mediated by the recently discovered G protein-coupled receptor 31 [33] either on islet cells or indirectly via immune cell activation through a mechanism that requires presence of 12-S-HETE in platelet-derived microparticles [34]. Last, but not least, a better understanding of the pathogenic roles of lipoxygenase in other insulin-sensitive tissues, such as adipose tissue, liver and muscle, as well as in cells of the immune system, will be critical to establish the best strategies for future therapies targeting the lipoxygenase pathway in diabetes. Along with these efforts, determination of the in vivo efficacy and safety of the newly discovered selective 12-LO inhibitors will be a major step forward towards identifying potent compounds to limit inflammation and to improve islet health in diabetes.
Abbreviations
- 12-S-HETE:
-
12(S)-Hydroxyeicosatetraenoic acid
- HFD:
-
High-fat diet
- 12-LO:
-
12-Lipoxygenase
- MAPK:
-
Mitogen-activated protein kinase
- NDGA:
-
Nordihydroguaiaretic acid
- NOX1:
-
NADPH oxidase-1
- pLOKO mice:
-
Pancreas-specific 12-Lo −/− mice
- PP:
-
Pancreatic polypeptide
- qHTS:
-
Quantitative high-throughput screening
- SAR:
-
Structure–activity relationship
- STZ:
-
Streptozotocin
References
Imai Y, Dobrian AD, Morris MA, Nadler JL (2013) Islet inflammation: a unifying target for diabetes treatment? Trends Endocrinol Metab 24:351–360
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) Islet-associated macrophages in type 2 diabetes. Diabetologia 52:1686–1688
Westwell-Roper CY, Ehses JA, Verchere CB (2014) Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 63:1698–1711
Eguchi K, Manabe I, Oishi-Tanaka Y et al (2012) Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15:518–533
Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA (2014) Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol 92:314–323
Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL (2011) Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res 50:115–131
Kuhn H, Banthiya S, van Leyen K (2015) Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta 1851:308–330
Imai Y, Dobrian AD, Weaver JR et al (2013) Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab 15(Suppl 3):117–129
Lieb DC, Brotman JJ, Hatcher MA et al (2014) Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J Clin Endocrinol Metab 99:E1713–E1720
Middleton MK, Rubinstein T, Pure E (2006) Cellular and molecular mechanisms of the selective regulation of IL-12 production by 12/15-lipoxygenase. J Immunol 176:265–274
Chen M, Yang ZD, Smith KM, Carter JD, Nadler JL (2005) Activation of 12-lipoxygenase in proinflammatory cytokine-mediated beta cell toxicity. Diabetologia 48:486–495
Nazarewicz RR, Zenebe WJ, Parihar A et al (2007) 12(S)-hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch Biochem Biophys 468:114–120
Nunemaker CS, Chen M, Pei H et al (2008) 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 295:E1065–E1075
Cole BK, Morris MA, Grzesik WJ, Leone KA, Nadler JL (2012) Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediat Inflamm 2012:851798
Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, Mirmira RG (2014) 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol Cell Biol 34:3735–3745
Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL (1999) Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest 103:1431–1436
McDuffie M, Maybee NA, Keller SR et al (2008) Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 57:199–208
Green-Mitchell SM, Tersey SA, Cole BK et al (2013) Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15 null mice, leading to protection against type 1 diabetes development. PLoS One 8:e56763
Grzesik WJ, Nadler JL, Machida Y, Nadler JL, Imai Y, Morris MA (2015) Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J Clin Endocrinol Metab 100:E387–E395
El-Gohary Y, Tulachan S, Wiersch J et al (2014) A smad signaling network regulates islet cell proliferation. Diabetes 63:224–236
Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223–1234
Butcher MJ, Hallinger D, Garcia E et al (2014) Association of proinflammatory cytokines and islet resident leukocytes with islet dysfunction in type 2 diabetes. Diabetologia 57:491–501
Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL (2010) 12-Lipoxygenase products reduce insulin secretion and β-cell viability in human islets. J Clin Endocrinol Metab 95:887–893
Weaver JR, Holman TR, Imai Y et al (2012) Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol 358:88–95
Whitman S, Gezginci M, Timmermann BN, Holman TR (2002) Structure-activity relationship studies of nordihydroguaiaretic acid inhibitors toward soybean, 12-human, and 15-human lipoxygenase. J Med Chem 45:2659–2661
Segraves EN, Shah RR, Segraves NL et al (2004) Probing the activity differences of simple and complex brominated aryl compounds against 15-soybean, 15-human, and 12-human lipoxygenase. J Med Chem 47:4060–4065
Suzuki H, Ueda T, Juranek I et al (2000) Hinokitiol, a selective inhibitor of the platelet-type isozyme of arachidonate 12-lipoxygenase. Biochem Biophys Res Commun 275:885–889
Deschamps JD, Kenyon VA, Holman TR (2006) Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem 14:4295–4301
Kenyon V, Rai G, Jadhav A et al (2011) Discovery of potent and selective inhibitors of human platelet-type 12- lipoxygenase. J Med Chem 54:5485–5497
Luci D, Jameson JB, Yasgar A et al (2010) Discovery of ML355, a potent and selective inhibitor of human 12-lipoxygenase. In: Probe reports from the NIH Molecular Libraries Program [Internet]. National Center for Biotechnology Information (US), Bethesda, MD
Rai G, Jadhav A, Schultz L et al (2010) Selective small molecule inhibitors of 12-human lipoxygenase (12-hLO). In: Probe reports from the NIH Molecular Libraries Program [Internet]. National Center for Biotechnology Information (US), Bethesda, MD
Taylor-Fishwick DA, Weaver J, Glenn L et al (2015) Selective inhibition of 12-lipoxygenase protects islets and beta cells from inflammatory cytokine-mediated beta cell dysfunction. Diabetologia 58:549–557
Guo Y, Zhang W, Giroux C et al (2011) Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem 286:33832–33840
Duchez AC, Boudreau LH, Bollinger J et al (2015) Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl Acad Sci U S A 112:E3564–E3573
Acknowledgements
The authors acknowledge the key collaborative efforts of R. Mirmira (Indiana University School of Medicine), T. Holman (University of California, Santa Cruz, CA, USA), and D. Maloney, A. Jadhav and associates at the National Center for Advancing Translational Science (NCATS), National Institutes of Health.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
The work in the authors’ laboratories pertinent to this review has been supported by the JDRF to JLN (from Sanofi/JDRF collaboration grant) and to DATF, Congressionally Directed Medical Research Programs to DATF, National Institutes of Health to JLN (R01-HL112605, R01-DK105588), YI (R01-DK090490) and ADD (R15-HL114062).
Duality of Interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
YI, ADD, MAM, DATF, and JLN drafted the article, revised it critically for important intellectual content, and approved the final version to be published.
Rights and permissions
About this article

Cite this article
Imai, Y., Dobrian, A.D., Morris, M.A. et al. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 59, 673–678 (2016). https://doi.org/10.1007/s00125-016-3890-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3890-y