
Abstract
Key message
The pepper mutants (‘221-2-1a’ and ‘1559-1-2h’) with very low pungency were genetically characterized. The Pun4 locus, responsible for the reduced pungency of the mutant fruits, was localized to a 208 Mb region on chromosome 6. DEMF06G16460, encoding 3-ketoacyl-CoA synthase, was proposed as a strong candidate gene based on the genetic analyses of bulked segregants, DEG, and expression analyses.
Abstract
Capsaicinoids are unique alkaloids present in pepper (Capsicum spp.), synthesized through the condensation of by-products from the phenylpropanoid and branched-chain fatty acid pathways, and accumulating in the placenta. In this study, we characterized two allelic ethyl methanesulfonate-induced mutant lines with extremely low pungency (‘221-2-1a’ and ‘1559-1-2h′). These mutants, derived from the pungent Korean landrace 'Yuwolcho,' exhibited lower capsaicinoid content than Yuwolcho but still contained a small amount of capsaicinoid with functional capsaicinoid biosynthetic genes. Genetic crosses between the mutants and Yuwolcho or pungent lines indicated that a single recessive mutation was responsible for the low-pungency phenotype of mutant 221-2-1a; we named the causal locus Pungency 4 (Pun4). To identify Pun4, we combined genome-wide polymorphism analysis and transcriptome analysis with bulked-segregant analysis. We narrowed down the location of Pun4 to a 208-Mb region on chromosome 6 containing five candidate genes, of which DEMF06G16460, encoding a 3-ketoacyl-CoA synthase associated with branched-chain fatty acid biosynthesis, is the most likely candidate for Pun4. The expression of capsaicinoid biosynthetic genes in placental tissues in Yuwolcho and the mutant was consistent with the branched-chain fatty acid pathway playing a pivotal role in the lower pungency observed in the mutant. We also obtained a list of differentially expressed genes in placental tissues between the mutant and Yuwolcho, from which we selected candidate genes using gene co-expression analysis. In summary, we characterized the capsaicinoid biosynthesis-related locus Pun4 through integrated of genetic, genomic, and transcriptome analyses. These findings will contribute to our understanding of capsaicinoid biosynthesis in pepper.
Similar content being viewed by others

Introduction
The Capsicum genus includes approximately 42 species and is believed to have originated in tropical and temperate Central and South America, Mexico, and the West Indies of North America (Barboza et al. 2019, 2020; Eshbaugh 1980; Walsh and Hoot 2001). Five Capsicum species have been extensively cultivated and domesticated throughout Europe, Africa, India, China, and South America: chili pepper (C. annuum), habanero pepper (C. chinense), tabasco pepper (C. frutescens), cayenne pepper (C. baccatum), and tree pepper (C. pubescens) (Barboza et al. 2020; Bosland 1994; Carrizo García et al. 2016). Pepper fruits are used in various ways, ranging from crunchy vegetables to seasonings. The spicy compound used for seasoning in pepper belongs to the capsaicinoids, unique alkaloid mixtures found only in the genus Capsicum (Aza-González et al. 2011). Capsaicinoids have numerous functions relevant in human health, including analgesic, anti-cancer, anti-inflammatory, anti-oxidative, and anti-obesity properties (Aza-González et al. 2011; Govindarajan and Sathyanarayana 1991; Liu and Nair 2010; Luo et al. 2011; Negulesco et al. 1987). Furthermore, capsaicinoids exert anti-fungal and anti-oomycete effects, including against Fusarium (Veloso et al. 2014). In light of these attributes, extensive research has explored the genetic mechanisms underlying capsaicinoid biosynthesis and regulation.
The existence of the enzyme capsaicin synthase (CS) was proposed based on the chemical structure of capsaicin, as an attempt to explain the condensation reaction leading to the formation of capsaicin (Nelson and Dawson 1923). The capsaicinoid biosynthetic pathway branches in two directions: the phenylpropanoid pathway for vanillylamine biosynthesis and the branched-chain fatty acid pathway for the production of 8-methyl-6-nonenoic acid (Bennett and Kirby 1968; Leete and Louden 1968). The hypothesis of a CS enzyme paved the way for identifying potential capsaicinoid biosynthetic enzymes and their corresponding encoding genes, by harnessing transcriptome and genomic information (Aluru et al. 2003; Curry et al. 1999; Kim et al. 2001). In the phenylpropanoid pathway, well-characterized and conserved precursor enzymes contribute to vanillylamine biosynthesis, including phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase (C4H), 4-coumaronyl-CoA ligase (4CL), coumarate 3-hydroxylase (C3H), caffeoyl-CoA O-methyltransferase (CCoAOMT), hydroxycinnamoyl-CoA hydratase/lyase (HCHL), and putative aminotransferase (pAMT) (Aza-González et al. 2011; Fujiwake et al. 1982; Mazourek et al. 2009; Stewart Jr et al. 2005; Sukrasno and Yeoman 1993). Similarly, several structural genes have been identified from the branched fatty acid pathway, including those encoding acyl carrier protein (ACL), acyl-ACP thioesterase (FatA), ketoacyl-ACP synthase (KAS), branched-chain amino acid aminotransferase (BCAT), and ketoacyl-ACP reductase (CaKR1) (Aluru et al. 2003; Koeda et al. 2019; Mazourek et al. 2009). Furthermore, genes regulating capsaicinoid biosynthesis, such as transcription factor genes from the R2R3-MYB (Han et al. 2019; Liu et al. 2022; Sun et al. 2019; Yu et al. 2023; Zhu et al. 2019) and WRKY families (Zhang et al. 2023; Zhu et al. 2019) participate in the regulatory mechanisms behind these two biosynthetic branches. Nonetheless, the functions of only a few genes in the pathway, such as Pun1 (also known as C), pAMT, Pun3 (also known as CaMYB31), and CaKR1, have been confirmed using natural loss-of-function variants. (Han et al. 2019; Koeda et al. 2019; Lang et al. 2009Stewart Jr et al. 2005). However, due to limitations in available natural variants, it is necessary to utilize artificially induced mutant populations to further elucidate the biosynthetic genes of capsaicinoids.
Several attempts have been made to create mutagenized populations within Capsicum species and look for phenotypes of interest, primarily using C. annuum cultivars as starting materials. One such early effort involved treating the blocky C. annuum cultivar ‘Maor’ with ethyl methanesulfonate (EMS), focusing primarily on mutants with altered shoot architecture (Paran et al. 2007). Subsequently, three different populations have been generated using EMS- or radiation-induced mutations by two separate Korean groups (Hwang et al. 2014; Jo et al. 2016; Siddique et al. 2020). In particular, our group generated EMS-induced populations using the Korean C. annuum landrace ‘Yuwolcho’ and the ornamental cultivar ‘Micro-Pep Red.’ We undertook a systematic characterization of phenotypes in this mutant population, including plant growth and development of leaves, flowers, and fruits, in addition to assessing capsaicinoid content (Hwang 2014; Hwang et al. 2014; Jeong et al. 2012b; Siddique et al. 2020). More recently, even reproductive phenotypes, including seed number, have attracted interest (Arisha et al. 2015; Tanaka et al. 2021). To harness the potential of the mutant resources generated through mutagenesis in genetic research, both forward and reverse genetic approaches can be employed. Forward genetics is the main method currently in use to uncover the genetic basis of specific traits. Conversely, reverse genetics is suitable for testing the contribution of a candidate gene to a phenotype of interest and exploring novel allelic variants. High-throughput sequencing techniques have greatly facilitated the identification of causal mutations in species with high-quality genomes, with two main possible approaches (Kim et al. 2014, 2017; Lee et al. 2022; Ou et al. 2018; Qin et al. 2014). For example, the causal mutation behind a phenotype of interest can be rapidly identified by bulked-segregant analysis (BSA) followed by sequencing, named ‘fast-forward genetics,’ which expedites the identification of candidate genes (Schneeberger and Weigel 2011). Through the above BSA-seq approach, a mapping interval can be accurately delimited to a small region while all sequence polymorphisms across the genome can be cataloged, thus enabling the swift identification of candidate genes (Klein et al. 2018).
In our previous studies, we generated an EMS-induced mutant population in the pungent Korean landrace Yuwolcho (Hwang et al. 2014; Jeong et al. 2012a). Among the resulting individuals, we screened 917 M2 lines for pungency-related phenotypes. Among the potential mutants, we selected two lines, ‘221-2-1a’ and ‘1559-1-2h,’ with significantly lower capsaicinoid content compared to Yuwolcho, for further investigation (Hwang 2014; Lee 2018). In this study, we performed a comprehensive analysis of the mutant phenotypes and determined the mode of inheritance of these two allelic mutations in different segregating populations. Subsequently, we employed BSA-seq to precisely map the mutant locus and narrow down the candidates. Furthermore, we explored the candidate genes responsible for the low-pungency phenotype of these two mutants using transcriptome analyses.
Materials and methods
Plant materials and allelism test
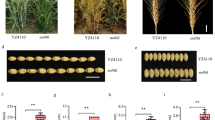
The Capsicum annuum accessions ‘Yuwolcho’ (wild type: WT), ‘Lam32,’ and ‘Micro-Pep Red’ (MR) were selected from the germplasm of the Horticultural Crops Breeding and Genetics Laboratory (Seoul National University, Seoul, Republic of Korea) (Fig. 1a). The C. annuum mutant lines ‘221-2-1a’ and ‘1559-1-2h’ showing extremely low pungency derived from an ethyl methanesulfonate (EMS)-induced Yuwolcho mutant population, were selected to construct three populations in this study (Hwang et al. 2014; Jeong et al. 2012a). The first population, ‘221Y,’ consisting of 133 F2 and 80 F3 plants, was derived from a cross between 221 and 2-1a and WT. The bulked samples of 18 low-pungent F3 lines selected from the 221Y population and 18 WT plants were used for bulked-segregant analysis by sequencing (BSA-seq). The second population, ‘221L,’ was derived from a cross between 221 and 2-1a and Lam32. A total of 283 F2 plants were obtained to delimit the candidate gene interval. The third population, ‘1559M’, was derived from a cross between 1559 and 1-2h and MR. Among the 136 F2 plants, 36 plants (18 pungent and 18 with low pungency) were selected for bulked-segregant RNA sequencing (BSR-seq).

Extent of phenotypic variation among pepper fruits used in this study. a Fruit morphologies of parental and control lines. MG, mature green stage; BK, breaker stage. Scale bars, 1 cm. b Capsaicinoid content of parental and control fruits. Yuwolcho, MicroPep_Red, and Lam32 are pungent cultivars; ECW is a non-pungent cultivar. F1, hybrid of 221-2-1a and 1559-1-2h. Capsaicinoid content is shown as capsaicin (CAP) and dihydrocapsaicin (DHCAP) concentrations in fruit placental tissues. At least three pepper fruits were used for replicated samplings. Values are means ± standard deviation (SD)
Allelism tests were conducted by crossing 1559-1-2h to C. annuum ‘ECW’, C. annuum ‘YCM334’, and C. chinense ‘No. 3341.’ Another test was conducted by crossing 221-2-1a to 1559-1-2h. The F1 plants from each cross were grown to evaluate the capsaicinoid content of their placental tissues.
Capsaicinoid content evaluation
Capsaicinoid content was evaluated using the Gibb’s reagent test and high-performance liquid chromatography (HPLC). Capsaicinoid extracts were prepared from at least three fruits at the mature green (MG) and breaker (BK) stages for each plant following the method of Han et al. (2013). For the capsaicinoid content of the 221Y F2, F3, and 221L F2 populations, Gibb’s reagent method was used to determine the presence of capsaicinoids in plants (Jeong et al. 2012b). Those fruits that contained no capsaicinoids based on the Gibb’s reagent test were re-analyzed by HPLC. In the case of the 1559M F2 population, all 136 F2 plants were analyzed by HPLC. The HPLC analysis was performed at the National Instrumentation Center for Environmental Management (NICEM; Seoul, Republic of Korea). Total capsaicinoid content was calculated as the sum of capsaicin and dihydrocapsaicin contents, and the phenotype (pungent or non-pungent) was determined from comparison to total capsaicinoid content in the control and parental lines each year.
Capsaicinoid biosynthetic gene (CBG) expression analysis
The expression of CBGs was analyzed by calculating the Log2 fold-change between the expression estimates for WT and 221-2-1a, using fragments per kilobase of transcripts per million mapped reads (FPKM) values derived from RNA sequencing (RNA-seq) analysis. Two separate RNA pools were obtained from at least three fruit placentas at 25 days post-anthesis (DPA) from seven plants. Total RNA was extracted from each plant using TaKaRa MiniBEST Plant RNA Extraction Kit (Takara Korea Biomedical, Seoul, Korea), and library construction and data analysis were followed the method of Byun et al. (2022) based on a NovaSeq 6000 instrument (Illumina, San Diego, CA, US). A total of 73 loci corresponding to 48 CBGs were selected from the C. annuum ‘CM334’ v1.6 genome based on their sequence similarity with genes annotated as capsaicin biosynthesis-related genes in previous studies (Kim et al. 2017; Mazourek et al. 2009). Among the CBGs, eight genes, Pun1 (AT3), Pun3 (CaMYB31), pAMT, BCAT homolog, BCKDH E1α homolog, KAS homolog, ACL homolog, and FatA homolog, were analyzed by reverse-transcription quantitative PCR (RT-qPCR) at five stages of fruit development (14-, 21-, 28-, 35-, and 42-DPA) for the confirmation of RNA-seq data. The RT-qPCR was performed with the same condition as described by Byun et al. (2022) using Rotor-Gene 6000 real-time PCR instrument (Qiagen, Hilden, Germany) and Actin was used as a reference gene for RT-qPCR.
MutMap analysis
MutMap analysis was carried out based on the whole-genome sequencing of a low-pungency bulk consisting of 18 F3 individuals derived from the 221Y population and a WT bulk from 18 WT plants. Two separate paired-end DNA libraries were constructed for WT and 18 F3 samples using a TruSeq DNA PCR-Free kit (Illumina, San Diego, CA, US) as per Illumina guidelines. The libraries were sequenced on an Illumina HiSeq 4000 instrument, producing paired-end reads of 100 bp. Based on the whole-genome sequencing data of WT, a Yuwolcho reference genome was constructed by replacing the polymorphic positions in the C. annuum Dempsey genome (Lee et al. 2022) with their corresponding nucleotides obtained from Illumina short reads of Yuwolcho. MutMap analysis was conducted using the MutMap pipeline version 1.4.4 from Iwate Biotechnology Research Center (IBRC; http://genome-e.ibrc.or.jp) (Abe et al. 2012). Coval software was used to filter low-quality reads based on the number of mismatches, and SNP indices were calculated at all SNP positions (Kosugi et al. 2013). The SNP-index plots were plotted in R. The effects of SNPs were annotated with SnpEff (Cingolani et al. 2012).
Genetic mapping of Pun4
A total of 153 genome-wide SNP markers distinguishing between WT and Lam32 were selected from 412 SNP marker sets, which have been designed for marker-assisted backcrossing (MABC) (Kang et al. 2014), to construct a linkage map of the 221L F2 population, consisting of 283 plants. The genetic map was constructed using a minimum logarithm of the odds (LOD) score of 3 and a maximum genetic distance of 30 centimorgan (cM) between consecutive markers using CARTHA GENE software (de Givry et al. 2005). For fine-mapping of Pun4 on chromosome 6, a systematic approach was followed, leading to the development of 11 high-resolution melting (HRM) markers (Table S1). For HRM, a Roter-Gene 6000 real-time PCR instrument (Qiagen, Hilden, Germany) and Rotor-Gene Q series software version 2.1.0 were used. To determine the physical locations of the markers, marker sequences were compared to the Dempsey, CM334 v1.6, and ‘Zunla-1’ v2.0 genomes by BLASTn (Kim et al. 2017; Lee et al. 2022; Qin et al. 2014).
De novo contig level assembly of Yuwolcho genome
High-quality genomic DNAs obtained from Yuwolcho leaf tissues were subjected to sequencing on the Pacific Biosciences (PacBio, CA, USA). Single-molecule real-time (SMRT) sequencing platform. The resulting high-fidelity (HiFi) reads were then de novo assembled into contigs using hifiasm (Cheng et al. 2021), and the quality of the assembly was assessed through benchmarking universal single-copy orthologs (BUSCO) analysis (Solanales odb10) at the National Instrumentation Center for Environmental Management (NICEM; Seoul, Republic of Korea). Following the identification of SNP marker positions used in genetic mapping on the assembled contigs, local gene prediction was carried out for a 6-Mb region within a contig near the Pun4 locus using MAKER (Cantarel et al. 2008). The predicted genes were subsequently functionally annotated through a BLASTp search against the Swissprot plant protein database.
Detection of differentially expressed genes (DEGs) and gene ontology (GO) enrichment analysis
DEGs were identified from the RNA-seq reads of WT and 221–2-1a and two separate RNA pools from the 1559M F2 population. In the case of WT and 221–2-1a, the same samples used for the CBG expression analysis above were analyzed, and the RNA pools of the 1559M F2 population were obtained from the placental tissues of at least three fruits at 25-DPA from 18 with the highest capsaicinoid content and 18 plants with the lowest capsaicinoid content, respectively. These two groups were then divided into three pools each, and RNA extraction, library construction, and sequencing of two separate RNA pools from the 1559M F2 population were conducted in the same manner as described earlier. DEGs between WT and 221–2-1a or the 1559M F2 pools were identified with the DESeq2 R package using RNA-seq reads aligned to the ‘Dempsey’ reference genome (Lee et al. 2022; Love et al. 2014). Among the DEGs, CBG homologs were annotated and their expression levels visualized using the EnhancedVolcano R package (Blighe et al. 2019). GO term enrichment analysis of DEGs was performed using agriGO v2.0 and the GO terms were grouped into higher groups using REVIGO (Supek et al. 2011; Tian et al. 2017). For agriGO v2.0, annotated GO terms were analyzed with single enrichment analysis (SEA); Fisher method was used as statistical test.
Weighted gene co-expression network analysis (WGCNA)
A gene co-expression network analysis was conducted using the R package WGCNA to identify genes whose expression pattern correlated with that of differentially expressed CBGs between WT and 221-2-1a (Langfelder and Horvath 2008). FPKM values for five different stages of WT placenta development (14-, 21-, 28-, 35-, and 42-DPA) were used for this analysis. The FPKM values for seven CBGs (BCAT, BCKDH E1α, ACL, KasI, KasIII, FatA, and AT3) that showed differential expression between WT and 221-2-1a were used as trait data input. Adjacency (network connectivity) values between each gene expression pattern were calculated using a ‘signed’ network type and a soft threshold power value of 65. Adjacency values were then translated into a topological overlap matrix (TOM) to calculate the corresponding dissimilarity (dissTOM = 1 − TOM). Gene clustering was performed based on dissTOM, and a tree plot was drawn using the cutreeDynamic function, with each branch displaying modules of clustered eigengenes (MEs). The correlation between MEs and trait data was calculated, and a correlation heatmap was drawn according to this calculation.
Quantitative trait locus (QTL)-seq analysis
QTL-seq analysis of the 1559M F2 population was conducted using the QTLseqr package (Mansfeld and Grumet 2018). The same data of two RNA pools from 1559M F2 population that used in DEG analysis were used. The RNA reads were aligned to the C. annuum Dempsey genome, and the significant single-nucleotide polymorphisms (SNPs) between the two groups were selected and filtered through the QTLseqr package.
Results
Characterization of two EMS mutants with extremely low pungency
We characterized the two extremely low-pungent mutant lines 221-2-1a and 1559-1-2h, derived from an EMS-mutagenized population in the Yuwolcho cultivar, to identify the genetic factor (s) that regulate fruit pungency. We performed HPLC analysis of placental tissues, which revealed that the 221-2-1a and 1559-1-2h mutants accumulated 17.7 ± 24.8 μg/g dry weight (DW) and 258.8 ± 285.7 μg/g DW of total capsaicinoids, respectively, much less than did the parental and control lines (20,979.2 ± 8195.5 μg/g DW) (Fig. 1b). To determine whether 221-2-1a and 1559-1-2h harbor a mutation in the same gene, we crossed the two mutants and measured the capsaicinoid content of their F1 progeny, using the placenta (Table 1). All these F1 plants had extremely low pungency, as evidenced by an average capsaicinoid content of 34 μg/g DW. We thus conclude that the mutants 221-2-1a and 1559-1-2 h carry a mutation in the same gene.
Inheritance of Pun4, a novel locus controlling pungency in Capsicum
We generated three segregating populations to identify the causal mutation behind the low-pungency phenotype of the mutants 221-2-1a and 1559-1-2 h. More precisely, we crossed 221-2-1a to the cultivar Yuwolcho (221Y population) or to the cultivar Lam32 (221L population); we also crossed 1559-1-2 h to the cultivar Micro-Pep Red (1559 M population). We evaluated the pungency of each plant based on Gibb’s reagent and HPLC analysis. Gibb’s reagent produced a blue color, which we compared to serially diluted standards to estimate capsaicinoid content. We considered a capsaicinoid content below 3,000 μg/g DW through HPLC analysis as low pungency, based on the distribution of capsaicinoid content within the 1559 M F2 population (Fig. S1). All F1 plants from all three crosses had pungent fruit, indicating that the two mutations are recessive. Pungency exhibited distinct segregation ratios in various F2 populations (Table 2). The segregation ratio for the 221L F2 and the 1559M F2 populations was 4:1 and 2:1, respectively, suggesting that the low-pungency phenotype of the mutants is caused by the mutation of one major gene with additional influence from minor factors.
Allelism test with pun1, pun3, and cakr1 mutants
The genes Pun1, Pun3, pAMT, and CaKR1 contribute to the control of capsaicinoid biosynthesis in Capsicum (Han et al. 2019; Koeda et al. 2019; Lang et al. 2009; Stewart Jr et al. 2005). We sequenced these genes in the mutants 221-2-1a and 1559-1-2 h but detected no mutations compared to their reference sequences. To validate these results, we conducted allelism tests by crossing the 1559-1-2 h mutant to Capsicum accessions harboring non-functional alleles of Pun1, Pun3, or CaKR1. All F1 hybrids from crosses between 1559 and 1-2h and ECW (pun1/pun1), 1559-1-2h and YCM334 (pun3/pun3), and 1559-1-2h and No. 3341 (cakr1/cakr1) had significantly pungent fruit, with an average capsaicinoid content over 3,000 μg/g DW (Table 1). These results suggest that the low-pungent mutants 221-2-1a and 1559-1-2 h harbor functional alleles of Pun1, Pun3, and CaKR1. Therefore, we named the locus defined by these two mutants as Pun4.
Expression of CBGs in WT and 221–2-1a
We identified 73 CBG-like genes based on their sequence similarity to genes annotated as capsaicin biosynthesis-related genes in previous studies (Kim et al. 2017; Mazourek et al. 2009). Of these 73 genes present in the CM334 v1.6 reference genome, we narrowed down the list to 48 CBGs that are expressed in the placentas of 25-DPA fruit and determined their expression levels in WT and 221-2-1a by RNA-seq (Table S2, Fig. 2a). Among the 48 CBGs, 42 gene were expressed to lower levels in the 221-2-1a mutant compared to WT. The expression of functionally identified genes, such as Pun1 (AT3), Pun3 (CaMYB31), pAMT, and CaKR1, was much lower in 221-2-1a relative to WT, as was the expression of genes associated with the branched-chain fatty acid biosynthesis pathway, including BCAT, BCKDH, ACL, MCAT, KasI, KasIII, and FAT. By contrast, we observed minor differences in the expression levels of genes involved in the phenylpropanoid pathway (Fig. 2b). We validated the RNA-seq analysis by conducting RT-qPCR on eight genes using RNA samples extracted from five different stages of fruit development (Fig. S2). All genes showed a significant decrease in expression in the mutant compared to the WT throughout the various stages.

Expression of CBGs in WT and the mutant 221-2-1a. a Heatmap representation of the expression levels of CBGs in WT and 221-2-1a. Values are shown as Log2(FPKM + 1). b Overview of the capsaicin biosynthesis pathway. Biosynthetic intermediates are shown in white ellipses, with the names of each structural gene encoding the enzyme responsible for the step shown by the arrows. The expression values are shown as Log2([FPKM of WT / FPKM of 221-2-1a] + 1), with black arrows indicating no difference between the two genotypes and red indicating higher expression in WT. Most of the genes showing decreased expression in the mutant, such as BCAT, BCKDH, ACL, MCAT, KasI, KasIII, and FAT, belong to the latter part of valine/leucine metabolism and fatty acid biosynthesis pathways. Thee pathways were modified from Mazourek et al. (2009)
Genetic mapping of Pun4 with genome-wide SNP markers
To determine the chromosomal location of the Pun4 locus, we constructed a linkage map based on 89 F2 plants from the 221L F2 population and 153 genome-wide SNP markers that are polymorphic between WT and Lam32. We obtained 12 linkage groups, using a maximum genetic distance between consecutive markers of 30 cM, resulting in a map with an average genetic distance of 6.8 cM between consecutive markers (Fig. S3a). We then used this genetic map to determine the location of the Pun4 locus, using the pungency phenotype of the 221L F2 population. We mapped the Pun4 locus to the middle of the linkage group LG6, between the markers CAPS_CONTIG.192 and KS20039B04, defining a genetic interval of 12.3 cM and a physical interval ranging from 209.05 Mb to 210.96 Mb on the chromosome 6 (Fig. S3b).
MutMap analysis for the detection of EMS-induced causal SNPs
We independently performed a MutMap analysis to identify sequence differences between WT and the mutant 221-2-1a that would be consistent with an EMS-induced mutation. We performed BSA-seq of two pools, one containing genomic DNA of WT fruits and one from F3 individuals of the 221Y population with low pungency. Of the clean reads produced from sequencing of these two pools, 91.7% and 68.5% of reads aligned to the Dempsey reference genome, respectively (Table S3). We then generated a pseudogenome for Yuwolcho by introducing the Yuwolcho-type SNPs into the Dempsey reference genome, followed by MutMap analysis with a 4-Mb sliding window size and 10-kb increment. Of the 12 chromosomes, we observed consecutive peaks with ΔSNP-indices above the 99% confidence interval of the sliding window average only for chromosome 6 (Fig. S4). We detected four peaks spanning 193.11–215.89 Mb on chromosome 6 with ΔSNP-indices of 1. Within these peak regions, we identified 2,560 SNPs, of which 216 potential sequence differences were consistent with an EMS mutation (G-to-A and C-to-T conversions) with a mutant allele frequency (AF) exceeding 0.6. Of these 216 sequence differences, we retained 41 based on their location within the 208–212-Mb region encompassing the mapping interval defined above (Fig. S5).
Fine-mapping of the Pun4 locus
To further delimit the mapping interval of Pun4, we designed 11 HRM markers to genotype 283 F2 plants from the 221L population. Of these 283 plants, we selected 30 recombinants, each exhibiting various recombination patterns within the mapping interval of 197.14–210.96 Mb on chromosome 6 (Fig. 3). We narrowed down the target region to a 66-kb interval through fine-mapping, from 207.71 to 208.37 Mb. In the Dempsey reference genome, this interval contains 11 genes and six sequence differences, based on the MutMap analysis. Only one of these differences was within a large intron within DEMF06G16330, based on the current genome annotation (Fig. 3) (Table S4). However, it is likely an intergenic region due to the misannotation caused by the high similarity between two genes (Fig. S6). We mapped all six EMS-type sequence differences onto the CM334 v1.6 and Zunla-1 v2.0 genomes, but no consensus was found regarding their location within the genic region or the 3-kb promoter region.

Fine-mapping of candidate locus on chromosome 6. Using 11 HRM marker sets in the mapping interval, the genotypes of 30 recombinants with different pungency phenotypes were used for genotyping. Red, pungent (Lam32 genotype); white, low-pungency phenotype (221-2-1a genotype); orange, pungent phenotype (heterozygous). The number of recombinants for each marker is given below the marker name. The black arrowheads denote EMS-induced SNPs in each genome sequences over this interval. Annotated genes are shown as black arrows in each reference genomes. The gray box shows a misannotated region
De novo contig level assembly of Yuwolcho genome
The genome of Yuwolcho was sequenced and assembled at the contig level using long reads from PacBio SMRT sequencing. A total of 5,141,472 HiFi reads were generated, resulting in approximately 70.8 Gb (about 23 × coverage) of read bases. The contig assembly produced 726 contigs, which were subsequently filtered to 244 contigs originating from organelles. The assembly process notably increased the N50 value (the size of the shortest contig fragment representing 50% of the total genome) from 14.06 kb to 95.29 Mb (Table S5), and BUSCO analysis indicated a high completeness of 98.2% for the assembly (Table S6). The contig labeled 'ptg000019l' contained HRM markers tightly linked with the Pun4 locus in genetic mapping (Table S1). Therefore, a 6-Mb region spanning approximately 205–211 Mb of the Dempsey reference genome was chosen for functional annotation. The functional annotation predicted a total of 173 proteins in this sequence, with 151 proteins annotated through BLASTp against the Swissprot plant protein database using a criterion of e-value < 1.
Identification of DEGs and GO term enrichment analysis
To identify DEGs associated with the Pun4 gene, we analyzed two datasets: specifically, we compared expression levels between WT and 221–2-1a, as well as between pools of pungent and low-pungency fruits from 1559M F2 individuals. We defined DEGs based on the criteria of p value < 0.05 and absolute Log2(fold-change) > 2. We identified 9186 DEGs between WT and 221-2-1a, of which 4448 were upregulated in the mutant and 4738 were downregulated in the mutant. Similarly, we identified 365 DEGs between the two 1559M pools, with 70 upregulated DEGs and 295 downregulated DEGs in the low-pungency pool. We discovered that nearly all CBGs were downregulated in the mutant and in the low-pungency pool, as illustrated by volcano plots (Fig. 4a–c). Comparison of the two datasets revealed 295 overlapping DEGs. For each set, we performed a GO term enrichment analysis using the SEA of AgriGO v2.0 and summarized the obtained GO terms by REVIGO, using a p value threshold of < 0.05. The two sets had 16 common GO terms (Fig. 4d), including ‘lipid biosynthetic process,’ ‘lipid metabolic process,’ ‘monocarboxylic acid metabolic process,’ and ‘organic acid metabolic process.’ These terms are related to lipid metabolism and may identify genes that serve in the steps of capsaicinoid biosynthesis.

DEGs related to pungency. a Venn diagram showing the overlap between the number of detected DEGs between WT and the mutant 221-2-1a and between the pungent pool and the low-pungency pool derived from the 1559M F2 population using fruit placenta. b, c Volcano plot of DEGs between WT and 221-2-1a (b) and between the two 1559M F2 pools (c). Blue circles represent DEGs with an absolute Log2(fold-change) < 2; red circles represent DEGs with an absolute Log2(fold-change) > 2 and p value < 0.05. Putative CBGs were annotated using black arrows in the plot. d Enriched GO terms among DEGs common to the WT vs 221-2-1a comparison and to the two extreme 1559M F2 pools. Each value indicates the −Log10(p value) (color figure online)
WGCNA using the expression of CBGs across fruit development
We performed WGCNA to refine the list of candidate genes showing similar expression patterns as seven CBGs that displayed significant differential expression between WT and the mutant among the identified DEGs. We determined gene expression levels in five pools of WT fruit tissues at different developmental stages and used these expression values to construct a co-expression network, resulting in 21 MEs. Among these, MEgreen showed the strongest correlation with the expression levels of the seven CBGs mentioned above between WT and the mutant (Fig. 5b). However, KasI, the only gene showing high expression in 14-DPA fruits (Fig. 5a), was highly correlated with MEbisque3. Although MEgreen was the most highly positively correlated ME with all seven CBGs (Fig. 5c), each of these CBGs exhibited significant correlations with different MEs, such as MEbisque3 (ACL, KasIII), MEcornsilk2 (BCAT), MEblack (BCKDH E1α), MEblue3 (KasI), and MEbrown1 (Pun1). From the co-expression network, we extracted 512 genes located on chromosome 6 that belong to these MEs for characterization.

Summary of WGCNA. Twenty-one MEs were identified, each represented by a color name. The expression levels of BCAT, BCKDH E1α, ACL, KasI, KasIII, FatA, and AT3 (Pun1) were used as trait input. a Dendrogram of samples based on the similarity in expression pattern of the seven CBGs. In the heatmap, intensity of red indicates higher expression. b Module–trait relationships, as indicated by Pearson’s correlation coefficients. c, d Correlation between MEs and traits, represented as heatmap (c) or dendrogram (d)
Integration of fine-mapping, BSA-seq, and RNA-seq to identify candidates
RNA-seq analysis and WGCNA helped us define 512 genes on chromosome 6 that are differentially expressed in the 221-2-1a mutant and whose expression is highly correlated with that of CBGs. Among these genes, 12 were located close to the final mapping interval (207.71–208.37 Mb). Of these 12 genes, we retained five as high-confidence candidate genes based their Log2(fold-change) values between WT and 221-2-1a (Table 3). We functionally annotated these five genes through BLASTp searches using the non-redundant (nr) database from the National Center for Biotechnology Information (NCBI) to predict gene function. Of these five genes, we speculate that DEMF06G16460 is a strong candidate for the Pun4 locus, as its encoded protein shows similarity to 3-ketoacyl-CoA synthases, which may function as a long fatty acid synthase downstream of the fatty acid elongation cycle that could start the middle of capsaicinoid biosynthesis to bypass. It is also possible that one or more of the final six sequence differences we detected from MutMap analysis is the causal mutation and may interact with the candidate gene.
Discussion
In this study, we characterized the two extremely low-pungency mutants 221-2-1a and 1559-1-2h from an EMS-mutagenized population and investigated the genetic characteristics of the causal Pun4 locus across three segregating populations. The expression of CBGs in the 221-2-1a mutant suggests a potential association between Pun4 and branched-chain fatty acid biosynthesis. Furthermore, we propose a strong candidate gene from genetic mapping and DEG analysis. Overall, we speculate that the newly identified Pun4 locus is directly or indirectly involved in the biosynthesis of branched-chain fatty acids during capsaicinoid biosynthesis.
Characterization of mutants in a new gene controlling capsaicin biosynthesis
The two mutants 221-2-1a and 1559-1-2h accumulate only 0.07% and 1.02%, respectively, of the capsaicinoid content in the WT. The mutant 221-2-1a almost completely lacks capsaicinoids, similar to ECW cultivar that harboring Pun1 loss-of-function allele (Fig. 1b). An allelism test confirmed that the two mutants harbor a mutation in the same causal locus, Pun4; the reason for their phenotypic differences is unknown. However, a similar phenomenon was observed in a previous study of pAMT alleles, with weaker alleles being caused by the insertion of transposable elements at various positions within the same intron of the pAMT gene (Tanaka et al. 2019).
All F1 plants derived from crosses between the mutant 1559-1-2h and non-pungent varieties (pun1, pun3, and cakr1) accumulated significantly more capsaicinoids than did the mutants (Fig. 1b) (Table 1), indicating that the Pun4 locus is unlikely to be allelic to these known genes. The much higher capsaicinoid content of F1 plants derived from the cross 1559-1-2h × No. 3341 (cakr1/cakr1) compared to the wild-type C. annuum accessions used in this study may reflect the high pungency of C. chinense, which was used as the paternal line. All F1 plants derived from crosses between each mutant (221-2-1a and 1559-1-2h) and pungent cultivars were also pungent (Table 2), suggesting that the Pun4 locus is dominant, similar to other functionally identified CBGs such as Pun1, Pun3, pAMT, and CaKR1 (Han et al. 2019; Koeda et al. 2019; Lang et al. 2009; Stewart Jr et al. 2005).
A combined analysis of linkage mapping and BSA-seq anchored the Pun4 locus to chromosome 6 around 208-Mb in the Dempsey reference genome, indicating one major gene (Fig. 3) (Fig. S5). However, the segregation ratios of two different F2 populations were inconsistent (Table 2), suggesting the segregation of one major dominant gene (Pun4) together with minor contributing factor(s). While pungency was treated as a qualitative trait, the strength of pungency is a quantitative trait that is affected by various factors, including environmental factors such as light, temperature, water availability, or nutrition (Naves et al. 2019). Therefore, further research is required to identify the specific factors contributing to the variation in pungency in the segregating populations.
Selection of candidate genes for CBGs based on transcriptome analysis
We conducted RNA-seq analysis of two datasets (between WT and 221-2-1a; between individuals from the 1559M F2 population with high pungency or low pungency). Independently, we measured gene expression in WT fruits across five developmental stages; we used these results as input for WGCNA and looked specifically for genes that are co-expressed with known or suspected CBGs. In the DEG analysis, we identified 9186 DEGs and 365 DEGs, respectively (Fig. 4a), with most CBGs being downregulated in the mutant (221-2-1a) and the low-pungency pool (Fig. 4b, c). Among the downregulated DEGs in 221-2-1a relative to its WT, genes showing at least a four-fold difference in expression included genes similar to BCAT, BCKDH, PYRUVATE DEHYDROGENASE (PDH), ACL, MCAT, KasIII, KasI, HYDROXYL-ACP DEHYDRATASE (DH), and FatA, which are thought to be associated with fatty acid biosynthesis, with the exception of a CINNAMYL ALCOHOL DEHYDROGENASE (CAD)-like gene and the AT3 gene (Pun1). Among the 11 genes, 5 (BCAT, BCKDH, ACL, FatA, and CAD) were common to both datasets, including DEMF06G16460, our candidate gene for Pun4. Based on these results, we hypothesize that Pun4 is involved in fatty acid biosynthesis. The changes in fatty acid biosynthetic gene expression may be correlated with the downregulation of the CAD homolog and the AT3 gene, which are located at the end of the phenylpropanoid pathway. Therefore, all these CBG homologs should be functionally investigated: BCAT (DEMF04G22720), BCKDH (DEMF06G13250), ACL (DEMF01G33980), FatA (DEMF06G29890), and CAD (DEMF02G11700).
WGCNA produced 21 distinct MEs, of which MEgreen showed high correlation with the expression levels of seven CBGs that were DEGs in the above analysis (Fig. 5a–c). However, each CBG belonged to distinct MEs, prompting us to select all MEs that contained at least one CBG. We therefore selected 746 (MEbisque3), 241 (MEcornsilk2), 51 (MEblack), 615 (MEblue3), and 61 (MEbrown1) genes. Of these genes, 20 mapped onto chromosome 6 and were among the DEGs in dataset 1 and 2. After comparing their expression patterns to those of seven CBGs, we retained 15 genes as candidates (Table S7). We functionally annotated these genes, revealing CBG-like genes (DEMF06G11720, DEMF06G17360, DEMF06G29890, and DEMF06G30030) and a transcription factor gene (DEMF06G23780). These genes may also contribute to capsaicin biosynthesis, and their function should be examined in detail.
QTL-seq analysis exhibits consistent results with other analyses
We conducted a BSR-seq analysis on two extreme pools derived from the 1559M F2 population to detect EMS-induced mutations within the mapping interval. While several chromosomes had strong discrete signals for Gprime analysis, only chromosome 6 exhibited consistently high Gprime values across the chromosome. Moreover, the defined interval overlapped with that obtained by genetic mapping (Fig. S7a). In the region of chromosome 6 after 100-Mb, we detected 1,245 EMS-induced mutations (comprising G-to-A and C-to-T conversions). We also detected several peaks exceeding the significance threshold on chromosomes 1, 2, 3, 4, 5, 7, 10, and 12, which likely reflect natural variation between WT and MR. Several of the Gprime peaks had a matching peak from QTL-seq analysis, although only two (on chromosomes 2 and 6) showed a ΔSNP index above the 99% confidence interval (CI) (Fig. S7b). We then filtered EMS-induced mutations located within the peak on chromosome 6, using 99% CI, leading to 338 sequence differences consistent with an EMS mutation. Of these, 12 were located in genic regions within the broad 208–212-Mb mapping interval. However, Sanger sequencing revealed that all of these differences could be attributed to polymorphisms between WT and MR rather than to EMS treatment. These findings indicate that the low-pungency phenotype of the mutant may not be attributed to sequence variation within genic regions.
Lower expression of fatty acid pathway-related genes results in low pungency
The capsaicinoid biosynthetic pathway consists of two main branches (Fig. 2b), each sharing certain components with other pathways. Phenylpropanoid metabolism might be connected to various secondary metabolite pathways, including the shikimate, quinate, flavonoid, and lignin pathways (Fraser and Chapple 2011). MYB24, a negative co-regulator of capsaicin and lignin biosynthesis, might play a role behind this connection of pathways (Yu et al. 2023). Fatty acid biosynthesis is, however, thought to be more complex. It initiates with acetyl-CoA, which serves as the precursor for the citrate cycle, and involves several branching pathways for biosynthesis. These biochemical steps include elongation cycles that can generate a multitude of fatty acid analogs (Mazourek et al. 2009). To date, no research has explored the interaction or co-regulation between the capsaicinoid pathway and the fatty acid cycle. We determined that fatty acid biosynthetic genes are downregulated in fruits with low pungency, suggesting a connection between fatty acid biosynthesis and the capsaicinoid pathway (Figs. 2, 4b, c ). Among the genes identified as candidates for Pun4, the candidate gene DEMF06G16460 encodes a putative 3-ketoacyl-CoA synthase potentially associated with fatty acid elongation and wax biosynthesis (Table 3) (Todd et al. 1999). Hence, we propose that Pun4 likely corresponds to this KCS homolog. KCS may play a role in a regulation of the production of a capsaicin pathway precursor and the regulation of long-chain fatty acids.
Comparison to previously studied pungency-related loci on chromosome 6 of pepper
According to previous studies, the four capsaicinoid biosynthesis genes Pun1, Pun3, pAMT, and CaKR1 are located on pepper chromosomes 2, 7, 3, and 10, respectively, based on the C. annuum genomes (Han et al. 2019; Koeda et al. 2019; Lang et al. 2009; Stewart Jr et al. 2005). In addition, various quantitative trait loci (QTLs) or genomic regions associated with capsaicinoid content in placenta or pericarp have been identified (Table 4) (Ben-Chaim et al. 2006; Blum et al. 2003; Cao et al. 2022; Han et al. 2018; Kondo et al. 2023; Lee et al. 2016; Yarnes et al. 2013). Among these genomic regions, two (SJ-dhc6 and punv) are located near Pun4. SJ-dhc6 was identified from a population derived from a cross between the non-pungent C. chinense cultivar ‘SNU11-001’ and the pungent C. chinense cultivar ‘Bhut Jolokia’ for capsaicinoid content in both placenta and pericarp. punv was identified by genome-wide association study (GWAS), with two polymorphisms in a gene (Capana06g001204) associated with pungency across a population of 347 cultivars from 12 Capsicum species (Cao et al. 2022; Park et al. 2019). We observed no sequence variation and expression difference that would alter the protein encoded by Capana06g001204 between WT and the pun4 mutants. Therefore, Capana06g001204 may be different from Pun4.
Genome context differences between Dempsey and Yuwolcho genome near the Pun4 locus
Considering the potential failure in discovering variation due to contextual differences within the mapping region of Dempsey and Yuwolcho, we generated a Yuwolcho assembly. This resulted in a contig-level assembly and confirmed contextual differences between Yuwolcho and Dempsey through gene prediction and functional annotation (Fig. 5). And, the BSA-seq data was aligned to the Yuwolcho assembly to obtain the SNP list within the predicted gene sequences (Table S8). However, three SNPs (ptg000019l:9,538,501, ptg000019l:9,560,873, ptg000019l:9,610,609) that homogeneously preserved the EMS-induced sequence were not present within the coding sequence. It is expected that the gene found in the delimited region obtained through mapping will not contain the Pun4 target gene, and that an external genetic factor will interact with the SNP found in the region.
Candidate gene selection and further studies
By combining the results of all analyses presented in this study, we have selected five genes located in the proximity of the Pun4 locus as high-confidence candidates, although none of them showed significant differences in their genic sequences between WT and the mutants. Enhancers can modulate the expression of genes located as far as 2–3 Mb away in metazoans, and this phenomenon has also been observed in crop plants, such as maize (Krivega and Dean 2012; Stam et al. 2002; Weber et al. 2016). Therefore, it is possible that one of the mutations may be within a long-distance cis-regulatory element region of the candidates, acting as an enhancer or silencer. Additionally, the SNPs in the mapping region may exist on the trans-regulatory genetic factors, including ncRNA or miRNA related to CBGs, which cannot be ruled out. Consequently, the five candidate genes could potentially serve as target genes for Pun4, acting as either enhancers or silencing factors. Furthermore, CBG homologs that are located on chromosome 6 and exhibit differential expression between WT and mutants may be considered as another set of potential genes that interact with the mutation(s) within the Pun4 locus (Table S7).
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30(2):174–178
Aluru MR, Mazourek M, Landry LG, Curry J, Jahn M, O’Connell MA (2003) Differential expression of fatty acid synthase genes, Acl, Fat and Kas Capsicum Fruit. J Exp Bot 54(388):1655–1664
Arisha MH, Shah SN, Gong Z-H, Jing H, Li C, Zhang H-X (2015) Ethyl methane sulfonate induced mutations in M2 generation and physiological variations in M1 generation of peppers (Capsicum annuum L.). Front Plant Sci 6:399
Aza-González C, Núñez-Palenius HG, Ochoa-Alejo N (2011) Molecular biology of capsaicinoid biosynthesis in chili pepper (Capsicum spp.). Plant Cell Rep 30:695–706
Barboza GE, Carrizo García C, Leiva González S, Scaldaferro M, Reyes X (2019) Four new species of Capsicum (Solanaceae) from the tropical Andes and an update on the phylogeny of the genus. PLoS ONE 14(1):e0209792
Barboza GE, de Bem Bianchetti L, Stehmann JR (2020) Capsicum carassense (Solanaceae), a new species from the Brazilian Atlantic forest. PhytoKeys 140:125
Ben-Chaim A, Borovsky Y, Falise M, Mazourek M, Kang B-C, Paran I, Jahn M (2006) QTL analysis for capsaicinoid content in Capsicum. Theor Appl Genet 113:1481–1490
Bennett D, Kirby G (1968) Constitution and biosynthesis of capsaicin. J Chem Soc C 442–446
Blighe K, Rana S, Lewis M (2019). EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R package version, 1(0).
Blum E, Mazourek M, O’connell M, Curry J, Thorup T, Liu K, Jahn M, Paran I (2003) Molecular mapping of capsaicinoid biosynthesis genes and quantitative trait loci analysis for capsaicinoid content in Capsicum. Theor Appl Genet 108:79–86
Bosland P (1994). Chiles: history, cultivation, and uses
Byun J, Kim T-G, Lee J-H, Li N, Jung S, Kang B-C (2022) Identification of CaAN3 as a fruit-specific regulator of anthocyanin biosynthesis in pepper (Capsicum annuum). Theor Appl Genet 135(7):2197–2211
Cao Y, Zhang K, Yu H, Chen S, Xu D, Zhao H, Zhang Z, Yang Y, Gu X, Liu X (2022) Pepper variome reveals the history and key loci associated with fruit domestication and diversification. Mol Plant 15(11):1744–1758
Carrizo García C, Barfuss MH, Sehr EM, Barboza GE, Samuel R, Moscone EA, Ehrendorfer F (2016) Phylogenetic relationships, diversification and expansion of chili peppers (Capsicum, Solanaceae). Ann Bot 118(1):35–51
Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (austin) 6(2):80–92
Curry J, Aluru M, Mendoza M, Nevarez J, Melendrez M, O’Connell MA (1999) Transcripts for possible capsaicinoid biosynthetic genes are differentially accumulated in pungent and non-pungent Capsicum spp. Plant Sci 148(1):47–57
de Givry S, Bouchez M, Chabrier P, Milan D, Schiex T (2005) CARHTA GENE: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 21(8):1703–1704
Eshbaugh W (1980) The taxonomy of the genus Capsicum (solanaceae). Phytologia 47:153–166
Fraser CM, Chapple C (2011). The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book, 9
Fujiwake H, Suzuki T, Iwai K (1982) Intracellular distributions of enzymes and intermediates involved in biosynthesis of capsaicin and its analogues in Capsicum fruits. Agric Biol Chem 46(11):2685–2689
Govindarajan V, Sathyanarayana M (1991) Capsicum—production, technology, chemistry, and quality. Part V. Impact on physiology, pharmacology, nutrition, and metabolism; structure, pungency, pain, and desensitization sequences. Crit Rev Food Sci Nutr 29(6):435–474
Han K, Jeong H-J, Sung J, Keum YS, Cho M-C, Kim J-H, Kwon J-K, Kim B-D, Kang B-C (2013) Biosynthesis of capsinoid is controlled by the Pun1 locus in pepper. Mol Breed 31:537–548
Han K, Lee HY, Ro NY, Hur OS, Lee JH, Kwon JK, Kang BC (2018) QTL mapping and GWAS reveal candidate genes controlling capsaicinoid content in Capsicum. Plant Biotechnol J 16(9):1546–1558
Han K, Jang S, Lee J-H, Lee D-G, Kwon J-K, Kang B-C (2019) A MYB transcription factor is a candidate to control pungency in Capsicum annuum. Theor Appl Genet 132:1235–1246
Hwang D, Jeong H-J, Kwon J-K, Kim H, Kang S-Y, Kang B-C (2014) Phenotypic variants among ethyl methanesulfonate M2 mutant lines in Capsicum annuum. Plant Genet Resour 12(S1):S141–S145
Hwang D (2014). Development of an EMS mutant population and isolation of non-pungent mutants in Capsicum annuum [Masters dissertation, Seoul National University]. Seoul.
Jeong H-J, Kwon J-K, Pandeya D, Hwang J, Hoang NH, Bae J-H, Kang B-C (2012a) A survey of natural and ethyl methane sulfonate-induced variations of eIF4E using high-resolution melting analysis in Capsicum. Mol Breed 29:349–360
Jeong H, Hwang D, Ahn J, Chun J, Han K, Lee W, Kwon J, Lee Y, Kang B (2012b) Development of a simple method for detecting capsaicinoids using Gibb’s reagent in pepper. Korean J Hortic Sci Technol 30(3):294–300
Jo YD, Kim SH, Hwang J-E, Kim Y-S, Kang HS, Kim SW, Kwon S-J, Ryu J, Kim J-B, Kang S-Y (2016) Construction of mutation populations by gamma-ray and carbon beam irradiation in chili pepper (Capsicum annuum L.). Hortic Environ Biotechnol 57:606–614
Kang J-H, Yang H-B, Jeong H-S, Choe P, Kwon J-K, Kang B-C (2014) Single nucleotide polymorphism marker discovery from transcriptome sequencing for marker-assisted backcrossing in Capsicum. Hortic Sci Technol 32(4):535–543
Kim M, Kim S, Kim S, Kim B-D (2001) Isolation of cDNA clones differentially accumulated in the placenta of pungent pepper by suppression subtractive hybridization. Mol Cells 11(2):213–219
Kim S, Park M, Yeom S-I, Kim Y-M, Lee JM, Lee H-A, Seo E, Choi J, Cheong K, Kim K-T (2014) Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46(3):270–278
Kim S, Park J, Yeom S-I, Kim Y-M, Seo E, Kim K-T, Kim M-S, Lee JM, Cheong K, Shin H-S (2017) New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol 18:1–11
Klein H, Xiao Y, Conklin PA, Govindarajulu R, Kelly JA, Scanlon MJ, Whipple CJ, Bartlett M (2018) Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3: Genes, Genomes, Genetics 8(11):3583–3592
Koeda S, Sato K, Saito H, Nagano AJ, Yasugi M, Kudoh H, Tanaka Y (2019) Mutation in the putative ketoacyl-ACP reductase CaKR1 induces loss of pungency in Capsicum. Theor Appl Genet 132:65–80
Kondo F, Umeda K, Sudasinghe SP, Yamaguchi M, Aratani S, Kumanomido Y, Nemoto K, Nagano AJ, Matsushima K (2023) Genetic analysis of pungency deficiency in Japanese chili pepper ‘Shishito’(Capsicum annuum) revealed its unique heredity and brought the discovery of two genetic loci involved with the reduction of pungency. Mol Genet Genom 298(1):201–212
Kosugi S, Natsume S, Yoshida K, MacLean D, Cano L, Kamoun S, Terauchi R (2013) Coval: improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS ONE 8(10):e75402
Krivega I, Dean A (2012) Enhancer and promoter interactions: long distance calls. Curr Opin Genet Dev 22(2):79–85
Lang Y, Kisaka H, Sugiyama R, Nomura K, Morita A, Watanabe T, Tanaka Y, Yazawa S, Miwa T (2009) Functional loss of pAMT results in biosynthesis of capsinoids, capsaicinoid analogs, in Capsicum annuum cv. CH-19 Sweet. Plant J 59(6):953–961
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9(1):1–13
Lee J, Park SJ, Hong SC, Han JH, Choi D, Yoon JB (2016) QTL mapping for capsaicin and dihydrocapsaicin content in a population of Capsicum annuum ‘NB 1’× Capsicum chinense ‘Bhut Jolokia.’ Plant Breed 135(3):376–383
Lee J-H, Venkatesh J, Jo J, Jang S, Kim GW, Kim J-M, Han K, Ro N, Lee H-Y, Kwon J-K (2022) High-quality chromosome-scale genomes facilitate effective identification of large structural variations in hot and sweet peppers. Hortic. Res. 9:uhac210
Lee J (2018). Genetic mapping of a nonpungent EMS mutant in pepper (Capsicum annuum L.) [Masters dissertation, Seoul National University]. Seoul
Leete E, Louden MC (1968) Biosynthesis of capsaicin and dihydrocapsaicin in Capsicum frutescens. J Am Chem Soc 90(24):6837–6841
Liu Y, Nair MG (2010) Capsaicinoids in the hottest pepper Bhut Jolokia and its antioxidant and antiinflammatory activities. Nat Prod Commun 5(1):1934578X1000500122
Liu Y, Zhang Z, Fang K, Shan Q, He L, Dai X, Zou X, Liu F (2022) Genome-wide analysis of the MYB-related transcription factor family in pepper and functional studies of CaMYB37 involvement in capsaicin biosynthesis. Int J Mol Sci 23(19):11667
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21
Luo X-J, Peng J, Li Y-J (2011) Recent advances in the study on capsaicinoids and capsinoids. Eur J Pharmacol 650(1):1–7
Mansfeld BN, Grumet R (2018) QTLseqr: An R package for bulk segregant analysis with next-generation sequencing. Plant Genome 11(2):180006
Mazourek M, Pujar A, Borovsky Y, Paran I, Mueller L, Jahn MM (2009) A dynamic interface for capsaicinoid systems biology. Plant Physiol 150(4):1806–1821
Naves ER, de Ávila Silva L, Sulpice R, Araújo WL, Nunes-Nesi A, Peres LE, Zsögön A (2019) Capsaicinoids: pungency beyond Capsicum. Trends Plant Sci 24(2):109–120
Negulesco J, Noel S, Newman H, Naber E, Bhat H, Witiak D (1987) Effects of pure capsaicinoids (capsaicin and dihydrocapsaicin) on plasma lipid and lipoprotein concentrations of turkey poults. Atherosclerosis 64(2–3):85–90
Nelson E, Dawson L (1923) The constitution of capsaicin, the pungent principle of Capsicum. III J Am Chem Soc 45(9):2179–2181
Ou L, Li D, Lv J, Chen W, Zhang Z, Li X, Yang B, Zhou S, Yang S, Li W (2018) Pan-genome of cultivated pepper (Capsicum) and its use in gene presence–absence variation analyses. New Phytol 220(2):360–363
Paran I, Borovsky Y, Nahon S, Cohen O (2007) The use of induced mutations to study shoot architecture in Capsicum. Isr J Plant Sci 55(2):125–131
Park M, Lee J-H, Han K, Jang S, Han J, Lim J-H, Jung J-W, Kang B-C (2019) A major QTL and candidate genes for capsaicinoid biosynthesis in the pericarp of Capsicum chinense revealed using QTL-seq and RNA-seq. Theor Appl Genet 132:515–529
Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111(14):5135–5140
Schneeberger K, Weigel D (2011) Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci 16(5):282–288
Siddique MI, Back S, Lee J-H, Jo J, Jang S, Han K, Venkatesh J, Kwon J-K, Jo YD, Kang B-C (2020) Development and characterization of an ethyl methane sulfonate (EMS) induced mutant population in Capsicum annuum L. Plants 9(3):396
Stam M, Belele C, Dorweiler JE, Chandler VL (2002) Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 16(15):1906–1918
Stewart C Jr, Kang BC, Liu K, Mazourek M, Moore SL, Yoo EY, Kim BD, Paran I, Jahn MM (2005) The Pun1 gene for pungency in pepper encodes a putative acyltransferase. Plant J 42(5):675–688
Sukrasno N, Yeoman M (1993) Phenylpropanoid metabolism during growth and development of Capsicum frutescens fruits. Phytochemistry 32(4):839–844
Sun B, Zhu Z, Chen C, Chen G, Cao B, Chen C, Lei J (2019) Jasmonate-inducible R2R3-MYB transcription factor regulates capsaicinoid biosynthesis and stamen development in Capsicum. J Agric Food Chem 67(39):10891–10903
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6(7):e21800
Tanaka Y, Asano T, Kanemitsu Y, Goto T, Yoshida Y, Yasuba K, Misawa Y, Nakatani S, Kobata K (2019) Positional differences of intronic transposons in pAMT affect the pungency level in chili pepper through altered splicing efficiency. Plant J 100(4):693–705
Tanaka Y, Mitani A, Shimizu N, Goto T, Yoshida Y, Yasuba K-I (2021) Characterization and bulk segregant analysis of a novel seedless mutant tn-1 of chili pepper (Capsicum annuum). Sci Hortic 276:109729
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z (2017) agriGO v2. 0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45(W1):W122–W129
Todd J, Post-Beittenmiller D, Jaworski JG (1999) KCS1 encodes a fatty acid elongase 3-ketoacyl-CoA synthase affecting wax biosynthesis in Arabidopsis thaliana. Plant J 17(2):119–130
Veloso J, Prego C, Varela M, Carballeira R, Bernal A, Merino F, Díaz J (2014) Properties of capsaicinoids for the control of fungi and oomycetes pathogenic to pepper. Plant Biol 16(1):177–185
Walsh BM, Hoot SB (2001) Phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two noncoding regions: the chloroplast atpB-rbcL spacer region and nuclear waxy introns. Int J Plant Sci 162(6):1409–1418
Weber B, Zicola J, Oka R, Stam M (2016) Plant enhancers: a call for discovery. Trends Plant Sci 21(11):974–987
Yarnes SC, Ashrafi H, Reyes-Chin-Wo S, Hill TA, Stoffel KM, Van Deynze A (2013) Identification of QTLs for capsaicinoids, fruit quality, and plant architecture-related traits in an interspecific Capsicum RIL population. Genome 56(1):61–74
Yu S, Zhang W, Zhang L, Wu D, Sun P, Huang C, Fu G, Deng Q, Wang Z, Cheng S (2023) MYB24 negatively regulates the biosynthesis of lignin and capsaicin by affecting the expression of key genes in the phenylpropanoid metabolism pathway in Capsicum chinense. Molecules 28(6):2644
Zhang L, Wu D, Zhang W, Shu H, Sun P, Huang C, Deng Q, Wang Z, Cheng S (2023) Genome-wide identification of WRKY gene family and functional characterization of CcWRKY25 in Capsicum chinense. Int J Mol Sci 24(14):11389
Zhu Z, Sun B, Cai W, Zhou X, Mao Y, Chen C, Wei J, Cao B, Chen C, Chen G (2019) Natural variations in the MYB transcription factor MYB31 determine the evolution of extremely pungent peppers. New Phytol 223(2):922–938
Funding
Open Access funding enabled and organized by Seoul National University. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT). (No. 2021R1A2C2007472).
Author information
Authors and Affiliations
Contributions
BCK and SB contributed to the study conception and design of the experiments; BCK and JKK supervised the experiments; SB, HC, KH, and DH prepared plant materials; SB and HC performed phenotype data collection; SB and HC were involved in genetic mapping and RT-qPCR; SB, JHL, and JMK analyzed high-throughput sequencing data; SB and BCK prepared the manuscript; and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Esther van der Knaap.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Back, S., Kim, JM., Choi, H. et al. Genetic characterization of a locus responsible for low pungency using EMS-induced mutants in Capsicum annuum L.. Theor Appl Genet 137, 101 (2024). https://doi.org/10.1007/s00122-024-04602-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04602-3