
Abstract
Key message
Incorporating feature-engineered environmental data into machine learning-based genomic prediction models is an efficient approach to indirectly model genotype-by-environment interactions.
Abstract
Complementing phenotypic traits and molecular markers with high-dimensional data such as climate and soil information is becoming a common practice in breeding programs. This study explored new ways to combine non-genetic information in genomic prediction models using machine learning. Using the multi-environment trial data from the Genomes To Fields initiative, different models to predict maize grain yield were adjusted using various inputs: genetic, environmental, or a combination of both, either in an additive (genetic-and-environmental; G+E) or a multiplicative (genotype-by-environment interaction; GEI) manner. When including environmental data, the mean prediction accuracy of machine learning genomic prediction models increased up to 7% over the well-established Factor Analytic Multiplicative Mixed Model among the three cross-validation scenarios evaluated. Moreover, using the G+E model was more advantageous than the GEI model given the superior, or at least comparable, prediction accuracy, the lower usage of computational memory and time, and the flexibility of accounting for interactions by construction. Our results illustrate the flexibility provided by the ML framework, particularly with feature engineering. We show that the feature engineering stage offers a viable option for envirotyping and generates valuable information for machine learning-based genomic prediction models. Furthermore, we verified that the genotype-by-environment interactions may be considered using tree-based approaches without explicitly including interactions in the model. These findings support the growing interest in merging high-dimensional genotypic and environmental data into predictive modeling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genotype-by-environment interaction (GEI) plays an essential role in plant breeding, resulting in differential changes in individual performance or rank-changing across environments (Falconer 1996; Tabery 2008; Bernardo 2014). Consequently, prediction frameworks that do not consider GEI have been shown to underperform in multi-environment trials (MET) (Burgueño et al. 2012; Jarquín et al. 2017; Gillberg et al. 2019). One approach often used in MET is the linear mixed model with a factor analytic structure modeling the variance-covariance between environments (Smith et al. 2001; de los Campos and Gianola 2007; Dias et al. 2018). Another alternative proposed based on the mixed model framework is to incorporate environmental data with a reaction norm model utilizing covariance structures that account for the genetic similarity between genotypes and the similarity among environmental conditions (Jarquín et al. 2014). These models were necessary steps toward developing a MET prediction framework. However, they are still limited in their utilization of environmental data due to the constraint of incorporating GEI only through covariance structures, which limits, for instance, the inclusion of high-order interactions in the model.
The high availability of environmental data in testing locations has catalyzed a thorough characterization of environmental effects over the observed phenotype. In addition to the traits commonly measured by plant breeders, data from weather stations, soil surveys, and public repositories have been recently integrated into genomic prediction (GP) models (Malosetti et al. 2016; Monteverde et al. 2019; Canella Vieira et al. 2022). These environmental data can be applied in enviromics studies by envirotyping the testing locations (Costa-Neto et al. 2021, 2022) or in a combined way with biological knowledge through crop growth models to increase prediction accuracy in GP (Heslot et al. 2014; Technow et al. 2015). In this case, increases of up to 11% in prediction accuracy have been observed (Heslot et al. 2014).
One approach that has been gaining momentum when modeling GEI is machine learning (ML) (Montesinos-López et al. 2019; Jubair et al. 2023). The flexibility of integrating high-dimensional and multilayered data makes ML a good alternative for plant breeding, especially as weather, soil, and other environmental information become more commonly used in GP models. The ML model can leverage this diversity of data to improve the learning process, resulting in higher prediction accuracy (Gong et al. 2019). How this information is processed to derive new features (i.e., the feature engineering stage) may also play an essential role in the model’s performance. However, applications of ML in GP are recent, and currently, there is no consensus on the best ML approach to combine environmental and genetic data when accounting for GEI.
Some of the recent ML methods utilized in the GEI context include neural networks for predicting yield, protein content, and oil content (Ray et al. 2023), convolutional neural networks (CNN) to predict grain yield using genetic, environmental, management, and historical (e.g. yield and weather) data (Washburn et al. 2021) and dense neural networks (DNN) using intermediate layers to allow interactions between the different data sets (Kick et al. 2023). Tree-based ML models such as Random Forest (Breiman 2001), XGBoost (Chen and Guestrin 2016), and LightGBM (Ke et al. 2017) are other examples of successful applications of ML in modeling GEI. However, none of these studies extensively explored feature engineering, which could be an effective approach to including environmental data in GP models. We hypothesized that by using feature engineering, we would be able to efficiently characterize the environment (i.e., envirotyping), which, combined with additive and non-additive genetic data, would result in high prediction accuracy. Therefore, we propose GP models that use feature-engineered environmental data, additive and non-additive genotypic data, or a combination of both. Our findings indicate that combining genotypic and environmental data in ML using our approach is an efficient strategy for predicting maize grain yield in multi-environment trials.
Materials and methods
Phenotypic information
This study used the MET data set from the Genomes to Fields (G2F) 2022 Maize Genotype by Environment Prediction Competition (Genomes to Fields 2023; Lima et al. 2023). Specifically, we used the trial information from 2019, 2020, and 2021, which consisted of 1,179 maize (Zea mays L.) hybrids evaluated in 72 environments (a combination of year and location), comprising 14 states in the USA (CO, DE, GA, IA, IL, IN, MI, MN, NC, NE, NY, SC, TX, and WI) and one city in Germany (Göttingen).
Trials from 2019 included two testers (LH195, PHT69), and trials from 2020 and 2021 included four testers (LH195, PHZ51, PHK76, and PHP02). The experimental design used in the trials was a modified Randomized Complete Block Design (RCBD), mainly with two replications per environment. The response variable used in this study consisted of grain yield in \({\rm Mg}~{\rm ha}^{-1}\) at 15.5% grain moisture. More details on 2019, 2020, and 2021 genetic material are available at Lopez-Cruz et al. (2023).
Single-environment trial models, adjusted using the package ASReml-R 4 (Butler et al. 2017), were fitted to generate the best linear unbiased estimates (BLUEs) for each hybrid in each environment as follows:
where \(Y_{ijklm}\) is the observed grain yield of the \(i^{th}\) hybrid of the \(j^{th}\) replicate, \(k^{th}\) block, \(l^{th}\) row, and \(m^{th}\) column; \(\mu \) is the intercept; \(H_i\) is the fixed effect of the \(i^{th}\) hybrid; \(R_j\) is the fixed effect of the \(j^{th}\) replicate; \(b(R)_{jk}\) is the random effect of the \(k^{th}\) block nested within the \(j^{th}\) replicate with \(b(R)_{jk} \sim N(0, \sigma ^2_b)\); \(r_l\) is the random effect of the \(l^{th}\) row, with \(r_l \sim N(0, \sigma ^2_r)\); \(c_m\) is the random effect of the \(m^{th}\) column, with \(c_m \sim N(0, \sigma ^2_c)\); and \(e_{ijklm}\) is the residual term associated with the \(ijklm^{th}\) experimental unit, with \(e_{ijklm} \sim N(0, \sigma ^2_e)\). BLUEs from model 1 were used as the response variable for further analysis (Fig. 1a).
For each location in 2019, 2020, and 2021, the coefficient of variation was calculated as follows:
where \(\sqrt{\sigma ^2_r}\) and \({\mu _{\hat{y}}}\) are the square root of the residual variance component and the mean of predicted values, respectively, from the equation (1).
The generalized heritability (Cullis et al. 2006) for each location in 2019, 2020, and 2021 was calculated as follows:
where \({\sigma ^2_g}\) is the genetic variance component, and \(\bar{V} (\Delta )\) is the mean pairwise prediction error variance. To estimate the heritability, we used a similar model as (1), where \(H_i\) was treated as a random effect.
Environmental information
Each location was equipped with a Spectrum WatchDog 2700 weather station, which collected information on variables such as rainfall, solar radiation, humidity, and air temperature every 30 min daily. Aggregations were employed to derive new environmental features. Within each environment, weather data was aggregated based on the season and various summary statistics were calculated, including the mean, maximum, minimum, and standard deviation of each weather variable (Supplementary Table S1).
Similarly, lagged yield features were created based on the historical grain yield. For each field location, we calculated summary statistics such as the mean, minimum, and percentiles of the grain yield in the previous year (Supplementary Table S1), i.e., when the observed yield was from 2021, these features were calculated based on the grain yield of 2020 for a given field location. When the observed yield was from 2020, grain yield data from 2019 was used instead.
In some environments, the field trials were close to each other. We used latitude and longitude to obtain bins representing nearby regions to reduce the noise around the varying locations. Bins were obtained as follows:
where \(x'\) is the new binned latitude or longitude, \(\lfloor . \rfloor \) is the floor operator, x is the latitude or longitude to be binned, and s is a step parameter to control the binning range. The greater the s, the lower the number of unique latitude and longitude values created. For example, latitudes equal to 39.785, 39.824, and 39.927 would all become 39.6 when using \(s=1.2\). We used \(s=1.2\) and \(s=3.6\) as step parameters for latitude and longitude, respectively.
The data set also included 765 environmental covariates (ECs) derived using an Agricultural Production Systems sIMulator (APSIM) crop model from an unpublished work available in (Lima et al. 2023) (see file “COMPETITION_DATA_README.docx” at https://doi.org/10.25739/tq5e-ak26). Briefly, this model used \(200 {\rm kg}~{\rm ha}^{-1}\) of NO3 fertilization at all locations, and phenological periods were estimated based on averages from the training set and are not specific to any hybrid. The ECs are formed by a combination of a variable, a phenological period, and a soil layer. Soil layers are specified as 1 through 10 (1 is the top layer), with each layer consisting of 20 cm in the soil column for a total of two meters deep. For example, the EC denoted as “SDR_pGerEme_1” is based on the variable Water Supply–Demand Ratio (“SDR”) during the phenological period from germination to emergence (“pGerEme”) within the soil layer from 0 to 20 cm below ground (soil layer “1”). All the variables and phenological periods utilized are available in the aforementioned file.
As there was a large number of ECs, we performed dimensionality reduction using a Singular Value Decomposition (SVD), utilizing the package scikit-learn 1.2.1 (Pedregosa et al. 2011) from Python 3.8 (Van Rossum and Drake 2009). After performing SVD, we kept the first 15 components, which explained 99.9% of the variance.
Finally, we also used soil variables such as available Nitrates in parts per million, amount of Nitrogen in pounds per acre, and percentage of calcium. In total, we ended up with 90 environmental features stored in a matrix called the environmental matrix, where each row represents a unique environment, and each column represents an environmental feature. The 90 environmental features comprised 6 categories: 63 weather-related, 15 derived from the ECs after dimensionality reduction, 6 derived from the lagged historical grain yield, three soil-based, two based on geographical coordinates, and one related to management (Supplementary Table S1). Details of the data preprocessing steps are illustrated in Fig. 1b.
Genetic information
The genotypic data was described in Lima et al. (2023). Briefly, variant calls for the 2014–2023 G2F materials were obtained using the Practical Haplotype Graph (PHG) (Bradbury et al. 2022). Hybrid genotypes were generated by combining information about their parent lines using the CreateHybridGenotypes plugin available in TASSEL 5 (Bradbury et al. 2007), which yielded a file with 4,928 individuals and 437,214 markers. We used VCFtools 0.1.15 (Danecek et al. 2011) to keep only the individuals evaluated in 2019, 2020, and 2021, which resulted in a data set with 1,179 individuals. Next, we excluded SNPs with minor allele frequency (MAF) less than 0.01 using VCFtools. Finally, we conducted a step of Linkage Disequilibrium (LD) pruning using PLINK 1.9 (Purcell et al. 2007) with the option “–indep-pairwise”, which is based on the squared Pearson’s correlation coefficient (\(r^2\)), along with a window size of 100, step size of 20, and \(r^2\) threshold of 0.9. The final file consisted of 1,179 hybrids and 67,083 markers, which was converted to a numeric format using the package simplePHENOTYPES 1.3.0 (Fernandes and Lipka 2020) to enable us to calculate genomic relationship matrices.
We used the package AGHmatrix (Amadeu et al. 2023) from R 4.2.2 (R Core Team 2023) to create additive (A) (VanRaden 2008) and dominance (D) (Vitezica et al. 2013) relationship matrices. Thus, the genetic information utilized for downstream analysis consisted of the A matrix (\(1179 \times 1179\)) and the D matrix (\(1179 \times 1179\)) (see Fig. 1c for an illustration of the genetic data preprocessing steps).
Genomic prediction models
All genomic prediction models were fitted with a Gradient Boosting Machine (GBM) model implemented in the LightGBM (Ke et al. 2017) framework from Python 3.8. The models utilized different inputs for training, and they were grouped into four categories, namely, environmental (E), genetic (G), genetic-and-environmental (G+E), and genotype-by-environment interaction (GEI). All models using genetic information were independently fitted with A or D genomic relationship matrices. All models included the field location (i.e., the environment name without the year) as a categorical variable.
Environmental (E)
The E model was fitted using the data set of 90 environmental features (Supplementary Table S1). This data set was connected to the phenotypic data through a left join using the “Env” (environment) column as the primary key and fitted to the LightGBM framework. As these 90 environmental features were calculated using only the environmental data, all their values are the same within each environment regardless of the hybrid. Therefore, all predictions are the same within a given environment. Figure 1d outlines the E model fitting steps.
Genetic (G)
The G model was adjusted using the genomic relationship matrices previously obtained. The G models are denoted as G(A) when using the A matrix and G(D) when using the D matrix. The first step before using the genomic relationship matrix is to obtain the first 100 components from the SVD (Supplementary Table S2). Next, these components are merged to the phenotypic data via a left join using the “Hybrid” (hybrid) column as the primary key to be fitted to the LightGBM framework (Fig. 1e).
Genetic-and-environmental (G+E)
The G+E model integrates genotypic and environmental information but does not include an explicit genotype-by-environment interaction effect. The possible G+E models are denoted as G(A)+E when using the A matrix and G(D)+E when using the D matrix. The environmental and genetic data were connected to the phenotypic data through a left join using the “Env” and “Hybrid” columns as primary keys, where the environmental and genetic features together are decomposed with the SVD to obtain the first 100 components (Supplementary Table S2). The resultant data was fitted to the LightGBM framework (Fig. 1f).
Genotype-by-environment interaction (GEI)
For fitting GEI models, we calculated Kronecker products between the environmental matrix and a given genomic relationship matrix as follows:
where \({\textbf{X}}\) is the new genotype-by-environment matrix,Footnote 1\({\textbf{E}}\) is the environmental matrix with n unique environments and m environmental features, and \({\textbf{G}}\) is the genomic relationship matrix (either A or D) with q unique hybrids. Thus, the possible GEI models are denoted as G(A)EI when using the A matrix and G(D)EI when using the D matrix.
The \({\textbf{X}}\) matrix proceeded to an SVD step using the first 100 components (Supplementary Table S2). Next, these components are combined with the phenotypic data through a left join using the “Env” and “Hybrid” columns as primary keys and fitted to the LightGBM framework (Fig. 1g). As the genotype-by-environment matrix (\({\textbf{X}}\)) contains both environment and hybrid information, a unique prediction is obtained for each hybrid \(\times \) environment combination.

Workflow for machine learning genomic prediction. Data preprocessing steps (panels a–c) and model fitting steps (panels d–g). a, \(\text {y}_{\rm ENV_1}\), \(\text {y}_{\rm ENV_2}\), \(\dots \), \(\text {y}_{\rm ENV_n}\) represent the response variable vector for the \(1 {\rm st}\), \(2 {\rm nd}\), \(\dots \), \(n {\rm th}\) environments. Single-environment trial models are adjusted for each environment using equation (1) to compute the Best Linear Unbiased Estimates (BLUEs). Then, the BLUEs are concatenated row-wise to form the phenotypic matrix, which contains the columns “Env” (environment), “Hybrid” (hybrid), and “y” (BLUE). The columns “Env” and “Hybrid” are used as primary keys in further left join operations. b, Each type of environmental data proceeds to a different preprocessing step: weather, soil, and historical yield data are aggregated based on various summary statistics to produce new features, longitude and latitude are binned to reduce the noise around the varying locations, Agricultural Production Systems sIMulator (APSIM) environmental covariates (ECs) are moved to a Singular Value Decomposition (SVD) step to reduce their dimensionality from 765 to 15 columns, yet explaining 99.9% of the variance, and management is used as is. All the resulting features are then concatenated column-wise to create the environmental matrix, which includes the column “Env” (environment), used as a primary key in further left join operations. c, The matrix of single nucleotide polymorphisms (SNPs), consisting of 4928 individuals and 437,214 markers, proceeds to several steps for filtering and quality control: filtering to keep only individuals phenotyped in 2019, 2020, and 2021, excluding SNPs with minor allele frequency (MAF) less than 0.01, and pruning SNPs based on a Linkage Disequilibrium (LD) threshold of \(r^2 =\) 0.9, a window size of 100, and a step size of 20. The resulting matrix retains 1179 individuals and 67,083 markers. Then, this matrix is passed to a numericalization step to convert genotypes to numbers (Fernandes and Lipka 2020) and moves to the calculation of genomic relationship matrices. Using VanRaden (2008) yields the additive (A) genomic relationship matrix, and Vitezica et al. (2013) produces the dominance (D) genomic relationship matrix, in which both matrices have dimensions \(1179 \times 1179\). d, E: environmental. The phenotypic and environmental matrices are combined through a left join using the “Env” column as the primary key and then fitted to the LightGBM framework. e, G: genetic. The genomic relationship matrix (A or D) is passed to an SVD step to keep only 100 components, merged to the phenotypic matrix via left join using the “Hybrid” column as the primary key, and then fitted to the LightGBM framework. f, G+E: genetic-and-environmental. The environmental matrix, the genomic relationship matrix (A or D), and the phenotypic matrix are connected through a left join using the “Env” and “Hybrid” columns as primary keys. The environmental and genetic features are passed to an SVD step to keep only 100 components, and the resultant data is fitted to the LightGBM framework. g, GEI: genotype-by-environment interaction. A Kronecker product between the environmental matrix and the genomic relationship matrix (A or D) is taken to form the genotype-by-environment matrix, which proceeds to an SVD step to keep only 100 components, linked to the phenotypic matrix via a left join using the “Env” and “Hybrid” columns as primary keys, and then fitted to the LightGBM framework
Factor analytic multiplicative mixed model (FA)
To compare our results with a commonly used genomic prediction model, we fitted a Factor Analytic Multiplicative Mixed Model (FA) using the package ASReml-R 4 (Butler et al. 2017). The BLUEs of each hybrid \(\times \) environment combination were used as the response variable when fitting the following statistical model:
where \(Y_{ij}\) is the BLUE of the \(j^{th}\) hybrid on the \(i^{th}\) environment; \(\mu \) is the intercept; \(E_i\) is the fixed effect of the \(i^{th}\) environment; \(h(E)_{ij}\) is the random effect of the \(j^{th}\) hybrid within the \(i^{th}\) environment, with \(h(E)_{ij} \sim N(0, \varvec{\Sigma }_g)\), \(\varvec{\Sigma }_g = \textbf{FA}_1 \otimes {\bf{G}}\), where \(\textbf{FA}_1\) is the factor analytic matrix of order 1 of environments, \({\bf{G}}\) is the additive genomic relationship matrix of hybrids, \(\otimes \) is the Kronecker product, and \(e_{ij}\) is the residual term associated with the \(ij^{th}\) experimental unit, with \(e_{ij} \sim N(0, \sigma ^2_e)\).
Cross-validation schemes
We performed three cross-validation (CV) schemes as done in Sukumaran et al. (2018). All the schemes used ten repetitions of k-fold cross-validation with \(k=5\). Thus, in all cases, the phenotypic information was divided into five subsets, and each subset was used once as the validation set, while the remaining four were used as the training set.
CV2
The CV2 scheme was concerned with predicting the performance of known hybrids in known environments but with unknown combinations of hybrid and environments (sparse testing).
The following steps were taken for each of the five folds: (1) sample 20% of all the environment-hybrid combinations to build the validation set, and (2) remove this 20% randomly chosen combinations from the training set (trials from 2020 and 2021).
CV1
The CV1 scheme involved predicting the performance of unknown hybrids in known environments. The models were trained using trials from 2020 and 2021, and the prediction was made using trials from 2021.
The steps taken for each of the five folds were: (1) sample 20% of all hybrids from 2021 trials to build the validation set, and (2) remove this 20% randomly chosen hybrids from the training set (trials from 2020 and 2021).
CV0
The CV0 scheme predicted the performance of known hybrids in a new year. The models were trained using data from 2019 and 2020, and the predictions were made on 2021 trials.
The procedure applied for each of the five folds was as follows: (1) sample 20% of the hybrids evaluated in 2021 to build the validation set; (2) take the phenotypic data from 2019 and 2020 for these 20% randomly chosen hybrids to include in the training set, and (3) sample an additional 60% hybrids evaluated either in 2019 or 2020 trials and add to the training set. This last step was done to obtain an 80 : 20 proportion for the training and validation sets as in the other cross-validation schemes utilized in this study.
Metrics
As usually done in GP studies, we used Pearson’s correlation coefficient between the observed and the predicted yield to assess the prediction accuracy of the models. Another metric utilized was the Coincidence Index (CI) (Hamblin and Zimmermann 1986), which calculates how well the top predicted hybrids overlap with the top observed hybrids. The Coincidence Index (CI) was calculated considering the top 20% hybrids as follows:
where T is the total number of hybrids to evaluate, B is the number of overlapping hybrids (i.e., hybrids common to both observed and prediction sets), and R is the expected number of hybrids selected by chance. The CI was another metric employed in this study because it provides an alternative to compare how different models perform in terms of a predefined percentage of selected individuals.
Prediction accuracy pairwise comparisons were conducted following Meng et al. (1992) to assess model performance differences. For example, the pairwise comparison “FA - G(A)+E” denotes the difference between Pearson’s correlation of BLUEs and predictions of the FA model and Pearson’s correlation of BLUEs and predictions of the G(A)+E model. This type of statistical test compares two overlapping correlations based on dependent groups because both correlations share one variable in common (i.e., the BLUEs) and the same set of samples. A significance level of 0.05 was adopted, and the Bonferroni correction was applied to reduce the type I error.
The mean proportion of overlapping testers between training and validation populations was calculated as follows:
where \(\bar{p}\) is the mean proportion of overlapping testers between training and validation populations, i is the \(i^{th}\) fold, \(i = \{1, \dots , 5\}\), j is the \(j^{th}\) repetition, \(j = \{1, \dots , 10\}\), \({|W_{ij} \cap Z_{ij}|}\) is the number of overlapping testers between training (W) and validation (Z) sets, and \({|W_{ij} \cup Z_{ij}|}\) is the number of testers in training and validation sets.
Code and data availability
A repository containing all the scripts and documentation on reproducing the results is available at https://github.com/igorkf/Maize_GxE_Prediction. All the initial data used in this study are available at https://doi.org/10.25739/tq5e-ak26 (Genomes to Fields 2023; Lima et al. 2023). All intermediate files and scripts as well as final predictions obtained from our models are deposited at https://zenodo.org/records/12702650. All the plots were created using the packages matplotlib 3.2.2 (Hunter 2007) and seaborn 0.12.2 (Waskom 2021).
Results
The average coefficient of variation across years was 10.8%, 17.1%, and 15.1%, with values ranging from 0.1% to 22.3%, 11.5% to 27.9%, and 9.0% to 26.7% across environments, in 2019, 2020, and 2021, respectively (Supplementary Figure S1).
The average heritability across years was 0.38, 0.39, and 0.47, with values ranging from 0 to 0.84, 0.01 to 0.63, and 0 to 0.82 across environments, in 2019, 2020, and 2021, respectively (Supplementary Figure S1).
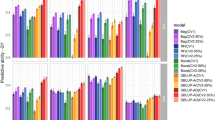
A great variation was also observed in prediction accuracy across models, especially when comparing different CV schemes. The mean prediction accuracy across CV schemes was 0.69, 0.68, and 0.42, with values ranging from 0.64 to 0.75, 0.62 to 0.73, and 0.28 to 0.49 across models in CV2, CV1, and CV0, respectively (Fig. 2).
Likewise, the mean CI showed variation across CV schemes (0.42, 0.48, and 0.31), with values ranging from 0.33 to 0.51, 0.34 and 0.66, and − 0.18 to 0.63 across models in CV2, CV1, and CV0, respectively (Fig. 3).

Prediction accuracy (the Pearson’s correlation between the observed and the predicted yield) of 5-fold cross-validation (CV) over ten repetitions for each model and CV scheme. The dashed line represents the mean prediction accuracy of the Factor Analytic Multiplicative Mixed Model (FA). G(.), G(.)+E, and G(.)EI are the genetic, genetic-and-environmental, and genotype-by-environment interaction models, respectively, with (.) being a genomic relationship matrix (A: additive or D: dominance)

Coincidence Index (CI) of 5-fold cross-validation (CV) over ten repetitions for each model and CV scheme. The dashed line represents the mean CI of the Factor Analytic Multiplicative Mixed Model (FA). G(.), G(.)+E, and G(.)EI are the genetic, genetic-and-environmental, and genotype-by-environment interaction models, respectively, with (.) being a genomic relationship matrix (A: additive or D: dominance). The CI was calculated considering the top 20% hybrids. This metric is used to compare how different models perform in terms of a predefined percentage of selected individuals
Population structure
Six groups were identified from a Principal Component Analysis (PCA), with the first two principal components explaining 29.6% of the variance. Using eq. (8), we noticed that, on average, a proportion of 100%, 90%, and 72% of overlapping testers was used between training and validation populations for the CV2, CV1, and CV0 schemes, respectively (Fig. 4).

Principal component analysis (PCA) of the population structure. The first two principal components (PC1 and PC2) explain 29.6% of the variance. The relative frequency of hybrids per tester is 23.7% (LH195), 23.9% (PHT69), 17.2% (PHZ51), 16.9% (PHK76), 16.7% (PHP02), and 1.6% (Other)
Depending on the cross-validation scheme, the genetic data alone can result in the worst prediction
For CV2 and CV1 schemes, the models that only used genetic information resulted in the worst performance in terms of prediction accuracy (Fig. 2).
For CV2, the prediction accuracy was statistically different among FA and G(D) (means 0.69 and 0.66) and among FA and G(A) (means 0.69 and 0.67). In both pairwise comparisons, FA exhibited better performance (Supplementary Table S3).
Likewise, for CV1, the prediction accuracy statistically differed between FA and G(D) (means 0.67 and 0.63) and between FA and G(A) (means 0.67 and 0.65), where, in both cases, FA showed better performance (Supplementary Table S3).
For CV0, the prediction accuracy ranged from 0.40 to 0.48 for model G(A) and from 0.39 to 0.47 for model G(D) with means 0.45 and 0.43, respectively. In contrast, FA resulted in a lower mean prediction accuracy of 0.41, with values ranging from 0.35 to 0.47. The pairwise comparisons FA and G(A) and FA and G(D) were statistically significant (Supplementary Table S3).
The models with only genetic information, particularly the dominance one, also showed a low coincidence index (CI) performance under CV2 and CV1 schemes (Fig. 3).
While the FA model had a mean CI of 0.41, the mean CI values for models G(D) and G(A) resulted in a prediction accuracy of 0.36 and 0.41, respectively, under the CV2 scheme.
Similarly, for CV1, the mean CI values were 0.46 for G(D), 0.52 for G(A), and 0.53 for FA.
For the CV0 scheme, G(A) showed the best mean CI (0.41), with values ranging from 0.26 to 0.55, while the model G(A)EI performed the worst with a mean CI of 0.14 and values ranging from − 0.16 to 0.50 (Fig. 3).
With the inclusion of environmental information in the G models, the mean prediction accuracy was at least equal to the FA model for all the CV schemes. For example, all G+E models yielded higher mean prediction accuracy than FA in CV1 and CV2 (Fig. 2). Furthermore,
G(A)+E had a significant boost in mean prediction accuracy over G(A) of \(10.0\%\) for CV2 and \(11.1\%\) for CV1 (Supplementary Table S3).
In the CV0 scenario, the models G(A)+E and G(A)EI both resulted in a mean prediction accuracy of 0.45, which is \(7.1\%\) greater than the mean prediction accuracy achieved by the FA, with both differences being statistically significant. Overall, regarding prediction accuracy, the G(A)+E model was superior or at least equal to all other models across all the CV schemes evaluated in this study (Supplementary Table S3).
Discussion
The success of modern breeding programs lies not only in the scale of data acquisition but also in the different types of information used in the decision-making process. In this study, we investigated the inclusion of several non-genetic types of data in genomic prediction models. We incorporated all of these data by taking advantage of the flexibility of machine learning models such as GBM. We compared them with a standard approach utilized for MET analysis.
Many trials were performed in the G2F initiative from 2019 to 2021. Each environment employed different sets of hybrids to compose the yield trials. In total, 1,179 unique hybrids were evaluated in this study, although many of the hybrids were not used in all the environments (Supplementary Figure S2). The number of common hybrids among environments shows significant variability, and few hybrids from the 2019 trials were tested in the 2020 and 2021 trials.
The influence of the genetic and environmental diversity in the prediction models
The genetic diversity of the population and the relatedness between the training and validation sets are critical factors in genomic prediction accuracy (Crossa et al. 2017; Fraslin et al. 2022). The number of testers used in the G2F initiative population changed over the years. For the subset of the data utilized in our research, two testers were used in 2019 and four testers in 2020 and 2021 (one of the testers from 2019 and three new testers). The genetic diversity was mostly driven by these five testers utilized (Fig. 4). Conversely, the magnitude of the environmental variability was considerably higher than what is typically observed in breeding programs, given that the environments include very distinct locations. The larger diversity of the environment compared with the genetic diversity could be the reason for the environmental model, which predicts the average of a given environment for all hybrids, to perform similarly to genomic models.
The importance of environmental information should not be underestimated
Several studies have indicated that environmental information helps to enhance the prediction of phenotypic performance (Heslot et al. 2014; Technow et al. 2015; Costa-Neto et al. 2021, 2022). When modeling GEI in maize with machine learning, Westhues et al. (2021) did not observe much gain when predicting plant height, but the grain yield prediction improved after including environmental information. The prediction accuracies from CV1 and CV2 indicate that, on average, the environmental model was just as good as the genetic models. However, accurately selecting the top individuals is more relevant than average prediction for breeding programs. As expected, using environmental variables alone was not as useful in selecting the top 20% individuals. However, in the G+E combination, the selection of the top 20% individuals was still equal or superior to all models evaluated. Our results are further evidence that environmental data should not be neglected in prediction models, especially when considering the G+E scenario.
The usefulness of feature engineering will depend on the approaches utilized
Several options exist for doing the feature engineering step in time series data, such as weather. We summarised weather conditions using the season as the grouping factor, which differed from previous studies in maize (Westhues et al. 2021; Kick et al. 2023). Although Westhues et al. (2021) utilized feature engineering without much success they derived features based on developmental stages. The authors argued that, due to phenotyping costs, the crop developmental stages through time were incorporated in their models using only three main developmental stages (vegetative, flowering, and grain filling), which could have negatively affected the efficiency of their features. We did not attempt to use the growing stages as a grouping factor. Kick et al. (2023) utilized k-means with dynamic time warping to cluster the weather and management time-series data, which enhanced the performance of models that only used genomic data. Also, we included the field location as a categorical factor in all the ML models to account for constant environmental effects (e.g., soil texture, management practices, etc.).
The usefulness of diverse data for accurate prediction
Including several data types in ML is often more straightforward than in linear models for GP. Because of this flexibility, our models benefited from utilizing a diverse set of information, including historical yield, ECs, soil properties, and climatic and geographical information. Although no attempt was made to fit individual models with each of the different types of data available, the initial results observed in the model fitting process (data not shown) indicated an advantage of keeping all the environmental information utilized in our final models. The advantages of utilizing historical information, for instance, have also been observed in linear models for GP (Rutkoski et al. 2015; Dias et al. 2020). However, we use it in a fundamentally different way as we utilize the historical yield from hybrids that are not necessarily related to the target hybrids to characterize each environment. ECs are another type of data utilized in our model that is becoming more common in GP models (Bustos-Korts et al. 2019; Onogi 2022; Jighly et al. 2022). Our results confirmed that ECs such as the ones derived from the APSIM crop model are useful features in improving prediction abilities. Thus, developing approaches that directly integrate crop growth models with GP could be the best alternative for improving prediction accuracy. In agreement with what has been observed in the literature (Westhues et al. 2021; Washburn et al. 2021; Kick et al. 2023), our results indicate that researchers should consider broadening GP to phenotypic prediction based on multiple types of data.
GEI for machine learning
Our GEI models used Kronecker products to explicitly create interactions between each hybrid in the genomic relationship matrix and each environment in the derived environmental matrix. However, the Kronecker product abruptly increases the dimensionality of the data set, and fitting models using the resulting data set is not feasible. When using the GEI models, the number of columns in the resulting data set is massive (\(>100,000\)). Moreover, the memory consumption for GEI models surpassed 200GB of RAM, which is usually available only at high-performance computing (HPC) clusters. Conversely, the G+E models were much more parsimonious and computationally efficient, given that the number of columns in the data set is around 1250–1300. Despite having much smaller dimensionality than GEI models, the G+E models still had many features, which increases the time for model fitting and the chance of overfitting, given that not all features are individually meaningful. Thus, to overcome the problem of high dimensionality with possible redundancy of information and large model-fitting time, we employed the SVD method to reduce the data set dimensionality before fitting the models (Fig. 1e, f, g; Supplementary Table S2).
As noted by Westhues et al. (2021), tree-based machine learning models do not require an explicit inclusion of genotype-by-environment interaction as input to the model, given that a high-order interaction between features is captured by construction (Friedman 2001). This fact was corroborated in this study, where the G+E model, which only integrates genetic and environment data through the concatenation of data sets, was better or at least similar to the performance of GEI models in all the CV schemes. The G+E model was much more parsimonious than the GEI model and was computationally efficient, requiring approximately 30 s to fit the model for one fold and one repetition (data not shown) on a general-purpose computational node with two Intel(R) Xeon(R) Gold 6130 CPU @ 2.10GHz processors, equipped with 32 cores and 192GB of RAM. This contributes to the need for more efficient computational strategies for integrating genomic and environmental data to expand GP models to new environments and germplasm, enhancing our comprehension of genotype-by-environment interactions (Rogers and Holland 2021).
Conclusion
This study demonstrates the massive importance of the environment in the outcome of a prediction model. As a means to incorporate such information into genomic prediction models, the ML framework offers great flexibility, especially when utilizing feature engineering. Our results illustrate that the feature engineering step presents a valuable envirotyping option, creating useful variables for ML-based genomic prediction models. As the amounts and diversity of data available in breeding programs increase, there will be more opportunities for utilizing feature engineering in breeding programs. We demonstrated that environmental features, in combination with genetic data, can significantly improve prediction accuracy over models using only genetic information. Furthermore, we confirmed that with tree-based methods, the genotype-by-environment interactions can be accounted for without explicitly including interactions in the model. Collectively, these results are promising, especially with the increasing interest in combining envirotyping and genotyping approaches for prediction purposes.
Notes
Because of its large size, the \({\textbf{X}}\) matrix was saved in the “arrow” format using the package arrow 12.0.0 (Richardson et al. 2023). This format was adopted for fast reading the \({\textbf{X}}\) matrix before fitting the models, given that the model needed to be fitted many times (i.e., across different cross-validation (CV) schemes, and repetitions).
Abbreviations
- ML:
-
Machine learning
- G+E:
-
Genetic-and-environmental
- GEI:
-
Genotype-by-environment interaction
- FA:
-
Factor analytic multiplicative mixed model
- MET:
-
Multi-environment trials
- GP:
-
Genomic prediction
- CNN:
-
Convolutional neural networks
- DNN:
-
Dense neural networks
- G2F:
-
Genomes to fields
- RCBD:
-
Randomized complete block design
- BLUEs:
-
Best linear unbiased estimates
- ECs:
-
Environmental covariates
- APSIM:
-
Agricultural production systems sIMulator
- SVD:
-
Singular value decomposition
- PHG:
-
Practical haplotype graph
- SNP:
-
Single-nucleotide polymorphism
- MAF:
-
Minor allele frequency
- LD:
-
Linkage disequilibrium
- A:
-
Additive
- D:
-
Dominance
- GBM:
-
Gradient boosting machine
- E:
-
Environmental
- G:
-
Genetic
- CV:
-
Cross-validation
- CI:
-
Coincidence index
- PCA:
-
Principal component analysis
- HPC:
-
High-performance computing
- PAR:
-
Photosynthetically active radiation
References
Amadeu RR, Garcia AAF, Munoz PR, Ferrão LFV (2023) AGHmatrix: genetic relationship matrices in R. Bioinformatics 39(7):btad445. https://doi.org/10.1093/bioinformatics/btad445
Bernardo RN (2014) Essentials of plant breeding. Stemma press, Woodbury
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Bradbury PJ, Casstevens T, Jensen SE, Johnson LC, Miller ZR, Monier B, Romay MC, Song B, Buckler ES (2022) The practical haplotype graph, a platform for storing and using pangenomes for imputation. Bioinformatics 38(15):3698–3702. https://doi.org/10.1093/bioinformatics/btac410
Breiman L (2001) Random forests. Mach Learn 45(1):5–32. https://doi.org/10.1023/A:1010933404324
Burgueño J, de los Campos G, Weigel K, Crossa J (2012) Genomic prediction of breeding values when modeling genotype × environment interaction using pedigree and dense molecular markers. Crop Sci 52(2):707–719. https://doi.org/10.2135/cropsci2011.06.0299
Bustos-Korts D, Boer MP, Malosetti M, Chapman S, Chenu K, Zheng B, van Eeuwijk FA (2019) Combining crop growth modeling and statistical genetic modeling to evaluate phenotyping strategies. Front Plant Sci. https://doi.org/10.3389/fpls.2019.01491
Butler D, Cullis B, Gilmour A, Gogel B, Thompson R (2017) Asreml-r reference manual version 4. VSN International Ltd, Hemel Hempstead
de los campos G, Gianola D (2007) Factor analysis models for structuring covariance matrices of additive genetic effects: a bayesian implementation. Genet Sel Evolut 39(5):481. https://doi.org/10.1186/1297-9686-39-5-481
Canella Vieira C, Persa R, Chen P, Jarquin D (2022) Incorporation of soil-derived covariates in progeny testing and line selection to enhance genomic prediction accuracy in soybean breeding. Front Genet. https://doi.org/10.3389/fgene.2022.905824
Chen T, Guestrin C (2016) Xgboost: A scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Association for Computing Machinery, New York, NY, USA, KDD ’16, p 785-794, https://doi.org/10.1145/2939672.2939785
Costa-Neto G, Crossa J, Fritsche-Neto R (2021) Enviromic assembly increases accuracy and reduces costs of the genomic prediction for yield plasticity in maize. Front Plant Sci. https://doi.org/10.3389/fpls.2021.717552
Costa-Neto G, Crespo-Herrera L, Fradgley N, Gardner K, Bentley AR, Dreisigacker S, Fritsche-Neto R, Montesinos-López OA, Crossa J (2022) Envirome-wide associations enhance multi-year genome-based prediction of historical wheat breeding data. G3 Genes [Genomes Genet] 13(2):313. https://doi.org/10.1093/g3journal/jkac313
Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de Losampos G, Burgueño J, González-Camacho JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S, Singh R, Zhang X, Gowda M, Roorkiwal M, Rutkoski J, Varshney RK (2017) Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci 22(11):961–975. https://doi.org/10.1016/j.tplants.2017.08.011
Cullis BR, Smith AB, Coombes NE (2006) On the design of early generation variety trials with correlated data. J Agric Biol Environ Stat 11(4):381–393. https://doi.org/10.1198/108571106X154443
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, Depristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, Mcvean G, Durbin R, Group GPA (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Dias KOG, Gezan SA, Guimarães CT, Parentoni SN, Guimarães PEdO, Carneiro NP, Portugal AF, Bastos EA, Cardoso MJ, Anoni CdO, de Magalhães JV, de Souza JC, Guimarães LJM, Pastina MM (2018) Estimating genotype × environment interaction for and genetic correlations among drought tolerance traits in maize via factor analytic multiplicative mixed models. Crop Sci 58(1):72–83. https://doi.org/10.2135/cropsci2016.07.0566
Dias KOG, Piepho HP, Guimarães LJM, Guimarães PEO, Parentoni SN, Pinto MO, Noda RW, Magalhães JV, Guimarães CT, Garcia AAF, Pastina MM (2020) Novel strategies for genomic prediction of untested single-cross maize hybrids using unbalanced historical data. Theor Appl Genet 133(2):443–455. https://doi.org/10.1007/s00122-019-03475-1
Falconer DS (1996) Introduction to quantitative genetics, 4th edn. Prentice Hall Harlow, Harlow
Fernandes SB, Lipka AE (2020) simplephenotypes: simulation of pleiotropic, linked and epistatic phenotypes. BMC Bioinform 21(1):491. https://doi.org/10.1186/s12859-020-03804-y
Fraslin C, Yáñez JM, Robledo D, Houston RD (2022) The impact of genetic relationship between training and validation populations on genomic prediction accuracy in atlantic salmon. Aquac. Rep. 23(101):033. https://doi.org/10.1016/j.aqrep.2022.101033
Friedman JH (2001) Greedy function approximation: a gradient boosting machine. Ann Stat 29(5):1189–1232. https://doi.org/10.1214/aos/1013203451
Genomes to Fields (2023) Genomes to fields 2022 maize genotype by environment prediction competition. https://doi.org/10.25739/tq5e-ak26
Gillberg J, Marttinen P, Mamitsuka H, Kaski S (2019) Modelling G × E with historical weather information improves genomic prediction in new environments. Bioinformatics 35(20):4045–4052. https://doi.org/10.1093/bioinformatics/btz197
Gong Z, Zhong P, Hu W (2019) Diversity in machine learning. IEEE Access 7:64323–64350. https://doi.org/10.1109/ACCESS.2019.2917620
Hamblin J, Zimmermann MJdO (1986) Breeding Common Bean for Yield in Mixtures. Wiley, Hoboken, pp 245–272. https://doi.org/10.1002/9781118061015.ch8
Heslot N, Akdemir D, Sorrells ME, Jannink JL (2014) Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions. Theor Appl Genet 127(2):463–480. https://doi.org/10.1007/s00122-013-2231-5
Hunter JD (2007) Matplotlib: a 2d graphics environment. Comput Sci Eng 9(3):90–95. https://doi.org/10.1109/MCSE.2007.55
Jarquín D, Crossa J, Lacaze X, Du Cheyron P, Daucourt J, Lorgeou J, Piraux F, Guerreiro L, Pérez P, Calus M, Burgueño J, de Losampos G (2014) A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor Appl Genet 127(3):595–607. https://doi.org/10.1007/s00122-013-2243-1
Jarquín D, Lemes da Silva C, Gaynor RC, Poland J, Fritz A, Howard R, Battenfield S, Crossa J (2017) Increasing genomic-enabled prediction accuracy by modeling genotype × environment interactions in kansas wheat. Plant Genome. https://doi.org/10.3835/plantgenome2016.12.0130
Jighly A, Thayalakumaran T, O’Leary GJ, Kant S, Panozzo J, Aggarwal R, Hessel D, Forrest KL, Technow F, Tibbits JFG, Totir R, Hayden MJ, Munkvold J, Daetwyler HD (2022) Using genomic prediction with crop growth models enables the prediction of associated traits in wheat. J Exp Bot 74(5):1389–1402. https://doi.org/10.1093/jxb/erac393
Jubair S, Tremblay-Savard O, Domaratzki M (2023) Gxenet: novel fully connected neural network based approaches to incorporate gxe for predicting wheat yield. Artif Intell Agric 8:60–76. https://doi.org/10.1016/j.aiia.2023.05.001
Ke G, Meng Q, Finley T, Wang T, Chen W, Ma W, Ye Q, Liu TY (2017) Lightgbm: A highly efficient gradient boosting decision tree. In: Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R (eds) Advances in Neural Information Processing Systems, Curran Associates, Inc., vol 30, https://proceedings.neurips.cc/paper_files/paper/2017/file/6449f44a102fde848669bdd9eb6b76fa-Paper.pdf
Kick DR, Wallace JG, Schnable JC, Kolkman JM, Alaca B, Beissinger TM, Edwards J, Ertl D, Flint-Garcia S, Gage JL, Hirsch CN, Knoll JE, de Leon N, Lima DC, Moreta DE, Singh MP, Thompson A, Weldekidan T, Washburn JD (2023) Yield prediction through integration of genetic, environment, and management data through deep learning. G3 Genes Genomes Genet 13(4):jkad006. https://doi.org/10.1093/g3journal/jkad006
Lima DC, Washburn JD, Varela JI, Chen Q, Gage JL, Romay MC, Holland J, Ertl D, Lopez-Cruz M, Aguate FM, de Loscampos G, Kaeppler S, Beissinger T, Bohn M, Buckler E, Edwards J, Flint-Garcia S, Gore MA, Hirsch CN, Knoll JE, McKay J, Minyo R, Murray SC, Ortez OA, Schnable JC, Sekhon RS, Singh MP, Sparks EE, Thompson A, Tuinstra M, Wallace J, Weldekidan T, Xu W, de Leon N (2023) Genomes to fields 2022 maize genotype by environment prediction competition. BMC Res Notes 16(1):148. https://doi.org/10.1186/s13104-023-06421-z
Lopez-Cruz M, Aguate FM, Washburn JD, de Leon N, Kaeppler SM, Lima DC, Tan R, Thompson A, De La Bretonne LW, de Loscampos G (2023) Leveraging data from the genomes-to-fields initiative to investigate genotype-by-environment interactions in maize in north america. Nat Commun 14(1):6904. https://doi.org/10.1038/s41467-023-42687-4
Malosetti M, Bustos-Korts D, Boer MP, van Eeuwijk FA (2016) Predicting responses in multiple environments: Issues in relation to genotype × environment interactions. Crop Sci 56(5):2210–2222. https://doi.org/10.2135/cropsci2015.05.0311
Meng XL, Rosenthal R, Rubin DB (1992) Comparing correlated correlation coefficients. Psychol Bull 111(1):172–175. https://doi.org/10.1037/0033-2909.111.1.172
Montesinos-López OA, Montesinos-López A, Tuberosa R, Maccaferri M, Sciara G, Ammar K, Crossa J (2019) Multi-trait, multi-environment genomic prediction of durum wheat with genomic best linear unbiased predictor and deep learning methods. Front Plant Sci. https://doi.org/10.3389/fpls.2019.01311
Monteverde E, Gutierrez L, Blanco P, Pérez de Vida F, Rosas JE, Bonnecarrère V, Quero G, McCouch S (2019) Integrating molecular markers and Environmental covariates to interpret genotype by environment interaction in rice (Oryza sativa L.) grown in subtropical areas. Genes Genomes Genet 9(5):1519–1531. https://doi.org/10.1534/g3.119.400064
Onogi A (2022) Integration of crop growth models and genomic prediction. Springer, New York, pp 359–396. https://doi.org/10.1007/978-1-0716-2205-6_13
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit-learn: machine learning in python. J Mach Learn Res 10(5555/1953048):2078195. https://doi.org/10.5555/1953048.2078195
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. https://doi.org/10.1086/519795
R Core Team (2023) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/
Ray S, Jarquin D, Howard R (2023) Comparing artificial-intelligence techniques with state-of-the-art parametric prediction models for predicting soybean traits. Plant Genome 16(1):e20263. https://doi.org/10.1002/tpg2.20263
Richardson N, Cook I, Crane N, Dunnington D, François R, Keane J, Moldovan-Grünfeld D, Ooms J, Apache Arrow (2023) arrow: Integration to ’Apache’ ’Arrow’. https://CRAN.R-project.org/package=arrow, r package version 12.0.0
Rogers AR, Holland JB (2021) Environment-specific genomic prediction ability in maize using environmental covariates depends on environmental similarity to training data. G3 Genes Genomes Genet 12(2):jkab440. https://doi.org/10.1093/g3journal/jkab440
Rutkoski J, Singh RP, Huerta-Espino J, Bhavani S, Poland J, Jannink JL, Sorrells ME (2015) Efficient use of historical data for genomic selection: a case study of stem rust resistance in wheat. Plant Genome. https://doi.org/10.3835/plantgenome2014.09.0046
Smith A, Cullis B, Thompson R (2001) Analyzing variety by environment data using multiplicative mixed models and adjustments for spatial field trend. Biometrics 57(4):1138–1147. https://doi.org/10.1111/j.0006-341X.2001.01138.x
Sukumaran S, Jarquin D, Crossa J, Reynolds M (2018) Genomic-enabled prediction accuracies increased by modeling genotype × environment interaction in durum wheat. Plant Genome 11(2):170112. https://doi.org/10.3835/plantgenome2017.12.0112
Tabery J (2008) R. a. fisher, lancelot hogben, and the origin(s) of genotype-environment interaction. J Hist Biol 41(4):717–761. https://doi.org/10.1007/s10739-008-9155-y
Technow F, Messina CD, Totir LR, Cooper M (2015) Integrating crop growth models with whole genome prediction through approximate bayesian computation. PLOS ONE. https://doi.org/10.1371/journal.pone.0130855
Van Rossum G, Drake FL (2009) Python 3 Reference Manual. CreateSpace, Scotts Valley. https://doi.org/10.5555/1593511
van Raden P (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91(11):4414–4423. https://doi.org/10.3168/jds.2007-0980
Vitezica ZG, Varona L, Legarra A (2013) On the additive and dominant variance and covariance of individuals within the genomic selection scope. Genetics 195(4):1223–1230. https://doi.org/10.1534/genetics.113.155176
Washburn JD, Cimen E, Ramstein G, Reeves T, O’Briant P, McLean G, Cooper M, Hammer G, Buckler ES (2021) Predicting phenotypes from genetic, environment, management, and historical data using cnns. Theor Appl Genet 134(12):3997–4011. https://doi.org/10.1007/s00122-021-03943-7
Waskom ML (2021) seaborn: statistical data visualization. J Open Source Softw 6(60):3021. https://doi.org/10.21105/joss.03021
Westhues CC, Mahone GS, da Silva S, Thorwarth P, Schmidt M, Richter JC, Simianer H, Beissinger TM (2021) Prediction of maize phenotypic traits with genomic and environmental predictors using gradient boosting frameworks. Front Plant Sci. https://doi.org/10.3389/fpls.2021.699589
Acknowledgements
This research was supported by the University of Arkansas System Division of Agriculture. This research was only possible through the resources provided by the Arkansas High-Performance Computing Center, which is funded through multiple National Science Foundation grants and the Arkansas Economic Development Commission. Kaio O. G. Dias was supported by the Minas Gerais State Agency for Research and Development (FAPEMIG). We want to thank Dr. Matt Murphy and Ashmita Upadhyay for their valuable feedback.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Contributions
IKF and SBF designed the research; IKF performed the model fitting and statistical analyses; IKF and SBF wrote the first draft; IKF, CCV, KOGD, and SBF revised drafts of the paper. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical statement
The experiments were performed according to the current laws of The United States of America.
Additional information
Communicated by Daniela Bustos-Korts.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernandes, I.K., Vieira, C.C., Dias, K.O.G. et al. Using machine learning to combine genetic and environmental data for maize grain yield predictions across multi-environment trials. Theor Appl Genet 137, 189 (2024). https://doi.org/10.1007/s00122-024-04687-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04687-w