
Abstract
Aims/hypothesis
Precise regulation of insulin secretion by the pancreatic beta cell is essential for the maintenance of glucose homeostasis. Insulin secretory activity is initiated by the stepwise breakdown of ambient glucose to increase cellular ATP via glycolysis and mitochondrial respiration. Knockout of Lkb1, the gene encoding liver kinase B1 (LKB1) from the beta cell in mice enhances insulin secretory activity by an undefined mechanism. Here, we sought to determine the molecular basis for how deletion of Lkb1 promotes insulin secretion.
Methods
To explore the role of LKB1 on individual steps in the insulin secretion pathway, we used mitochondrial functional analyses, electrophysiology and metabolic tracing coupled with by gas chromatography and mass spectrometry.
Results
Beta cells lacking LKB1 surprisingly display impaired mitochondrial metabolism and lower ATP levels following glucose stimulation, yet compensate for this by upregulating both uptake and synthesis of glutamine, leading to increased production of citrate. Furthermore, under low glucose conditions, Lkb1 −/− beta cells fail to inhibit acetyl-CoA carboxylase 1 (ACC1), the rate-limiting enzyme in lipid synthesis, and consequently accumulate NEFA and display increased membrane excitability.
Conclusions/interpretation
Taken together, our data show that LKB1 plays a critical role in coupling glucose metabolism to insulin secretion, and factors in addition to ATP act as coupling intermediates between feeding cues and secretion. Our data suggest that beta cells lacking LKB1 could be used as a system to identify additional molecular events that connect metabolism to cellular excitation in the insulin secretion pathway.
Similar content being viewed by others


Introduction
The initiating steps in the glucose-stimulated insulin secretion (GSIS) pathway involve the stepwise breakdown of glucose through glycolysis and oxidation of the glycolytic product, pyruvate, in mitochondria. ATP-sensitive potassium channels (KATP channels) on the plasma membrane sense the increase in ATP and consequently close [1–4], triggering membrane depolarisation and opening of voltage-sensitive L-type Ca2+ channels [4, 5]. The secretory response is biphasic; within minutes a readily releasable pool (RRP) of granules docked at the plasma membrane is released [6–8]. A second phase, which requires recruitment and trafficking of insulin granules to the plasma membrane, follows [9]. The central role of the KATP channel in beta cell function is highlighted by deficiencies in both first and second phase GSIS in islets from sulfonylurea 1 (SUR1)−/− and ATP-sensitive inward rectifier potassium channel (KIR6.2)−/− mice [10]. In humans, inappropriate inhibition or activation of KATP channel activity arising from mutations in KIR6.2 and SUR1 underlie congenital hyperinsulinaemia or neonatal diabetes, respectively [11–13].
Our understanding of precisely how glucose is coupled to insulin output is still incomplete. Several lines of evidence have indicated that the secretory response is more intricate than the classical model outlined above would suggest. For example, an increase in cytosolic ATP and closure of KATP channels does not always correlate with insulin output in GSIS [10]. Beta cells can also secrete insulin independently of KATP channel closure by an unclear mechanism that still requires calcium influx and membrane depolarisation [10, 14]. Furthermore, KATP channel activity can be recorded from on-cell patches on beta cells exposed to glucose-free solutions, despite the fact that the intracellular ATP concentration under similar conditions would predict the channels to be closed [15]. Finally, the presence of depolarising agents and the allosteric KATP channel activator diazoxide can further enhance insulin secretion in response to glucose [16]. As a result, additional factors that drive metabolism-dependent secretion, such as mitochondrial metabolites (NADPH, GTP, phosphoenol pyruvate [PEP], α-ketoglutarate [α-KG]), and other secondary glucose-derived metabolites such as glutamate [17, 18] must exist.
We previously demonstrated that Lkb1 adult beta cell knockout (LABKO) mice are resistant to hyperglycaemia induced by a high-fat diet, owing in large part to a doubling of beta cell mass and enhanced insulin stores via mTOR activation [19]. However, targeting liver kinase B1 (LKB1) in the beta cell for the treatment of diabetes would be undesirable as knockout of Lkb1 in adult mice has a deleterious effect on haematopoietic stem cell maturation [20–22]. We and others have identified non-overlapping functions in beta cells for LKB1 targets, including MAP/microtubule affinity-regulating kinase 2 (MARK2), salt-inducible kinase 2 (SIK2), brain-specific kinase 2 (BRSK2) as well as AMP-activated protein kinase (AMPK) itself [19, 23–28], underscoring the potential for identifying the minimal target(s) of LKB1 responsible for enhancing insulin secretion. Here, we systematically interrogated insulin secretion pathway components to identify the signals responsible for enhanced insulin secretion in Lkb1 knockout islets.
Methods
Animals
Control LoxP/LoxP (L/L) and LABKO mice were obtained from mating homozygous Lkb1 floxed mice to Pdx1-CreER tam mice all on an FVB/n background as described previously [19] (electronic supplementary material [ESM] Methods).
Antibodies and short hairpin RNA sequences
Antibodies and short hairpin RNA (shRNA) sequences are included in ESM Methods.
GSIS and calcium experiments
GSIS and calcium experiments were performed as described previously [19] (ESM Methods).
Electron microscopy and insulin immunogold labelling
Pancreases were fixed in 2.6% glutaraldehyde overnight and processed as described previously [29] (ESM Methods).
Seahorse measurements
Measurements of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were performed as described previously [23] (ESM Methods).
Mitochondrial membrane potential and content measurements
Mitochondrial inner membrane potential was measured in dispersed islet cells using tetramethylrhodamine ethyl ester (TMRE) and normalised to Mitotracker green (MTG; Invitrogen, Burlington, ON, Canada) intensity (ESM Methods).
NAD(P)H autofluorescence imaging
Islets were incubated in 2 mmol/l glucose for 1 h and loaded into microfluidic devices and placed in a heating chamber on a Zeiss LSM710 microscope (Zeiss Canada, Toronto, ON, Canada) and imaged as described previously [30].
NEFA quantification in islets
NEFA levels in chloroform extracts from 50 islets per mouse were determined using a colorimetric assay (Abcam, Toronto, ON, Canada).
Membrane potential recordings and whole cell patch-clamp
Plasma membrane potential and voltage-dependent K channel activity was measured using a perforated patch technique in whole cell configuration as described previously [31, 32] (ESM Methods).
Metabolomics
106 MIN6 cells per condition were infected with lentivirus expressing non-silencing control or Lkb1 shRNA. At 72 h post-infection, cells were starved for 1 h in KRBH + 1 mmol/l glucose prior to pulsing. Cells were pulsed with 10 mmol/l [U-13C6]glucose or 2 mmol/l [U-13C5]glutamine for the indicated times and compared with starved and unlabelled carbon source samples. GC-MS analysis was performed as described previously [33] (ESM Methods).
Measurement of ATP/ADP nucleotides
Cells were processed for extraction of cell nucleotides as described previously [34] (ESM Methods).
Results
Loss of LKB1 in mouse beta cells increases insulin granule size and plasma membrane docking
We previously reported that islets lacking LKB1 in the beta cell compartment led to a twofold increase in insulin content per cell [19]. As the proximity of insulin granules to the plasma membrane dictates the amplitude of first phase insulin release, we evaluated the size of the RRP in LABKO islets. Electron microscopy revealed that the number of insulin granules docked at the plasma membrane was 60% greater in LABKO beta cells, which correlated well with the 1.9-fold increase in first phase GSIS (Fig. 1a, b). Insulin immunogold labelling revealed a greater than twofold increase in the number of insulin-positive particles per granule, and a greater than threefold increase in the number of large granules (>900 nm diameter; ESM Fig. 1).

Loss of LKB1 in mouse beta cells increases insulin granule size and plasma membrane docking. (a, b) The number of docked insulin granules quantified in L/L control (black bar) and LABKO (white bar) beta cells as the number of granules found within 100 nm of the plasma membrane (docked granules highlighted by white arrows; *p > 0.05, 10–20 fields per mouse were quantified, n = 3 per genotype); scale bar, 500 nm. (c) Insulin secretion (as % of total insulin content) measured during perifusion of isolated L/L control (black circles) and LABKO (white circles) islets at increasing glucose concentrations and 45 mmol/l KCl. Data are ±SEM, n = 3 per genotype, representative of three independent experiments. PM, plasma membrane
LKB1 regulates glucose sensing
We previously observed an enhanced rate of calcium influx in response to glucose in LKB1 knockdown (LKB1i) MIN6 cells, raising the possibility that LKB1 regulates sensitivity to feeding cues. To determine if glucose responsiveness is enhanced in the absence of LKB1, we performed islet perifusion experiments. A glucose ramp (2.8 to 16.7 mmol/l) revealed an increase in maximal release at near saturating glucose concentrations in the GSIS response curve, with the response from LABKO islets to 8 mmol/l glucose equal to that of L/L control islets to 16.7 mmol/l glucose (Fig. 1c). Taken together, these data suggest that, in addition to having an increase in insulin cargo per cell, LABKO beta cells are also more sensitive to glucose.
LKB1 depletion increases sensitivity to the anti-diabetic drug tolbutamide
To determine if the KATP channel was functionally altered in cells lacking LKB1, we monitored insulin secretion in response to tolbutamide, which belongs to a class of sulfonylurea drugs used to treat type 2 diabetes. Binding of tolbutamide to the SUR1 subunit of the KATP channel results in channel closure, membrane depolarisation and insulin secretion. Following treatment of MIN6 cells expressing control non-silencing shRNA with 200 μmol/l tolbutamide, we observed an eightfold stimulation of insulin secretion in 1 mmol/l (low) glucose and 17-fold stimulation in the presence of 20 mmol/l (high) glucose (Fig. 2a). Interestingly, following knockdown of LKB1, MIN6 cells in low glucose responded to tolbutamide as though they were cultured in stimulatory glucose concentrations (Fig. 2a). This effect was also seen in perifused LABKO islets, which unlike islets from control (L/L) animals, secreted insulin in response to tolbutamide in low glucose (Fig. 2b). Treatment of LABKO islets with mitiglinide, a secretagogue that closes the KATP channel without binding to SUR1, produced similar results (Fig. 2c). To determine if closure of the KATP channel was required for the observed sensitisation, we stimulated LABKO islets with increasing amounts of tolbutamide in the presence of diazoxide, which opens the KATP channel and prevents membrane depolarisation and insulin secretion [35]. As shown in Fig. 2d, both LABKO and control islets responded to tolbutamide in the presence of diazoxide, indicating that if LKB1 acts on the KATP channel it likely does so in a SUR1-independent manner, a mechanism that has been reported for glucose-stimulated secretion (reviewed in [36]).

LKB1 depletion increases sensitivity to the glucose-lowering drug tolbutamide. (a) Insulin secretion measured in MIN6 cells expressing non-silencing control shRNA (NSi, black bars) or Lkb1-targeting shRNA (LKB1i, white bars) following tolbutamide treatment. Effect of combined treatment with 200 or 500 μmol/l tolbutamide with 20 mmol/l glucose (G) is shown. (b) Insulin secretion in perifused L/L (black circles) and LABKO (white circles) islets following tolbutamide (50–200 μmol/l) treatment (n = 3 per genotype). (c) Insulin secretion in L/L (black bars) and LABKO (white bars) islets following static incubations in mitiglinide (10–250 μmol/l) and 250 μmol/l in the presence of 16.7 mmol/l glucose (n = 3–4 per genotype). (d) Insulin secretion from perifused L/L control (black circles) and LABKO (white circles) islets during addition of increasing concentrations of tolbutamide in the presence of diazoxide (200 μmol/l) and following a 10 min washout period in 2.8 mmol/l glucose (n = 3 per genotype). All data are ±SD (*p < 0.05) and representative of three independent experiments in (a) and (c) and representative of two independent experiments in (b) and (d). Insulin secretion in (a–d) is expressed as per cent of total insulin content
Enhanced membrane depolarisation and calcium influx in LABKO beta cells
The sensitivity to tolbutamide in LABKO cells suggested that closure of KATP channels could occur at a lower threshold, either because there are fewer channels present, or because the channels are more primed for closure at steady state. Whole cell patch-clamp analysis showed that LABKO beta cells fire action potentials (APs) at sixfold lower concentration of tolbutamide than control beta cells (100 μmol/l for control L/L, 15 μmol/l for LABKO; Fig. 3a, b), and that the frequency of APs in LABKO beta cells increased twofold to fivefold overall at either concentration of drug (Fig. 3b). Surprisingly, both resting membrane potential and the magnitude of depolarisation in response to glucose treatment were similar in control and LABKO beta cells, suggesting that KATP channel closure is still dynamically regulated (Fig. 3c; ESM Fig. 2a). Furthermore, the KATP channel density was the same in L/L and LABKO beta cells (Fig. 3d). Importantly, there was no change in expression of KATP channel subunits in MIN6 LKB1i cells or LABKO islets, and no enhancement in KATP channel closure in LABKO islets following glucose treatment (ESM Fig. 2b). These data indicate that loss of LKB1 enhances cellular excitability through promoting calcium influx and not through regulating KATP channel closure.

Enhanced membrane depolarisation in LABKO beta cells is calcium-influx-dependent. (a) Representative traces of AP firing in L/L and LABKO beta cells under perforated whole cell patch conditions following tolbutamide treatment. (b) Quantification of 11 and 12 beta cells from L/L (white bars) and LABKO (black bars) mice were pooled from three independent experiments showing AP frequency in response to tolbutamide; **p < 0.01. (c) Quantification of resting membrane potential of L/L (white bar; n = 11) vs LABKO (black bar; n = 12) beta cells in 2.8 mmol/l glucose. (d) Mean voltage-activated K+ current from whole cell patch-clamped beta cells from L/L (white circles; wild-type; n = 8 cells) and LABKO (black circles; KO; n = 10 cells) mice. Data are ±SD. (e) Ca2+ influx following treatment with tolbutamide (50–200 μmol/l), using the ratio of 380/340 excitation of Fura-2-acetoxymethyl ester (Fura2AM) dye in control shRNA (black circles) and LKB1i (white circles) MIN6 cells. Treatment with depolarising agent (45 mmol/l KCl) to compare total cytosolic Ca2+ capacity is shown. Data are ±SEM and are pooled averages from three independent experiments with n = 3 replicates each
Consistent with the increased tolbutamide-induced membrane depolarisation, maximal calcium response to tolbutamide in MIN6 cells was achieved at a tenfold lower concentration after silencing LKB1 (Fig. 3e), while calcium levels under basal conditions were unaffected (ESM Fig. 3a). A similar effect was observed in dispersed LABKO beta cells (ESM Fig. 4a). Treatment with the L-type specific calcium channel inhibitor diltiazem blocked this influx, indicating that the extracellular milieu and not intracellular stores was the source of calcium ions (ESM Fig. 4a, b). Taken together, these data reveal that LABKO beta cells secrete more insulin in part due to sensitisation of calcium influx.
LKB1 is required for mitochondrial respiration in beta cells
Mitochondrial oxidative phosphorylation using the glycolytic end-product pyruvate generates ATP that closes KATP channels and amplifies the insulin secretory response. The accelerated glucose responsiveness and calcium influx seen in LKB1 knockdown cells (Fig. 1b) prompted us to test whether loss of LKB1 enhances mitochondrial function. In control MIN6 cells and control (L/L) islets, the OCR increased in response to glucose (Fig. 4a, b). By contrast, LKB1 knockdown MIN6 cells and LABKO islets displayed a global reduction in OCR, to levels at or below which control MIN6 cells and L/L islets OCR respired following treatment with oligomycin, an inhibitor of mitochondrial F1Fo-ATP synthase (Fig. 4a, b). The effects on both respiration and insulin secretion were specific to loss of LKB1, as reconstitution of LKB1 in LABKO islets restored the OCR (Fig. 4c) and reduced GSIS in both primary mouse islets (Fig. 4b) and LABKO islets (ESM Fig. 5a). These data indicate a surprising uncoupling of insulin secretory capacity and mitochondrial bioenergetics in beta cells lacking LKB1. MIN6 cells, unlike primary islets [37], will metabolise pyruvate and consequently secrete insulin. Interestingly, provision of pyruvate or the anaplerotic substrate glutamine (see below) individually or in combination restored OCR in both basal and uncoupled states in cells lacking LKB1 (Fig. 4c, d; ESM Fig. 5b, c). Normal OCR was restored when green fluorescent protein (GFP)-tagged LKB1 was introduced into LABKO islets (Fig. 4e) or by overexpression of a constitutively active AMPK (AMPK-T172D), indicating that loss of AMPK activity at least in part resulted in the respiratory defect (ESM Fig. 5d). Consistent with this, overexpression of LKB1 in control islets inhibited secretion (Fig. 4f).

LKB1 is required for mitochondrial respiration in beta cells. (a, b) OCR normalised to total μg of protein, in (a) non-silencing control shRNA (NSi) and LKB1i MIN6 cells and (b) dissociated L/L and LABKO islets. In (a–d), control cells are represented by black circles and LKB1i/LABKO by white circles. Treatments were as follows: 16.7 mmol/l glucose (G) and 5 μmol/l oligomycin (O). (c, d) OCR normalised to total μg of protein, in (c) NSi and LKB1i MIN6 cells and (d) dissociated L/L and LABKO islets in the presence of 2 mmol/l pyruvate and 4 mmol/l glutamine. F, FCCP; R, rotenone. (e) OCR normalised to total μg of protein measured in L/L (black symbols) and LABKO (white symbols) islets infected with Adeno-GFP (circles) or Adeno-LKB1 (squares). OCR at baseline and following 16.7 mmol/l glucose (G) and 5 μmol/l oligomycin (O) are shown. Western blotting of protein extracts from parallel samples is shown. KO, LABKO. (f) Insulin secretion from wild-type mouse islets overexpressing empty vector (EV)-GFP or LKB1-GFP in low glucose (2.8 mmol/l; white bars) or high glucose (16.7 mmol/l; black bars). Endogenous (endog.) LKB1 is indicated. Data are ±SEM from n = 2 mice per condition. Western blot for protein levels is shown. (g) Fold change of ATP/ADP ratio in NSi (black bars) and LKB1i (white bars) MIN6 cells after glucose stimulation (20 mmol/l). Data are ±SEM of two independent experiments. (h) Mitochondrial membrane potential in dissociated L/L (black circles) and LABKO (white circles) islets using relative fluorescence intensity (RFU) of TMRE:MTG following treatment with 16.7 mmol/l glucose (G) and 50 μmol/l carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP). Data are pooled from n = 3 per genotype, with AUC inset expressed in arbitrary area units, and are ±SEM
The reduction in medium ECAR in LABKO cells (ESM Fig. 5b) suggested that impaired generation of a metabolic intermediate may underlie the defect in mitochondrial respiration. Indeed, we observed a reduced ATP/ADP ratio in MIN6 cells (Fig. 4g) as well as lower ATP levels and mitochondrial membrane potential (ΔΨ) in LABKO islets (Fig. 4h; ESM Fig. 6). These changes occurred without a reduction in mitochondrial mass (ESM Fig. 7). These data indicate that if provided with sufficient fuel, the electron transport chain (ETC) in LKB1-deficient cells can support electron transport.
Increased glutamine flux in beta cells lacking LKB1
To determine if impaired bioenergetics in LABKO cells was due to a defect in generation of mitochondrial fuel, we assessed the metabolic flux of glucose in LABKO islets by GC-MS. Following timed pulses with 10 mmol/l [13C6]glucose, there was an overall trend towards increased total pyruvate in MIN6 cells silenced for Lkb1, derived from both labelled (13C) and unlabelled (12C) sources (Fig. 5a, b). A significant increase in pyruvate was only observed in the relative abundance of [12C]pyruvate after 120 min, consistent with a reduction in its rate of production from glucose. We also observed a significant increase in unlabelled citrate in the absence of LKB1 at all time points (Fig. 5b). However, the proportion of 13C-labelling of citrate from glucose was similar in cells lacking LKB1, suggesting that tricarboxylic acid (TCA)-derived citrate from glucose was reduced, in a similar fashion to pyruvate. Following Lkb1 silencing, glucose flux to α-KG was also decreased, suggesting the excess pyruvate and citrate have an alternate fate besides cycling through the TCA cycle.

Increased glutamine flux in beta cells lacking LKB1. (a) Schematic showing the fate of 13C and 12C glucose labels into downstream TCA intermediates. Quantification of relative abundance (amount of metabolite in LKB1i cells compared with the corresponding amount in non-silencing control shRNA [NSi] cells) of 13C (black bars) and 12C (white bars) in pyruvate (b), citrate (c), α-KG (d) and glutamine (e) isolated from MIN6 cells infected with NSi (N) or LKB1i (L) shRNA lentivirus following a 10 or 120 min pulse with [13C6]glucose. (f) Schematic showing the fate of 13C and 12C in metabolites downstream of glutamine. Relative 13C (black bars) and 12C (white bars) levels of [13C]- and [12C]glutamine (g), glutamate (h), α-KG (i) and citrate (j) isolated from MIN6 cells transfected with NSi (N) or LKB1i (L) siRNA cells following a 10 or 60 min pulse with [13C5]glutamine. Samples in (b–j) are from n = 4 independent experiments, data are ±SEM, *p < 0.05; **p < 0.01; ***p < 0.001
Interestingly, levels of glucose-derived glutamine were elevated in LKB1 knockdown cells (Fig. 5b), which can be used as an alternative cellular carbon source for anabolic processes. To identify the fate of glutamine, we pulsed MIN6 cells with 2 mmol/l [13C5]glutamine for 10 and 60 min and measured incorporation into several intermediate metabolites using GC-MS (Fig. 5c). In cells silenced for Lkb1, there was a trend towards increased glutamine uptake and levels of glutamate, as well as a significant early increase in α-KG levels (Fig. 5d). Furthermore, while pyruvate levels were unaffected, citrate production from glutamine was significantly increased at all time points (Fig. 5d and data not shown). Taken together, these data indicate that in beta cells lacking LKB1, the breakdown of glucose is enhanced, resulting in an increase in citrate and glutamine abundance. That this occurs in the absence of a corresponding increase in glucose-derived TCA intermediates indicates that citrate is being redirected away from the TCA cycle and used for other biosynthetic pathways. Consistent with this observation, labelled carbons from glutamine contribute less to α-KG than to citrate, suggesting that this pool of citrate is shunted away from α-KG production.
Enhanced acetyl-CoA carboxylase 1 activity is required for insulin in LABKO islets
To determine if changes in reductive potential accompany the enhanced insulin secretion phenotype in cells lacking LKB1, we measured levels of NAD(P)H, a proposed coupling factor for insulin secretion, in both mitochondrial and cytosolic fractions. While we observed glucose-dependent increases in NAD(P)H levels, these were unaffected by silencing Lkb1 (Fig. 6a). Recent evidence indicates that in cancer cells, increased citrate can be generated in a reverse reaction that uses glutamine as a substrate known as reductive carboxylation [38]. Reductive carboxylation provides carbon for fatty acid synthesis and drives the accumulation of long chain fatty acids [38]. We therefore measured levels of NEFA (of longer than eight carbons) in LABKO islets and found they were increased twofold compared with control islets (Fig. 6b). Using labelled glucose and glutamine during 24 h to trace NEFA in LKB1i cells, we were unable to identify which NEFA was increased in LKBi cells, mainly due to the very small proportion of NEFA being produced from both glucose and glutamine (data not shown). However consistent with the overall increase of NEFA LABKO islets, phosphorylation of acetyl-CoA carboxylase 1 (ACC1; at the AMPK site Ser79 [39]) was reduced by 50% in LABKO islets, indicative of inappropriate ACC1 activation under low glucose conditions, which we attribute to a loss of AMPK activity (Fig. 6c). These observations suggest that ACC1-dependent lipid accumulation may be important for insulin secretion; indeed, knockdown of ACC1 blocked insulin secretion in MIN6 cells and in mouse islets even when Lkb1 was silenced (Fig. 6d; ESM Fig. 8). Taken together, these data show that LKB1 functions to restrain glucose-stimulated calcium influx and insulin secretion by attenuating ACC1 activity and the generation of glutamine-derived citrate (see model in Fig. 7).

Depletion of LKB1 activates ACC1 activity and insulin secretion. Autofluorescence intensity of (a) cytosolic and (b) mitochondrial NAD(P)H pools at 705 nm in indicated concentrations of glucose. Control, black bars; LKB1i/LABKO, white bars. (c) Measurement of NEFA levels from L/L (black bar) and LABKO (white bar) islets cultured in RPMI. (d) Western blots showing p-ACC1, total ACC1, p-AMPK, total AMPK, LKB1 and heat-shock protein 90 (HSP90) protein levels in L/L and LABKO islets cultured in low or high glucose (G). Quantification of p-ACC1:total ACC1 levels in L/L (black bars) and LABKO (white bars) is shown. (e) Insulin secretion from MIN6 cells cultured in low or high glucose after silencing Lkb1 (LKB1i, dark grey bars) or Acc1 alone (ACC1i, white bars) or together (light grey bars) compared with non-silencing control shRNA (NSi) (black bars). Western blot showing ACC1 knockdown is shown. Data are ±SEM, where *p < 0.01 pooled from three independent experiments

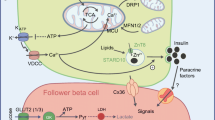
Model showing the role of LKB1 in the regulation of glucose-derived metabolites in pancreatic beta cells. LKB1/AMPK blocks ACC1, increasing the pool of citrate available for anaplerosis. In the absence of LKB1, ACC1 activity is elevated, promoting fatty acid accumulation and GSIS. ACL, ATP citrate lyase
Discussion
LKB1 depletion uncouples glucose metabolism from calcium influx and insulin secretion
We and others previously reported that deletion of Lkb1 in adult mouse beta cells leads to increased insulin secretion [19, 26]. Here, we report that LKB1 acts to attenuate KATP channel closure and calcium influx. This is evidenced by an enhanced closure of KATP channels, calcium influx and insulin secretion in response to tolbutamide in LABKO beta cells, even in the absence of glucose. Given the reduction in energy status in LABKO beta cells under both basal and stimulated conditions, the enhancement in insulin secretion is paradoxical. Taken together, our data are consistent with the notion that LKB1 regulates both exocytotic events as well as metabolic amplification of calcium influx through an alternate coupling factor that is not ATP and involves citrate and ACC1.
Metabolic rewiring in LABKO beta cells
That impaired mitochondrial respiration in LABKO islets can be restored with constitutively active AMPK indicates the LKB1–AMPK pathway may directly control mitochondrial bioenergetics. While provision of additional substrate can restore full ETC activity in beta cells lacking LKB1, following glucose stimulation there is a reduction by half in their bioenergetic capacity. Furthermore, in the absence of LKB1, glucose usage favours citrate and glutamine production, and glucose-derived glutamine is used to generate citrate, contributing to higher citrate levels. This metabolic shift towards glutaminolysis may help to maintain both the higher basal mitochondrial membrane potential and NADPH levels in LABKO β cells in which respiration is attenuated. Glutamine itself is known to amplify GSIS and enhance KATP channel-independent release [40], and may serve as a coupling factor to amplify metabolism-secretion coupling in LABKO beta cells. This idea is consistent with recent work using beta cells in which Lkb1 is deleted (using the Ins1–Cre system), wherein loss of LKB1 or AMPK results in a metabolic switch favouring glutamine metabolism [27].
Taken together, it appears that in cells lacking LKB1, fuel is redirected away from the TCA cycle to favour biosynthetic pathways such as amino acid synthesis, or NEFA and lipid generation for de novo granule generation, or perhaps for use in a signalling role. As ACC1 has been shown to be required for pyruvate cycling and generation of TCA intermediates [41, 42], we conclude that enhanced insulin secretion from LABKO beta cells is due at least in part to enhanced ACC1 activity and increased citrate production from glutamine. Taken together, these observations support the idea that activating ACC1 or elevating citrate levels may be beneficial for enhancing insulin secretion in settings of hypoinsulinaemia.
Abbreviations
- ACC1:
-
Acetyl-CoA carboxylase 1
- AMPK:
-
AMP-activated protein kinase
- AP:
-
Action potential
- ECAR:
-
Extracellular acidification rate
- ETC:
-
Electron transport chain
- GFP:
-
Green fluorescent protein
- GSIS:
-
Glucose-stimulated insulin secretion
- α-KG:
-
α-ketoglutarate
- KATP :
-
ATP-sensitive potassium channel
- KIR6.2:
-
ATP-sensitive inward rectifier potassium channel
- LABKO:
-
Lkb1 adult beta cell knockout
- LKB1:
-
Liver kinase B1
- LKB1i:
-
LKB1 knockdown
- L/L:
-
LoxP/LoxP
- MTG:
-
Mitotracker green
- OCR:
-
Oxygen consumption rate
- RRP:
-
Readily releasable pool
- shRNA:
-
Short hairpin RNA
- SUR1:
-
Sulfonylurea 1
- TCA:
-
Tricarboxylic acid
- TMRE:
-
Tetramethylrhodamine ethyl ester
References
Maechler P, Li N, Casimir M, Vetterli L, Frigerio F, Brun T (2010) Role of mitochondria in beta-cell function and dysfunction. Adv Exp Med Biol 654:193–216
Tarasov A, Dusonchet J, Ashcroft F (2004) Metabolic regulation of the pancreatic beta-cell ATP-sensitive K+ channel: a pas de deux. Diabetes 53(Suppl 3):S113–S122
Inagaki N, Gonoi T, Clement JP et al (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
MacDonald PE, Joseph JW, Rorsman P (2005) Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci 360:2211–2225
Arkhammar P, Nilsson T, Rorsman P, Berggren PO (1987) Inhibition of ATP-regulated K+ channels precedes depolarization-induced increase in cytoplasmic free Ca2+ concentration in pancreatic beta-cells. J Biol Chem 262:5448–5454
Olofsson CS, Gopel SO, Barg S et al (2002) Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B cells. Pflugers Arch 444:43–51
Barg S, Eliasson L, Renstrom E, Rorsman P (2002) A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes 51(Suppl 1):S74–S82
Jewell JL, Oh E, Thurmond DC (2010) Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol 298:R517–R531
Tsushima RG (2011) Second-phase insulin secretion gets cool. Am J Physiol Endocrinol Metab 301:E1070–E1071
Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J (2000) Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem 275:9270–9277
Pratt EB, Yan FF, Gay JW, Stanley CA, Shyng SL (2009) Sulfonylurea receptor 1 mutations that cause opposite insulin secretion defects with chemical chaperone exposure. J Biol Chem 284:7951–7959
Tammaro P, Girard C, Molnes J, Njolstad PR, Ashcroft FM (2005) Kir6.2 mutations causing neonatal diabetes provide new insights into Kir6.2-SUR1 interactions. EMBO J 24:2318–2330
Aittoniemi J, Fotinou C, Craig TJ, de Wet H, Proks P, Ashcroft FM (2009) Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B Biol Sci 364:257–267
Seino Y, Miki T, Fujimoto W et al (2013) Cephalic phase insulin secretion is KATP channel independent. J Endocrinol 218:25–33
Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49:424–430
Sato Y, Henquin JC (1998) The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: in search of the underlying mechanism. Diabetes 47:1713–1721
Gheni G, Ogura M, Iwasaki M et al (2014) Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep 9:661–673
Feldmann N, del Rio RM, Gjinovci A, Tamarit-Rodriguez J, Wollheim CB, Wiederkehr A (2011) Reduction of plasma membrane glutamate transport potentiates insulin but not glucagon secretion in pancreatic islet cells. Mol Cell Endocrinol 338:46–57
Fu A, Ng AC, Depatie C et al (2009) Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab 10:285–295
Gurumurthy S, Xie SZ, Alagesan B et al (2010) The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 468:659–663
Nakada D, Saunders TL, Morrison SJ (2010) Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468:653–658
Gan B, Hu J, Jiang S et al (2010) Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 468:701–704
Sakamaki JI, Fu A, Reeks C et al (2014) Role of the SIK2-p35-PJA2 complex in pancreatic beta-cell functional compensation. Nat Cell Biol 16:234–244
Nie J, Liu X, Lilley BN et al (2013) SAD-A kinase controls islet beta-cell size and function as a mediator of mTORC1 signaling. Proc Natl Acad Sci U S A 110:13857–13862
Chen XY, Gu XT, Saiyin H et al (2012) Brain-selective kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic beta-cells. J Biol Chem 287:30368–30375
Granot Z, Swisa A, Magenheim J et al (2009) LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metab 10:296–308
Kone M, Pullen TJ, Sun G et al (2014) LKB1 and AMPK differentially regulate pancreatic beta-cell identity. FASEB J 28:4972–4985
Sun G, Tarasov AI, McGinty JA et al (2010) LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. Am J Physiol Endocrinol Metab 298:E1261–E1273
Norton M, Ng AC, Baird S et al (2014) ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci Signal 7:ra10
Lam AK, Silva PN, Altamentova SM, Rocheleau JV (2012) Quantitative imaging of electron transfer flavoprotein autofluorescence reveals the dynamics of lipid partitioning in living pancreatic islets. Integr Biol (Camb) 4:838–846
Spigelman AF, Dai X, MacDonald PE (2010) Voltage-dependent K(+) channels are positive regulators of alpha cell action potential generation and glucagon secretion in mice and humans. Diabetologia 53:1917–1926
Allister EM, Robson-Doucette CA, Prentice KJ et al (2013) UCP2 regulates the glucagon response to fasting and starvation. Diabetes 62:1623–1633
Faubert B, Boily G, Izreig S et al (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17:113–124
Yang MS, Gupta RC (2003) Determination of energy charge potential in the C6 glioma and the HepG-2 cell culture. Toxicol Mech Methods 13:97–101
Sturgess NC, Kozlowski RZ, Carrington CA, Hales CN, Ashford ML (1988) Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br J Pharmacol 95:83–94
Komatsu M, Takei M, Ishii H, Sato Y (2013) Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig 4:511–516
Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA (2012) Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes 61:1719–1725
Metallo CM, Gameiro PA, Bell EL et al (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481:380–384
Fullerton MD, Galic S, Marcinko K et al (2013) Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19:1649–1654
Li C, Buettger C, Kwagh J et al (2004) A signaling role of glutamine in insulin secretion. J Biol Chem 279:13393–13401
Zhang S, Kim KH (1998) Essential role of acetyl-CoA carboxylase in the glucose-induced insulin secretion in a pancreatic beta-cell line. Cell Signal 10:35–42
Ronnebaum SM, Joseph JW, Ilkayeva O et al (2008) Chronic suppression of acetyl-CoA carboxylase 1 in beta-cells impairs insulin secretion via inhibition of glucose rather than lipid metabolism. J Biol Chem 283:14248–14256
Acknowledgements
We would like to acknowledge members of the Screaton laboratory for helpful discussions and commentary on the manuscript, T. Alves and R. Kibbey (Department of Internal Medicine, Yale University, New Haven, CT, USA) for assistance with metabolomics studies and helpful discussions, P. Rippstein (University of Ottawa Heart Institute, Ottawa, Canada) for assistance with Electron Microscopy, J. McBane (University of Ottawa, Ottawa, ON, Canada) for assistance with islet isolations. Adenovirus expressing AMPK-CA was a kind gift of J. Dyck (Department of Pediatrics, University of Alberta, Edmonton, AB, Canada).
Funding
This work was supported by grants to RAS from the Canadian Institutes of Health Research (CIHR; MOP #111186), Canadian Foundation for Innovation (CFI), and by a Studentship from the Canadian Diabetes Association to AF. RAS acknowledges the Canada Research Chair program for support.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AF researched data and wrote the manuscript. KR, BF and ABH researched data and edited the manuscript. CR, XQD, KSS, SO and JVR researched data and revised the article. OYAD, MBW and PEM designed methods, supervised data acquisition and edited the manuscript. RJ contributed to project design, supervised data acquisition and edited the manuscript. RAS designed the project and wrote the manuscript. All authors approved the final version. RAS is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 145 kb)
ESM Figure 1
(PDF 426 kb)
ESM Figure 2
(PDF 642 kb)
ESM Figure 3
(PDF 75 kb)
ESM Figure 4
(PDF 149 kb)
ESM Figure 5
(PDF 1223 kb)
ESM Figure 6
(PDF 139 kb)
ESM Figure 7
(PDF 12014 kb)
ESM Figure 8
(PDF 1745 kb)
Rights and permissions
About this article

Cite this article
Fu, A., Robitaille, K., Faubert, B. et al. LKB1 couples glucose metabolism to insulin secretion in mice. Diabetologia 58, 1513–1522 (2015). https://doi.org/10.1007/s00125-015-3579-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3579-7