
Abstract
Key message
Fon race 2 resistant QTLs were identified on chromosomes 8 and 9. Families homozygous for resistance alleles at a haplotype of three KASP markers had 42% lower disease severity than those with susceptible alleles in an independent, interspecific validation population confirming their utility for introgression of Fusarium wilt resistance.
Abstract
Fusarium oxysporum f. sp. niveum (Fon) race 2 causes Fusarium wilt in watermelon and threatens watermelon production worldwide. Chemical management options are not effective, and no resistant edible watermelon cultivars have been released. Implementation of marker-assisted selection to develop resistant cultivars requires identifying sources of resistance and the underlying quantitative trait loci (QTL), developing molecular markers associated with the QTL, and validating marker-phenotype associations with an independent population. An intraspecific Citrullus amarus recombinant inbred line population from a cross of resistant USVL246-FR2 and susceptible USVL114 was used for mapping Fon race 2 resistance QTL. KASP markers were developed (N = 51) for the major QTL on chromosome 9 and minor QTL on chromosomes 1, 6, and 8. An interspecific F2:3 population was developed from resistance donor USVL246-FR2 (C. amarus) and a susceptible cultivar ‘Sugar Baby’ (Citrullus lanatus) to validate the utility of the markers for introgression of resistance from the wild crop relative into cultivated watermelon. Only 16 KASP markers segregated in the interspecific C. amarus/lanatus validation population. Four markers showed significant differences in the separation of genotypes based on family-mean disease severity, but together explained only 16% of the phenotypic variance. Genotypes that inherited homozygous resistant parental alleles at three KASP markers had 42% lower family-mean disease severity than homozygous susceptible genotypes. Thus, haplotype analysis was more effective at predicting the mean disease severity of families than single markers. The haplotype identified in this study will be valuable for developing Fon race 2 resistant watermelon cultivars.
Similar content being viewed by others

Introduction
Watermelon is an important specialty crop in the USA but domestic production has decreased over the last few decades. Watermelons were produced on 102,400 acres in 2021, with a gradual annual decrease from the maximum domestic production of 220,000 acres in 1992 (NASS 2022). Correspondingly, imports have steadily increased to meet steady domestic demand (USDA 2020). One of the major reasons for decreased acreage and production is yield losses from soil-borne diseases, especially Fusarium wilt. Fusarium wilt is caused by four races of the fungal pathogen Fusarium oxysporum f. sp. niveum (Fon), i.e., race 0, race 1, race 2, and most recently, race 3 (Martyn 2014; Petkar et al. 2019). Fon race 2 (FonR2) is the most common race in South Carolina soils (Keinath et al. 2020) and has spread worldwide over the last three decades (Bruton et al. 2008; Egel et al. 2007; Gonzalez‐torres et al. 1993). Commercial cultivars with resistance to Fon races 0 and 1 are widely available; however, there are no edible cultivars with resistance to races 2 or 3. Integrated disease management strategies incorporating the usage of these resistant cultivars have greatly controlled losses due to race 1 (Martyn 2014). Discontinuation of the highly effective fumigant methyl bromide due to a mandate by the Montreal Protocol and Clean Air Act (1998) eliminated the most effective chemical management for Fusarium wilt. Other chemicals such as prothioconazole and thiophanate-methyl were shown to reduce the severity of watermelon Fusarium wilt caused by FonR2; however, they do not prevent economic losses (Everts et al. 2014). Management practices such as cover cropping with Vicia villosa, Trifolium incarnatum, Secale cereale and Brassica juncea reduced Fusarium wilt on triploid watermelon by 2 to 21% (Himmelstein et al. 2014). Grafting susceptible watermelon scions onto resistant rootstocks reduces disease severity and is currently the most effective management strategy against FonR2 (Keinath and Hassell 2014), but increases the cost of cultivation. Developing resistant edible Citrullus lanatus cultivars coupled with an effective integrated disease management plan will increase profits for growers through decreased yield losses and lower cost of chemical controls.
Apart from two Indian C. lanatus accessions with higher seedling survivability, no resistance has been reported in cultivated watermelons (Pal et al. 2023). Screening of the USDA Citrullus amarus (C. amarus) plant introduction (PI) collection identified a few accessions with high levels of resistance to FonR2 (Wechter et al. 2012). C. amarus and C. lanatus are easily crossable allowing exchange of genes between two species (Ren et al. 2015). Inbred lines, USVL246-FR2 and USVL252-FR2, were developed from the two most resistant C. amarus PIs (Wechter et al. 2016). QTL mapping with bi-parental early generation (F2:3) C. amarus population identified one major (on chromosome 9) and four minor QTLs associated with FonR2 resistance (Branham et al. 2017). The need to phenotype numerous individuals for each F3 family to estimate the true genetic effect of the respective F2 for a quantitative trait makes performing such large bioassays resource prohibitive. This limitation can lead to under- or over-estimating discovered QTL effects compared with studies using recombinant inbred line (RIL) populations (Bernardo 2020). Theoretically, 50% of the markers in F2 populations are heterozygous. Double crossover and single crossover events between neighboring markers inherited from different homozygous parents (resistant and susceptible) within segregating individuals cannot be distinguished from non-crossover events leading to sparse genetic maps or inaccurate genetic distances. Conversely, all the genotyped parental polymorphisms can be utilized to estimate genetic distances in a RIL population (Paran et al. 1995). In Branham et al. (2019a, b), QTL mapping with a RIL population provided improved resolution and more reliable QTL effects for Fon race 1 resistance compared to the F2:3 population from which the RIL was derived. Further, lower resolution and often over-estimation of QTL effects with F2:3 populations decrease the effectiveness of marker development for marker-assisted breeding (MAS) (Austin and Lee 1996). We have generated an intraspecific C. amarus RIL population through single-seed descent of the F2:3 population previously used for mapping FonR2 resistance (Branham et al. 2017). The RIL population was used in this study to identify/verify QTL to be targeted for MAS and to develop kompetitive allele-specific primers (KASP) tightly linked to FonR2 resistance.
Given the availability of molecular markers strongly correlated with resistance, MAS can be implemented effectively in quantitative disease resistance breeding (Yeo et al. 2022; Ganaparthi et al. 2023a). However, use of MAS for trait improvement has yielded ambiguous results in different breeding programs (Damien et al. 2019). Accurate estimation of correlation between developed molecular markers and phenotype in elite genetic backgrounds for planned introgressions plays a critical role in the success of MAS or marker-assisted backcross selection (MABS) projects (Cobb et al. 2019). Thus, before using molecular markers in MAS or MABS for quantitative trait improvement, validation of the developed molecular markers in elite genetic backgrounds is warranted. The objectives of this study were to (1) validate and improve the resolution of FonR2 resistance QTL from USVL246-FR2 in an intraspecific RIL population, (2) develop KASP markers tightly linked to the targeted QTL, (3) validate the developed KASP markers with an interspecific population between USVL246-FR2 (C. amarus) and the watermelon cultivar ‘Sugar Baby’ (C. lanatus), and (4) identify markers and/or haplotype(s) that can be used for MAS or MABS.
Materials and methods
Plant materials and growth conditions
An F8:9 RIL population (N = 200 lines) segregating for FonR2 resistance, developed from resistant parent USVL246-FR2 and susceptible parent USVL114 (Branham et al. 2019a, b), was used for QTL mapping. An interspecific F2:3 population was developed from a cross of USVL246-FR2 with the FonR2 susceptible cultivar ‘Sugar Baby’ (C. lanatus) for KASP marker validation. Eight F1 plants from the cross were self-pollinated by hand, and 161 randomly selected F2 seeds were germinated in 50-cell propagation trays (Hummert International, Earth City, MO). At the four-leaf stage, seedlings were transplanted into 6-L pots. Plants were maintained in the greenhouse at 25–35 °C. The second true leaf from each F2 plant was collected for DNA extraction using the CTAB method (Shu et al. 2018). Each F2 plant was selfed by hand-pollination to produce seeds for their respective F3 families. F3 families were phenotyped for disease response to artificial inoculation with FonR2 and family means were used as phenotypes for their respective F2 individuals.
Disease inoculations and evaluations
FonR2 cultures were prepared using the optimized protocol described by Wechter et al. (2012). FonR2 isolate B05-30cvd, obtained through single spore isolation, was grown on one-fourth Difco potato dextrose agar (Becton, Dickson & Co. Sparks, MD) for a week at 25 °C under 12-h diurnal light/dark cycle with fluorescent lighting. Five 1-cm disks were cut from the growing margin, added to 250 mL of potato dextrose broth, and placed on a rotary shaker at 200 rpm for two weeks at 25 °C temperature with 16/8-h light/dark cycle with fluorescent lighting. On the 15th day, the spore suspension was filtered through two layers of cheesecloth and a layer of Mira cloth (EMD chemicals, San Diego, CA) to remove hyphae. Spores were quantified by microscopy using a hemacytometer, and the final concentration was adjusted to 106 cfu/mL with sterile distilled water. Five liters of the diluted spore suspension was added to 22 L of tri-mix (perlite (THERM-O-ROCK EAST, INC.): vermiculite (Palmetto vermiculite, SC): metromix 360 potting soil (Sun Gro Horticulture Inc) in 1:1:1 ratio). Spores were homogeneously distributed in the tri-mix using an electric concrete mixer. Ten seeds each of the 200 RILs, along with their parents, F1 and race differentials (Burton et al. 2008) were evaluated in two tests with two replications (five seeds per each rep) per test. Seeds were seeded into inoculated tri-mix in 50-cell propagation trays and were grown in a growth chamber with LED lights (4 red: 1 blue) at 25 °C. All the plants grown in growth chambers were subjected to a 16:8 light-to-dark photoperiod. Similarly, 10 seeds per each of the 161 F2:3 families were evaluated in two tests into inoculated soil and were grown in a growth chamber with LED lights (4 red:1blue) maintained at 25 °C. Plants in each test were rated on a 1–5 scale on the 28th day after planting in inoculated soil as described by Ganaparthi et al. (2023b). Completely healthy plants were rated as 1 and plants with turgid stems but with chlorotic or necrotic spots either on cotyledons or true leaves were given a rating of 2. Plants with chlorotic or necrotic spots on both cotyledons and true leaves with turgid stem were rated as 3. Completely wilted plants were rated as 4 and completely necrotic plants were rated as 5.
Statistical analysis
Best linear unbiased estimates (BLUEs) for each test separately and the two tests combined were obtained for each RIL using the lmer function in the R package lme4 (R core team 2022) and were utilized in the mapping experiment.
The model utilized for obtaining BLUEs was:
where Y represents the BLUEs of each accession, \({g}_{i}\) is the fixed effect of the ith genotype and \({r}_{j}\) is the random effect of the jth rep, \({t}_{k}\) is the random effect of the kth test, \({g}_{i}: {t}_{k}\) is the interaction between ith genotype and kth test, and \({e}_{ij}\) is the random error variance. FonR2 resistance broad-sense heritability among the mapping population was estimated using the formula (Piepho and Möhring 2007):
where \({\sigma }_{g}^{2}\) is the variance due to genotype, \({\sigma }_{p}^{2}\) is the phenotypic variance, \({\sigma }_{gt}^{2}\) is the variance of the genotype-by-test interaction, \(m\) is the number of tests and \(r\) is the total number of replicates. Homogeneity of test and replication variances were tested with Bartlett’s test. Disease severity means for each F2:3 family in the validation population were utilized in identifying significant molecular markers or haplotypes correlated with FonR2 resistance.
Genetic map construction and QTL mapping
The SNPs (N = 2,143) and genetic map of the RIL population were obtained from Branham et al. (2019a, b) and were utilized for the initial mapping of FonR2 QTL(s) with the R package qtl (Broman et al. 2003). KASP markers were developed from the identified QTL (described below) and used to genotype the RIL population. KASP SNPs with identical segregation patterns were binned and then combined with the GBS SNPs to make a new genetic map (in Rqtl) for QTL mapping to improve resolution.
Linkage groups were formed with the formlinkagegroup function using a maximum recombination frequency of 0.35 and with a minimum LOD score of 7. Markers that did not fit into any of the 11 linkage groups were removed from the genetic map. Using the Kosambi mapping function, genetic distance between the marker pairs were calculated based on the recombination fraction between them (Kosambi 1943). Multiple QTL mapping (MQM) with Haley–Knott regression (Haley and Knott 1992) was used to identify potential genomic regions imparting FonR2 resistance in the RIL population. The optimal model with the highest penalized LOD score was determined with the stepwise qtl function (Broman and Sen 2009). The LOD value significance threshold was determined with 1,000 permutations using scantwo function with penalties at \(\alpha =0.05\). LOD profile figures were generated with the scanone function, and the addqtl function was used to add single additional QTL to visualize forward model selection. The Lodint function with argument ‘expandtomarkers = T’ identified the markers flanking each significant QTL for the 1.5 LOD interval. USVL246-FR2 genome annotations (Wu et al. 2023) were obtained from the CuGenDBv2 database (Yu et al. 2022) and used to identify the candidate genes within the 1.5 LOD interval of significant QTL.
KASP marker development and DNA amplification conditions
KASP markers were developed for all the significant FonR2 resistance QTL (Supplementary Table 1). The physical position of SNPs within the 1.5 LOD intervals of significant QTL were obtained from USVL246-FR2 v1 reference gene annotations (Wu et al. 2023). To design KASP suitable for interspecific introgression into C. lanatus, flanking genomic sequence (60 bp up- and downstream) were obtained and queried using BLAST against the reference genome of 97,103 v 2.5 (Guo et al. 2013), a C. lanatus elite line susceptible to FonR2. BLAST searches where all flanking nucleotides were monomorphic and the targeted SNP was polymorphic between resistant and susceptible individuals were selected so that KASP could be \used for interspecific introgression. Alternate alleles of the selected SNPs, along with respective flanking sequences, were sent to either LGC Genomics (Hoddesdon herts, UK) or Integrated DNA Technologies (Beverly, MA, USA) for designing primers. A total of 51 KASP markers across the major and minor resistance QTL intervals were developed from SNPs identified between the parents and sequence similarity to the C. lanatus reference genome. PCR reactions for amplification included 2.5 μL of Master mix (LGC Genomics, Alexandria, MN), 0.07 μL of primer mix (two fluorophore labeled allele-specific forward and a reverse primer), and 15–20 ng of sample DNA brought up to a 5 μL reaction with DI water. A touchdown PCR protocol was programmed on a standard thermal cycler beginning with a 15 min activation step at 94 °C followed by 11 alternate cycles of denaturation and annealing, 20 s cycles at 94 °C for denaturation, and initial annealing at 61 °C reduced by 0.6 °C each cycle for 60 s. These touchdown cycles were followed by 26 additional alternate cycles of denaturation and anneling at 94 °C for 20 s and 55 °C for 60 s, respectively. A Stratagene Mx300P (Agilent technologies, Santa Clara, CA) quantitative PCR system was used to quantify fluorescence for allele discrimination. Samples were clustered with MxPro v4.10 software (Agilent technologies, Santa Clara, CA) based on normalized HEX and FAM readings. Parents and the RIL population were genotyped with all the KASP markers, and only markers that showed polymorphism between the parents were utilized in genotyping the USVL246-FR2 x ‘Sugar Baby’ population. KASP markers significantly associated with resistance were identified with analysis of variance (ANOVA) of each marker on family-mean disease severity. R2 explained by all significant markers on regression with family mean was reported as the phenotypic variance explained. All possible haplotypes with significantly associated markers were identified. The significance of all families homozygous for the resistant or susceptible parent haplotype group was assessed with a Welch two-sample t test.
Results
Genetic map
A total of 2,185 SNPs were obtained for the intraspecific RIL population after combining variants from GBS and KASP. After dropping markers with identical segregation patterns, 2,177 SNPs were used for genetic map construction. Three markers did not fit into any linkage groups and were removed from the analysis as they are likely to be genotyping errors. Thus, 2,174 markers were left for further analysis. The total genetic map length was 1,192.9 cM with an average spacing of 0.6 cM between markers (Supplementary Table 1). Linkage groups were named according to the chromosomes of the USVL246-FR2 C. amarus reference genome (Wu et al. 2023). The maximum spacing between markers was 21.3 cM on chromosome 8. Chromosome 10 spanned 137 cM and was the largest chromosome, while chromosome 4 was the shortest with a length of 84 cM.
FonR2 resistance in the intraspecific RIL population and interspecific F2:3 population
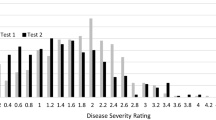
Disease severity variances for test 1 and test 2 of the intraspecific population were non-homogenous (P-value is 4.38e-09). Pearson correlation between BLUEs of test 1 and test 2 was 0.41. Replications within test 1 were non-homogenous (P-value is 0.0028), while replications within test 2 were homogenous. The coefficient of variance (CV) for test 1, test 2 and across the two tests was 0.17, 0.16 and 0.13, respectively. The mean disease severity of the RIL population across the two tests was normally distributed (Fig. 1). The mean disease severity of resistant and susceptible parents of the mapping population was 2.0 and 4.4, respectively. The mean disease severity of the RILs ranged from 2.1 to 4.5 across two tests, and the mean disease severity of the F1 was 3.5. Overall mean disease severity of the RIL population was 3.6. Broad sense heritability of disease severity in the RIL population was 0.57. Analysis of variance determined genotype and test as highly significant (Table 1).

Histograms showing the disease severity distributions for: a USVL246-FR2 X USVL-114 RIL population across two tests, b USVL246-FR2 X Sugar Baby F3 families inoculated with FonR2. Parental and F1 means are marked by vertical dashed lines. Numbers on each bar indicates number of RILs or F3 families in specific range of disease severity
The mean disease severity of the interspecific F2:3 population was 3.2, which was lower than the RIL population (3.6). Pearson correlation between means of test 1 and test 2 was 0.54. The lowest family mean of disease severity in the F2:3 population was 1.3, and the highest was 4.9 indicating transgressive segregation towards the resistant parent (Rieseberg et al. 1999) (Table 1). Broad sense heritability of disease severity in the F2:3 interspecific population was 0.32.
QTL mapping
The optimal multiple QTL mapping (MQM) model with the highest penalized LOD score was determined for each test and across tests. MQM of BLUEs across two tests identified three QTLs as significantly associated with FonR2 resistance in the intraspecific RIL population. Of the three QTLs, two were identified on chromosome 9 and one on chromosome 8 (Fig. 2a). The QTL with the highest LOD score was identified at 62.5 cM on chromosome 9, explaining 20.7% of the phenotypic variance with an additive effect of 0.22 (Table 2). Two minor QTLs, on chromosomes 9 and 8, explained 9.4 and 7.5% of the phenotypic variance, respectively. No epistatic interaction between QTL was identified. MQM of test 1 BLUEs identified 5 QTL associated with FonR2. The major QTL on chromosome 9 had a peak LOD score (8.85) at 65.6 cM and explained 12.0% of the phenotypic variance. Four minor QTLs were identified on chromosomes 9, 8, 6 and 1. Together, the minor QTL explained 21.6% of the total phenotypic variance. However, MQM with BLUEs obtained from test 2 identified only two significant QTL, both on chromosome 9 and collocated with the two QTL identified on chromosome 9 from test 1. KASP markers were developed for the major QTL on chromosome 9 and the minor QTL on chromosomes 8, 6 and 1 (Table 2) to pyramid resistance QTL for FonR2 resistance after their validation (Fig. 3).

Logarithm of odds (LOD) scores for QTL associated with best linear unbiased estimates (BLUEs) obtained from disease severity a Across two tests b Test 1 c Test 2 after inoculation with FonR2. The horizontal dashed line indicates the genome-wide significance threshold

Effect plots showing the mean and standard errors for best linear unbiased estimate of Fusarium oxysporum f. sp. niveum race 2 disease severity for genotypic class: AA homozygous resistant parent alleles, BB homozygous susceptible parent alleles. Panels depict the genotype of the SNP with highest LOD score for each quantitative trait loci identified across two tests: a SNP at QTL qFon2-9.2; b SNP at QTL qFon2-9.1; c SNP at QTL qFon2-8; d SNP at qFon2-1
KASP marker validation
Four KASP markers on chromosome 9, eight on chromosome 1, six on chromosome 6 and eight on chromosome 8 were polymorphic between the parents, these markers were used for genotyping the interspecific F2 population. Nevertheless, only three KASP markers for each QTL on chromosome 9 and chromosome 1 and five markers each for QTL on chromosome 6 and chromosome 8 showed useful polymorphism among interspecific population. ANOVA of each KASP marker on family-mean disease severity identified four markers (primer_q1_1, primer_q8_7, primer_q9_2 and primer_q9_16) as significantly associated with FonR2 disease severity. However, all significant markers combined explained only 16% of the phenotypic variance. To identify more meaningful association between markers and family-mean disease severity, all possible haplotype combinations with significant markers and their associated phenotypes were tested (Supplementary Table 1). While some haplotypes exhibited significant differences with Welch two-sample t test, they were not useful in discriminating resistant/susceptible families. One haplotype combination consisting of three KASP markers (q8-7, q9-2 and q9-16) was highly significant (P-value = 5.21e-05) and exhibited strong association (R2 = 0.42) between alleles and family-mean phenotype. Seven families that inherited resistant parental alleles at these three loci, had a mean disease severity of 2.51 and six families with susceptible parental alleles had a mean disease severity of 4.35 (Fig. 4). The mean disease severity of 13 heterozygous families for the haplotype was 3.23. Only one family inherited resistant parental alleles at all four significant loci and had a mean disease severity of 2.64.

Violin plot showing mean disease severity distribution of F2:3 families that inherited resistant (AA), resistant and susceptible (AB) and susceptible (BB) parent alleles at primer_q8_7, primer_q9_2 and primer_q9_16 with their respective group median and confidence intervals. X-axis represents parent alleles inherited at haplotype loci
Discussion
Sources of resistance to Fusarium wilt caused by FonR2 are limited (Wechter et al. 2012). The genetic architecture of resistance from a few of these sources was determined through QTL mapping with bi-parental mapping populations (Branham et al. 2017, 2020; Meru and McGregor 2016; Ren et al. 2015). QTL mapping studies using USVL252-FR2 as the resistance source identified QTL on chromosomes 1, 5, and 8 (Branham et al. 2020). Two QTLs, on chromosomes 9 and 10, were associated with FonR2 resistance using PI296341-FR as the resistance source (Ren et al. 2015). In a study by Meru and McGregor (2016) using WGA147 as the resistance donor, a single resistance QTL was identified on chromosome 10. Quantitative trait nucleotides (QTNs) were also identified with a genome-wide association study of Fonr2 resistance in the USDA C. amarus collection (Ganaparthi et al. 2023b). Ganaparthi et al. (2023b) identified five QTNs on chromosomes 1, 5, 9 and 10 associated with FonR2 resistance.
Here, we identified a total of five QTLs associated with FonR2 resistance, including two on chromosome 9 (qFon2-9.1 and qFon2-9.2), and one each on chromosomes 1 (qFon2-1), 6 (qFon2-6), and 8 (qFon2-9.1). QTLs qFon2-1 and qFon2-9.1 collocate with those identified from other sources, while QTLs qFon2-6, qFon2-8 and qFon2-9.2 are novel. QTLs qFon2-1 and qFon2-6 were only significantly associated with resistance in test 1, therefore they were excluded from further consideration. None of the QTLs identified by (Branham et al. 2020) collocate with QTLs identified in the current study. The QTL identified on chromosome 9 by Ren et al. (2015) is physically close to the qFon2-9.1 in the current study. The QTN identified on chromosome 9 through GWAS is 8.4 Mb upstream of qFon2-9.2 (Ganaparthi et al. 2023b). Thus, two QTL (i.e., qFon2-9.2 and qFon2-8) for FonR2 resistance identified in this study are likely to be unique to USVL246-FR2. Along with FonR2 resistance, USVL246-FR2 is also resistant to Fusarium wilt caused by race 1. The same RIL population was also used to map Fon race 1 resistance QTL (Branham et al. 2019a, b). The major resistance QTL qFon2-9.2 overlaps for races 1 and 2 in this population (15.6 to 26.1 Mb and 15.85 to 25.9 Mb, respectively). Therefore, QTL qFon2-9.2 either provides broad-spectrum Fusarium wilt resistance or the resistance QTLs for Fon races 1 and 2 are tightly linked.
The RIL population (USVL246-FR2 by USVL114) utilized in this study was derived from a previously published F2:3 population (Branham et al. 2017) through single-seed descent. FonR2 resistance was associated with a single major QTL on chromosome 9 and four minor QTLs in both populations, however, differences between the results were found. In comparison with the mapping performed on the F2:3 population (Branham et al. 2017), the estimated effect of the major QTL was smaller, the QTL interval identified on chromosome 8 was narrowed by 8.8 Mb, and fewer QTLs were identified with the RIL population. The major QTL discovered with the F2:3 population, qFon2-9, explained 43% of the phenotypic variance. Two QTL (qFon2-9.1 and qFon2-9.2) were identified on either side of qFon2-9 in the RIL population and explained 30% of the phenotypic variance. QTL qFon2-9 could be a ghost QTL: a spurious QTL that can arise when two QTLs are on the same chromosome (Martinez and Curnow 1992; Ronin et al. 1999; Stange et al. 2013). Lower recombination among the F2:3 population may have rendered the two QTLs closer in the genetic map causing them to appear as a single QTL with a large effect between the two QTL intervals.
Two hypotheses could explain the detection of more QTLs with the F2:3 than the RIL population. First, F2:3 populations include QTL with over-dominance effects, but those same QTL will not be identified using a RIL population. Another possibility is that lower recombination in the F2:3 population resulted in clustering of genomic regions with a small effect, causing r/qtl to detect a QTL at these clusters. Such clustered genomic regions can break apart due to higher recombination; thus, the minor effect QTL may not be detected with the RIL population (Austin and Lee 1996). In crops where hybrids are commercially grown and traits targeted for improvement exhibit both additive effects and over-dominance, mapping of QTL employing both F2:3 and RIL populations could be beneficial in devising breeding strategies for trait improvement. Finally, because of true replication due to homogeneity within each RIL, phenotype estimates in a RIL population are better than the estimates obtained for the segregating families in early generation populations. This improved precision in phenotypic estimation of genotypes along with a higher recombination rate allows for narrowed QTL intervals and lower phenotypic variances explained by significant QTL with RIL populations compared to F2:3 populations (Austin and Lee 1996). These two hypotheses also explain the higher heritability among RILs (0.57) compared with the F2:3 (0.32) population.
Marker validation studies with a C. lanatus population are required to demonstrate the effectiveness of resistance introgression into a cultivated watermelon background using MAS or MABS. Also, validation of markers developed for respective QTL in an independent population aid in construction of an effective model for genomic selection (Jannink et al. 2010; Poland and Rutkoski 2016; Rutkoski et al. 2014; Zhang et al. 2021). Although the FonR2 KASP markers were checked for introgression utility by genomic comparison of the wild (C. amarus) and cultivated (C. lanatus) genomes, only a few (N = 16) of the markers developed were polymorphic in the segregating interspecific population. Four of the polymorphic markers were significantly associated with FonR2 resistance in the interspecific validation population, including two markers within qFon2-9.2, one within qFon2-8 and one near qFon2-1. The realized effects of these QTL (16%VP) are smaller than the estimated effect with the RIL population. Haplotype analysis identified a combination of three significant markers, from QTLs qFon2-8, qFon2-9.1 and qFon2-9.2, with improved association with disease severity as compared to single markers. Families with resistant parental alleles at these three loci exhibited 42% less disease severity than families with susceptible parental alleles, suggesting that the two QTLs are complementary. Few families inherited resistant parental alleles at these three loci and only a single genetic background (‘Sugar Baby’) was used to confirm the haplotype effect on lowering disease severity. Future work will focus on validating the utility of the haplotype in different genetic backgrounds (cultivars).
Functionally relevant candidate genes were found within each QTL. Ethylene receptors and transcription factors are essential in ethylene-mediated plant response to biotic stress (Müller and Munné-Bosch 2015). Over-expression of ethylene or endogenous application of ethephon induced resistance to Fusarium oxysporum f. sp. cucumerinum (Foc) in cucumber (Dong et al. 2020). Five ethylene transcription factors were identified within qFon2-8. Receptor-like kinases (RLKs) are well-known resistance genes (Goff and Ramonell 2007). RNA-seq data suggested that overexpression of lectin receptor-like kinases conferred resistance to Fusarium root rot in tomato (Yue et al. 2022). Transcriptome data showed evidence for RLK-induced Fusarium wilt resistance in cotton and Arabidopsis by recognition of Fusarium through its cell wall extract (Babilonia et al. 2021). Genes predicted to encode RLKs were identified in the QTL intervals of qFon2-9.1 (N = 1 gene) and qFon2-9.2 (N = 7 genes).
In summary, we used an intraspecific C. amarus RIL population to map four QTL associated with resistance to FonR2. KASP markers were developed across the QTL and validated in an interspecific F2:3 population of the resistant C. amarus line, USVL246-FR2, by the C. lanatus cultivar ‘Sugar Baby.’ Although four KASP markers across three chromosomes (1, 8 and 9) were significantly associated with resistance, a combination of three of these markers (on chromosomes 8 and 9) explained more of the phenotypic variance. Future work will focus on using this haplotype to introgress FonR2 resistance into multiple elite backgrounds for development of improved watermelon cultivars.
Data availability
The datasets generated during and/or analysed during the current study are included in the supplementary tables.
References
Austin DF, Lee M (1996) Comparative mapping in W2:3 and W6:7 generations of quantitative trait loci for grain yield and yield components in maize. Springer-Verlag
Babilonia K, Wang P, Liu Z, et al (2021) A nonproteinaceous Fusarium cell wall extract triggers receptor-like protein-dependent immune responses in Arabidopsis and cotton. New Phytologist 230:275–289. https://doi.org/10.1111/NPH.17146
Bernardo R (2020) Breeding for quantitative traits in plants (3rd ed.). Stemma press
Branham SE, Levi A, Farnham MW, Patrick Wechter W (2017) A GBS-SNP-based linkage map and quantitative trait loci (QTL) associated with resistance to Fusarium oxysporum f. sp. niveum race 2 identified in Citrullus lanatus var. citroides. Theor Appl Genet 130:319–330. https://doi.org/10.1007/s00122-016-2813-0
Branham SE, Levi A, Katawczik ML, Wechter WP (2019a) QTL mapping of resistance to bacterial fruit blotch in Citrullus amarus. Theor Appl Genet 132:1463–1471. https://doi.org/10.1007/s00122-019-03292-6
Branham SE, Levi A, Wechter WP (2019b) QTL mapping identifies novel source of resistance to fusarium wilt race 1 in Citrullus amarus. Plant Dis 103:984–989. https://doi.org/10.1094/PDIS-09-18-1677-RE
Branham SE, Patrick Wechter W, Ling KS et al (2020) QTL mapping of resistance to Fusarium oxysporum f. sp. niveum race 2 and Papaya ringspot virus in Citrullus amarus. Theor Appl Genet 133:677–687. https://doi.org/10.1007/s00122-019-03500-3
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890. https://doi.org/10.1093/bioinformatics/btg112
Broman KW, & Sen B (2009) Statistics for Biology and Health A Guide to QTL Mapping with R/qtl. http://www.springer.com/series/2848
Bruton BD, Fish WW, Langston DB (2008) First report of fusarium wilt caused by fusarium oxysporum f. sp. niveum race 2 in Georgia watermelons. Plant Dis 92(6):983
Cobb JN, Biswas PS, Platten JD (2019) Back to the future: revisiting MAS as a tool for modern plant breeding. Theor Appl Genet 132:647–667
Dong J, Wang Y, Xian Q et al (2020) Transcriptome analysis reveals ethylene-mediated defense responses to Fusarium oxysporum f. Sp. Cucumerinum infection in Cucumis sativus L. BMC Plant Biol. https://doi.org/10.1186/s12870-020-02537-7
Egel DS, Harikrishnan R, Martyn R (2005) First report of Fusarium oxysporum f. sp. niveum race 2 as causal agent of Fusarium wilt of watermelon in Indiana. Plant Dis 89(1):108
Everts KL, Egel DS, Langston D, Zhou XG (2014) Chemical management of Fusarium wilt of watermelon. Crop Prot 66:114–119. https://doi.org/10.1016/J.CROPRO.2014.09.003
Ganaparthi VR, Adhikari S, Marais F et al (2023a) The use of PI 277012-derived Fusarium head blight resistance QTL in winter wheat breeding. Heliyon. https://doi.org/10.1016/j.heliyon.2023.e15103
Ganaparthi VR, Rennberger G, Wechter WP, Levi A, Branham SE (2023b) Genome-wide association mapping and genomic prediction of Fusarium wilt race 2 resistance in the USDA Citrullus amarus. Plant Dis. https://doi.org/10.1094/PDIS-02-23-0400-RE
Goff KE, Ramonell KM (2007) The role and regulation of receptor-like kinases in plant defense. Gene Regul Syst Bio 1:117762500700100. https://doi.org/10.1177/117762500700100015
Gonzalez-torres R, Meléro-vara JM, Gómez-vázquez J, Díaz RMJ (1993) The effects of soil solarization and soil fumigation on Fusarium wilt of watermelon grown in plastic house in south-eastern Spain. Plant Pathol 42:858–864. https://doi.org/10.1111/J.1365-3059.1993.TB02671.X
Guo S, Zhang J, Sun H, Salse J, Lucas, WJ, Zhang H, Zheng Y, Mao L, Ren Y, Wang Z, Min J, Guo X, Murat F, Ham BK, Zhang Z, Gao S, Huang M, Xu Y, Zhong S, Xu Y (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45:51–58.https://doi.org/10.1038/ng.2470
Haley CS, Knott SA (1992) A simple regression mhod for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69:315–324
Himmelstein JC, Maul JE, Everts KL (2014) Impact of five cover crop green manures and actinovate on fusarium wilt of watermelon. Plant Dis 98:965–972. https://doi.org/10.1094/PDIS-06-13-0585-RE
Jannink JL, Lorenz AJ, Iwata H (2010) Genomic selection in plant breeding: from theory to practice. Brief Funct Genomic Proteomic 9:166–177. https://doi.org/10.1093/bfgp/elq001
Keinath AP, Hassell RL (2014) Suppression of fusarium wilt caused by fusarium oxysporum f sp niveum race 2 on grafted triploid watermelon. Plant Dis 98(10):1326–1332
Keinath AP, DuBose VB, Katawczik MM, Patrick Wechter W (2020) Identifying races of fusarium oxysporum f. sp. niveum in South Carolina recovered from watermelon seedlings, plants, and field soil. Plant Dis 104:2481–2488. https://doi.org/10.1094/PDIS-11-19-2385-RE
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Martinez O, Curnow RN (1992) Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Springer-Verlag
Martyn RD (2014) Fusarium wilt of watermelon: 120 years of research. Horticultural reviews, vol 42. John Wiley & Sons Inc, Hoboken, New Jersey, pp 349–442
Meru G, McGregor CE (2016) A genetic locus associated with resistance to fusarium oxysporum f. sp. niveum race 2 in Citrullus lanatus-type watermelon. J Am Soc Horticult Sci 141:617–622. https://doi.org/10.21273/JASHS03890-16
Montreal protocol and clean sir act (1998)https://www.epa.gov/ods-phaseout/accelerated-phaseout-class-i-ozonedepletingsubstances#:~:text=In%20revisions%20made%20to%20the,bromide%20and%20its%20phaseout%20schedule. (Accessed on 25 mar 2024)
Müller M, Munné-Bosch S (2015) Focus on ethylene: ethylene response factors: a key regulatory hub in hormone and stress signaling. Plant Physiol 169:32. https://doi.org/10.1104/PP.15.00677
NASS (2022)https://www.nass.usda.gov/Publications/AgCensus/2022/index.php (Accessed on 25 mar 2024)
Pal S, Rao ES, Reddy DL, Dayanandhi E (2023) QTL-seq identifies and QTL mapping refines genomic regions conferring resistance to Fusarium oxysporum f. sp. niveum race 2 in cultivated watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai]. Sci Hortic. https://doi.org/10.1016/j.scienta.2023.112180
Paran I, Goldman I, Tanksley SD, Zamir D (1995) Recombinant inbred lines for genetic mapping in tomato. Springer-Verlag
Petkar A, Harris-Shultz K, Wang H et al (2019) Genetic and phenotypic diversity of Fusarium oxysporum f. Sp. Niveum populations from watermelon in the southeastern United States. PLoS ONE. https://doi.org/10.1371/journal.pone.0219821
Piepho HP, Möhring J (2007) Computing heritability and selection response from unbalanced plant breeding trials. Genetics 177:1881–1888. https://doi.org/10.1534/genetics.107.074229
Platten JD, Cobb JN, Zantua RE (2019) Criteria for evaluating molecular markers: Comprehensive quality metrics to improve marker-assisted selection. PLoS ONE 14(1):e0210529. https://doi.org/10.1371/journal.pone.0210529
Poland J, Rutkoski J (2016) Advances and challenges in genomic selection for disease resistance. Annu Rev Phytopathol 54:79–98. https://doi.org/10.1146/annurev-phyto-080615-100056
R Core Team (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Ren Y, Jiao Di, Gong G et al (2015) Genetic analysis and chromosome mapping of resistance to Fusarium oxysporum f. sp. niveum (FON) race 1 and race 2 in watermelon (Citrullus lanatus L.). Mol Breed. https://doi.org/10.1007/s11032-015-0375-5
Rieseberg LH, Archer MA, Wayne RK (1999) Transgressive segregation, adaptation and speciation. Heredity 83:363–372
Ronin YI, Korol AB, Nevo E (1999) Single-and Multiple-Trait Mapping Analysis of Linked Quantitative Trait Loci: Some Asymptotic Analytical Approximations
Rutkoski JE, Poland JA, Singh RP et al (2014) Genomic selection for quantitative adult plant stem rust resistance in wheat. Plant Genome. https://doi.org/10.3835/plantgenome2014.02.0006
Shu Y, Wan-Ting J, Ya-Ning Y, Yu-Han F (2018) An Optimized CTAB Method for Genomic DNA Extraction from Freshly-picked Pinnae of Fern Adiantum capillus-veneris L. Bio Protoc. https://doi.org/10.21769/BIOPROTOC.2906
Stange M, Friedrich Utz H, Schrag TA et al (2013) High-density genotyping: an overkill for QTL mapping? lessons learned from a case study in maize and simulations. Theor Appl Genet 126:2563–2574. https://doi.org/10.1007/s00122-013-2155-0
USDA (2020) https://www.ers.usda.gov/data-products/chart-gallery/gallery/chart-detail/?chartId=98750. (Accessed on 25 mar 2024)
Wechter WP, Kousik C, Mcmillan M, Levi A (2012). Identification of Resistance to Fusarium oxysporum f. sp. niveum Race 2 in Citrullus lanatus var. citroides Plant Introductions. In HORTSCIENCE (Vol. 47, Issue 3). http://www.ams.med.uni-goettingen.de/
Wechter WP, Mcmillan M, Farnham MW, Levi A (2016). Watermelon germplasm lines USVL246-FR2 and USVL252-FR2 tolerant to Fusarium oxysporum f. sp.niveum race 2. In HORTSCIENCE (Vol. 51, Issue 8). http://www.ams.med.uni-goettingen.de/
Wu S, Sun H, Gao L et al (2023) A Citrullus genus super-pangenome reveals extensive variations in wild and cultivated watermelons and sheds light on watermelon evolution and domestication. Plant Biotechnol J 21:1926–1928. https://doi.org/10.1111/PBI.14120
Yeo SM, Hong J, Hossain MR et al (2022) Genotyping by Sequencing (GBS)-Based QTL Mapping for Bacterial Fruit Blotch (BFB) in Watermelon. Genes (basel). https://doi.org/10.3390/genes13122250
Yu J, Wu S, Sun H et al (2022) CuGenDBv2: an updated database for cucurbit genomics. Nucleic Acids Res. https://doi.org/10.1093/nar/gkac921
Yue ZL, Tian ZJ, Zhang JW et al (2022) Overexpression of lectin receptor-like kinase 1 in tomato confers resistance to fusarium oxysporum f. sp. radicis-lycopersici. Front Plant Sci 13:836269. https://doi.org/10.3389/FPLS.2022.836269/BIBTEX
Zhang W, Boyle K, Brule-Babel A et al (2021) Evaluation of genomic prediction for fusarium head blight resistance with a multi-parental population. Biol (basel). https://doi.org/10.3390/biology10080756
Acknowledgements
Clemson University is acknowledged for their generous allotment of compute time on the Palmetto Cluster. This material is based on work supported by the National Science Foundation under Grant Nos. MRI# 2024205, MRI# 1725573, and CRI# 2010270. Disclaimer: Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Funding
Open access funding provided by the Carolinas Consortium. This work was supported by the USDA National Institute of Food and Agriculture (grant number 2020–51181-32139).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by VRG, SEB and WPW. The first draft of the manuscript was written by VRG and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial interests to disclose. Corresponding author Dr. Sandra E. Branham serves as an associate editor for Theoretical and Applied Genetics.
Additional information
Communicated by Sanwen Huang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ganaparthi, V.R., Wechter, P., Levi, A. et al. Mapping and validation of Fusarium wilt race 2 resistance QTL from Citrullus amarus line USVL246-FR2. Theor Appl Genet 137, 91 (2024). https://doi.org/10.1007/s00122-024-04595-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04595-z