
Abstract
Key message
Genetic characterization of a major spot form net blotch susceptibility locus to using linkage mapping to identify a candidate gene and user-friendly markers in barley.
Abstract
Spot form net blotch (SFNB), caused by the necrotrophic fungal pathogen Pyrenophora teres f. maculata (Ptm), is an economically important foliar diseases in barley. Although various resistance loci have been identified, breeding for SFNB-resistant varieties has been hampered due to the complex virulence profile of Ptm populations. One resistance locus in the host may be effective against one specific isolate, but it may confer susceptibility to other isolates. A major susceptibility QTL on chromosome 7H, named Sptm1, was consistently identified in many studies. In the present study, we conduct fine mapping to localize Sptm1 with high resolution. A segregating population was developed from selected F2 progenies of the cross Tradition (S) × PI 67381 (R), in which the disease phenotype was determined by the Sptm1 locus alone. Disease phenotypes of critical recombinants were confirmed in the following two consecutive generations. Genetic mapping anchored the Sptm1 gene to an ⁓400 kb region on chromosome 7H. Gene prediction and annotation identified six protein-coding genes in the delimited Sptm1 region, and the gene encoding a putative cold-responsive protein kinase was selected as a strong candidate. Therefore, providing fine localization and candidate of Sptm1 for functional validation, our study will facilitate the understanding of susceptibility mechanism underlying the barley-Ptm interaction and offers a potential target for gene editing to develop valuable materials with broad-spectrum resistance to SFNB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare L.) is an important cereal crop in the grass family Poaceae. It is the fourth largest cereal crop in both yield and cultivation area globally, after wheat, rice, and corn (Poehlman 1987). Barley is primarily grown for animal feed and malt which is important in the brewing and distilling industries. In some developing countries, it is also a staple food for human consumption. However, barley production is seriously threatened by various diseases. One of the important foliar diseases in barley is net blotch (NB) caused by the fungal pathogen Pyrenophora teres, resulting in up to 10–40% yield losses when susceptible varieties are grown (Mouchacca 1999).
Net blotch occurs in two forms, spot form (SFNB) and net form (NFNB) caused by P. teres f. maculata (Ptm) and P. teres f. teres (Ptt), respectively. Although hybrids can be obtained in the laboratory, Ptm and Ptt are genetically distinct, and they induce different symptoms on susceptible hosts (Liu et al. 2011). Diagnostic markers have been developed to differentiate the two forms of P. teres (Leisova et al. 2006). Moreover, Ptt infects as a necrotroph and grows mostly in the apoplast. In contrast, Ptm initially forms intracellular vesicles near the penetration site before switching to intercellular growth resulting in necrosis (Lightfoot and Able 2010). The early laten phase indicated that Ptm may secrete additional effectors to suppress host defense responses at the initial infection stage (Whisson et al. 2007). Both forms of P. teres secreted phytotoxins or necrotrophic effector responsible for the necrosis and chlorosis, while Ptt produces significantly more toxins in the culture medium (Lightfoot and Able 2010; Sarpeleh et al. 2007, 2008). Many of these host specific toxins or necrotrophic effectors are proteinaceous (Sarpeleh et al. 2007). Hijacking the host defense in an inverse gene-for-gene manner, necrotrophic effectors manipulate host susceptibility proteins or targets to induces programmed cell death (PCD) for necrotrophs to acquire nutrients from destroyed cells (Friesen and Faris 2021).
SFNB has been increasingly damaging in barley growing regions (Liu et al. 2011). Harnessing genetic resistance is an effective and sustainable means for disease control. Nevertheless, due to the sexual recombination in Ptm populations, the rapid evolution of effectors diversifies the virulence profiles. As a result, host reactions to Ptm are complex and controlled by various quantitative trait loci (QTL). Additionally, a QTL effective against certain isolates may be susceptible to others. Genome-wide association and linkage mapping studies have identified only a few major QTL which have been consistently detected using various Ptm isolates, including Rpt4 (on chromosome 7H), Rpt5 (6H), Rpt6 (5H), Rpt7 (4H), and Rpt8 (4H) (Alhashel et al. 2021; Daba et al. 2019; Franckowiak and Platz 2013; Friesen et al. 2006; Grewal et al. 2008; Manninen et al. 2006; Raman et al. 2003; Richards et al. 2016; Tamang et al. 2019; Vatter et al. 2017; Yun et al. 2005). These QTL provide valuable resources for breeding broad-spectrum resistance to SFNB. However, the identity and functional mechanisms of the genes underlying these QTL have been elusive.
The Rpt4 locus is strikingly important among the major QTL. This locus confers broad-spectrum resistance/susceptibility to Ptm including some isolates with unique virulence profiles, and it is effective against multiple Ptt isolates as well (Alhashel et al. 2021; Daba et al. 2019; Duellman 2015; Grewal et al. 2008; Wonneberger et al. 2017). Furthermore, although Rpt4 was identified as a dominant seedling resistance, it also contributed to adult plant resistance (APR) (Williams et al. 1999). The broad specificity of Rpt4 was confirmed by Tamang et al. (2019) using six geographically distinct isolates, but segregation ratios in the biparental population suggested that Rpt4 conditioned dominant susceptibility to SFNB (Tamang et al. 2019). Recent research in barley-Ptm interactions identified two major virulence loci located on Ptm Chr1 and Chr2, respectively, with the Ptm virulence on Chr2 targeting a dominant susceptibility gene at the Rph4 locus on barley 7H (Skiba et al. 2022). An inverse gene-for-gene association was demonstrated by the host and pathogen genetics in the barley-Ptm pathosystem (Skiba et al. 2022).
In the present study, we conducted genetic and physical mapping to identify the gene underlying the Rph4 locus. To avoid misperception, the gene is designated Susceptibility to Ptm 1 (Sptm1) hereafter. Genetic mapping delimited the Sptm1 gene within a ⁓400 kb region on 7H. A total of six protein-coding genes were identified in the Sptm1 region. Of those, one gene encoding a putative protein kinase was selected as a promising candidate for functional validation. Therefore, our research lays a foundation to isolate this agronomically and genetically important Sptm1 gene, which will facilitate our understanding of the molecular mechanisms regulating the barley-Ptm interactions and provide a target for gene manipulation to develop SFNB-resistant resources.
Materials and methods
Ptm isolate and plant materials
Ptm isolate Cel-A17 (CA17) collected in Montana State was used to map Sptm1 in this study. Using a recombinant inbred line (RIL) population derived from the cross between Tradition (six-rowed, susceptible) and PI 67,381 (two-rowed, resistant). Tamang et al. (2019) identified a total of three QTL against CA17 including the susceptibility gene Sptm1 on 7H. The other two QTL were located on 2H (QRptm-2H-1–31) and 3H (QRptm-3H-81–88) (Tamang et al. 2019). Using SNP markers flanking these three loci, we identified six plants from 200 Tradition × PI 67,381 F2 lines that were heterozygous for Sptm1 and homozygous recessive for QRptm-2H-1–31 and QRptm-3H-81–88. Segregating populations used to map Sptm1 were developed by selfing the selected six F2 plants. A total of 702 F2:3 plants were used for genetic mapping. Critical recombinants were assessed in the next generation with at least 40 F3:4 plants, and the derived homozygous F3:4 recombinant representing immortal critical recombinant (ICR) were used to increase seeds for further phenotyping. At least 30 ICRs (F4:5) were used to confirm the phenotype for each original F2:3 recombinant.
Inoculum preparation and phenotyping
The CA17 inoculum preparation, inoculation, and phenotyping were conducted as described by Neupane et al. (2015). Briefly, spores were collected with sterilized distilled water from V8-PDA culture plates (150 ml V8 juice, 10 g Difco PDA, 3 g CaCO3, 10 g agar, and 850 ml H2O). The spore concentration was adjusted to 2000 spores/ml with two drops of Tween-20 per 100 ml added. Barley segregants together with Tradition and PI 67,381 were individually grown in super-cell cones placed in RL98 trays. Inoculation was performed when the second leaf was fully expanded (∼2 weeks) using an air sprayer at 15 to 20 psi. The inoculated plants were kept in a mist chamber at 100% relative humidity for 24 h under continuous light, and then moved to a growth chamber under a 12 h/12 h—light/dark cycle at 21 °C. Disease reactions were assessed 7 days post inoculation (DPI) using a 1–5 rating scale with 1 being highly resistant and 5 being highly susceptible (Neupane et al. 2015).
Barley genotypes used for pangenome sequencing were also tested for disease responses with at least eight plants for each line (Jayakodi et al. 2020). Two plants in each cone were scored collectively as a single replicate, and at least four independent replicates for each genotype were conducted. The average value of all replicates was used as the phenotypic score.
Genotyping, marker development, and linkage mapping
The CTAB protocol was used to extract DNA (Murray and Thompson 1980). Around 100 mg of leaf tissue were collected from plants at the three-leaf stage. DNA concentration was quantified using a NanoDrop spectrophotometer (NanoDrop 8000, Thermo Fisher Scientific) according to the manufacturer’s instructions. The parental lines Tradition and PI 67,381 were genotyped using the barley 50 k iSelect SNP Array to identify markers (Bayer et al. 2017). GenomeStudio V2.0 (Illumina) was used for genotype calling with the de novo calling algorithm. The called SNPs, together with those flanking Sptm1, QRPtm-2H-1–31 (2H), and QRPtm-3H-81–88 (3H) reported by Tamang et al. (2019), were converted to semi-thermal asymmetric reverse PCR (STARP) markers (Table S1) (Long et al. 2017). PCR protocol and conditions were followed as previously described (Long et al. 2017). Amplicons were assayed on a 6% polyacrylamide gel stained with GelRed™ (MilliporeSigma), which was imaged using a Typhoon™ FLA 9500 variable mode laser scanner (GE Healthcare Life Sciences, Marlborough, MA). Simple sequence repeats (SSRs) markers were also developed using the barley cv. Morex v3 reference assembly (Mascher et al. 2017).
More SNPs were obtained to saturate the Sptm1 genetic region by genome re-sequencing of Tradition and PI 67,381. Paired-end sequencing of the genomic libraries was performed on an Illumina Novaseq 6000 system with 150 bp paired ends. All the Illumina paired end reads were cleaned with bbduk using the following parameters, ktrim = r; K = 23; mink = 11 and hdist = 1 (Bushnell 2021). The cleaned reads are aligned to the Morex v3 reference genome, and alignment files were then sorted and indexed using SAMtools (Danecek et al. 2021; Langmead et al. 2009). Filtered with a minimum mapping quality score of 30 and a minimum reads depth coverage of 4, raw single nucleotide variants and indels were called using SAMtools and bcftools v1.14, (Li 2011). SNP and indel variants were annotated with BEDtools (Quinlan and Hall 2010).
Genetic map was constructed using JoinMap 3.0 (Stam 1993). All markers used for genetic mapping of Sptm1 are listed in Table S2.
Physical mapping and sequence analysis of candidate genes in the Sptm1 region
The programs FGENESH and Pfam 32.0 were used to perform gene prediction and annotation. The predicted gene structure was verified using BaRTv1.0, a high-quality, non-redundant barley reference transcripts database (Barley Reference Transcripts–BaRTv1.0) (Rapazote-Flores et al. 2019). We extracted the fasta sequences of the annotated genes from the Morex v3 genome assembly corresponding to the Sptm1 region (Chr7H:592,631,221–593,037,317). With extra 1000 bp flanking coding region for each of the annotated gene. Genome re-sequencing reads of parental lines were mapped to the Morex v3 genome, and the aligned bam files were subset to the Sptm1 region using bcftools to generate variant call format files (VCFs). The generated variant files were visualized along with the bam files using Integrative Genomics Viewer (IGV) to identify sequence polymorphisms between alleles.
Results
Population development and phenotype evaluation
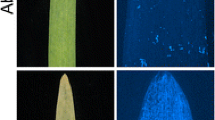
Tamang et al. (2019) identified 3 QTL associated with susceptibility to Ptm isolate CA17 using RILs of Tradition (S) × PI 67,381 (R), including Sptm1, QRptm-2H-1–31 (2H) and QRptm-3H-81–88 (3H). Using the SNP markers flanking these three QTL (Table S1), we excluded the genetic effect of QTL on 2H and 3H, and the disease phenotype in the resulting F2:3 population was controlled by Sptm1 only. Besides the resistant and susceptible F2:3 extremes, we identified a large number of plants showing intermediate disease types. F2:3 plants carrying homozygous PI 67,381 allele (n = 15), indicated by Sptm1-flanking SNP markers, exhibited an average disease reaction of 1.38 which was not significantly different from the score of PI 67,381 (1.5) (Table 1). F2:3 plants carrying homozygous Tradition allele (n = 15) showed typical SFNB symptoms 7 DPI with large necrotic lesions surrounded by a chlorotic halo on infected leaves, although their average disease type (3.3) was lower that of Tradition (4.5) (Fig. 1, Table 1). Therefore, based on the disease types of homozygous segregants, plants were considered to be resistant, intermediate, and susceptible if they displayed a reaction type ≤ 1.5, ≥ 2 but < 3, and ≥ 3, respectively. Phenotyping of an initial 178 F2:3 plants identified 35 resistant, 97 intermediate, and 46 susceptible. The segregation ratio fits 1:2:1 (χ2 = 2.80, df = 2, P = 0.25), suggesting that the disease reaction is controlled by a single gene with dosage effect.

Phenotypic responses of parental lines Tradition and PI 67,381 and homozygous F2:3 plants to Ptm isolate CA17. Typical SFNB symptom with large necrotic lesions was shown in Tradition homozygous susceptible F2:3 plant, while PI 67,381 and homozygous resistant F2:3 plant were incompatible with the pathogen
Genetic and physical mapping
SNP markers on 7H were identified through genotyping of parental lines using the barley 50 k SNP array (Table S2). Genetic mapping of Sptm1 was first conducted with the 178 F2:3 plants. Using 81 resistant and susceptible F2:3 extremes, we initially delimited the Sptm1 gene to a 6 Mb region flanked by SNP markers M24 and M33 (Fig. 2A). Plants including intermediate individuals genotyped as recombinants by M24 and M33 were saved for seed increase. All recombinants went through phenotype confirmation using both F3:4 plants and the derived homozygous ICRs. Relying on the ICRs of critical recombinants TP1-15 (the 15th plant in batch 1 tested), TP1-169, and TP1-25, the Sptm1 region was narrowed down to 1.2 Mb flanked by M28 and M29 (Fig. 2A). To increase the mapping resolution, we enlarged the population to 702 F2:3 individuals (Fig. 2B). More SNPs were called using short-read sequencing of Tradition and PI 67,381 to saturate the Sptm1 region (Table S2). A total of 20 recombinants were identified between M28 and M29. Taking advantage of the same phenotyping strategy involving F3:4 plants and ICRs, the Sptm1 gene was finally delimited to an ⁓400 kb region (Chr7H: 592,631,221–593,037,317) flanked by M69 and M87 based on three critical recombinants TP2-64, TP3-32, and TP5-78 (Fig. 2B).

Fine mapping of Sptm1. Genetic mapping was conducted sequentially with 178 (A) and 524 (B) F2:3 individuals representing 356 and 1048 gametes, respectively. Phenotypes of critical recombinants were first confirmed with F3:4 plants from which ICRs were selected. At least 30 ICRs for each recombinant were also tested to verify the disease response. The ICRs used to delimit the Sptm1 gene are shown by a combination of differential boxes. Black box represents homozygous susceptible genotype, and empty for homozygous resistant. Numbers below the linkage group indicate the number of recombination breakpoints separating the marker from Sptm1. A total of six protein-coding genes were identified in the Sptm1 region spanning ~ 400 kb (C). The maps are drawn to scale. M, marker; ICR, immortal critical recombinant; R, resistant; S, susceptible; G, gene
Gene annotation and prediction identified a total of six putative protein-coding genes according to the Morex v3 genome assembly (Fig. 2C, Table 2). Of those, four genes (G1–G4) encode either hypothetical or uncharacterized proteins (Table 2). The coding product of G5 (HORVU.MOREX.r3.7HG0735550) is homologous to human protein Werner Syndrome Exonuclease (WEX) with an exonuclease domain. G6 (HORVU.MOREX.r3.7HG0735560) encodes a protein with homology to Cold-responsive Protein Kinase 1 (CRPK1, At1G16670) in Arabidopsis thaliana (Table 2) (Liu et al. 2017). Because protein kinases play crucial roles in various signal transduction cascades, particularly in plant–microbe interactions, we focused on G6 for further analysis.
Allelic polymorphisms between resistant and susceptible G6 alleles
The coding region of G6 contains 6 exons and 5 introns, encoding a protein of 384 amino acids (aa) with a molecular weight of 42.8 kDa, composed of the catalytic domain of serine/threonine-specific and tyrosine-specific protein kinases (Fig. 3A). Allelic sequences of G6 were obtained by mapping WGS reads to the Morex v3 genome reference. A total of five SNPs, three in exons and two in introns, were identified between Tradition and PI 67,381 alleles, resulting in only one aa substitution D19N at the N-terminus (Fig. 3A–B). All SNPs were confirmed by Sanger sequencing as well.

Sequence analysis of G6. The coding region of G6 contains 6 exons and 5 introns (A), encoding a protein of 384 aa (B). Exons are shown as empty boxes, and black line for introns. The SNPs identified between Tradition and PI 67,381 alleles are indicated by red arrows. The catalytic domain of protein kinase is highlighted in blue. The aa substitutions identified among allele products are highlighted in red (B). Three G6 isoforms were identified using pangenome references and Bowman, and the D19N substitution is associated with disease type (C)
The pangenome derived from 20 barley accessions representing the global diversity provides an important tool to reveal the hidden allelic variations (Jayakodi et al. 2020). To investigate if the protein haplotype is associated with disease phenotype, we assessed 17 available pangenome references and a transformable variety Bowman. The result showed that 17 accessions including Morex were susceptible or intermediately susceptible, but ZDM02064 (Chiba) was the only line resistant to CA17 (Fig. 3C, Table S3). Two protein isoforms were identified among the susceptible lines, distinguished by an aa substitution at the distal C-terminus, G368S. Most susceptible lines (13 lines) carry the same protein isoform as Tradition (Fig. 3C). Notably, although the aa substitution D19N is outside of the conserved functional domain (Fig. 3B), the resistant line ZDM02064 shares the same protein haplotype as the resistant parental line PI 67,831 (Fig. 3C), which therefore strengthens the candidacy for this protein kinase gene.
Discussion
SFNB has been increasingly damaging for barley production. Genetic resistance has been identified in barley and its wild relatives, and the introgression of resistance from various sources into barley may facilitate achievement of more effective and durable resistance. However, the complex and quantitative nature of host responses has posed a major challenge to deploying effective and durable resistances (Wang et al. 2015), which also limits the understanding of molecular mechanisms controlling barley-Ptm interactions. Conferring broad spectrum recognition to both Ptm and Ptt, the Sptm1 locus provides a valuable resource for breeders and geneticists in barley improvement and genetic studies (Alhashel et al. 2021; Skiba et al. 2022; Tamang et al. 2019; Williams et al. 1999, 2003). In the present research, we conducted genetic and physical mapping toward cloning of the molecular determinant of Sptm1. Using ICRs derived from selected F2 lines of Tradition × PI 67,381, we precisely anchored the Sptm1 gene to an ⁓400 kb region on 7H, and G6 (HORVU.MOREX.r3.7HG0735560) homologous to AtCRPK1 was identified as a promising candidate. In addition, the SNP identified between resistant and susceptible alleles can be used as a diagnostic marker to assist breeding selection.
In a joint genetic analysis of Ptm virulence and host susceptibility, Skiba et al. (2022) reported that the Sptm1 allele of Hockett on 7H interacted with the virulence locus on Ptm Chr2 in a inverse gene-for-gene pattern. Of the six putative genes in the Sptm1 region, G6 is the only one whose coding product are known to be involved in protein–protein interactions and signal transduction. Although a transmembrane domain for signal sensing is missing in G6, several kinases containing only the catalytic domain have been identified to function in responses to plant pathogens, such as the tomato bacterial speck resistance gene Pto (Martin et al. 1993), wheat powdery mildew resistance gene Pm21 (Cao et al. 2011), wheat stripe rust resistance gene Yr15 (Klymiuk et al. 2018), wheat stem rust resistance gene Sr60 (Chen et al. 2020), barley stem rust resistance gene Rpg1 (Brueggeman et al. 2002), and wheat septoria nodorum blotch susceptibility gene Snn3 (Zhang et al. 2021). It is noteworthy that Pto, lacking a transmembrane domain, interacts directly with the corresponding avirulence factor avrPto (Frederick et al. 1998). Therefore, under these scenarios, G6 encoding a protein kinase was designated a strong candidate for Sptm1.
An intriguing question is how a protein homologous to AtCRPK1 is involved in plant–microbe interactions. Loss-of-function mutation in AtCRPK1 results in increased cold tolerance in Arabidopsis thaliana (Liu et al. 2017). Located on the plasma membrane, AtCRPK1 phosphorylated 14–3–3 proteins, and the phosphorylated 14–3–3 proteins translocate from cytosol to the nucleus where they destabilize the key cold-responsive C-repeat-binding factor (CBF) proteins. In line with this, overexpression of 14–3–3 enhanced freezing tolerance, while mutations in 14–3–3 improved freezing tolerance. The prominent role of CBF proteins in cold acclimation has been extensively characterized, but there is no precedent for CBFs being involved in plant responses to biotic stress (Shi et al. 2018; Yamaguchi-Shinozaki and Shinozaki 2006). In contrast, 14–3–3 proteins, acting as sensors for the phosphorylation status at specific sites, play significant roles in plant–pathogen interaction as the targets of pathogen effectors or interacting with defense-related proteins (Oh and Martin 2011; Teper et al. 2014; Yang et al. 2009). Therefore, 14–3–3 proteins may be the potential linker between the putative CRPK encoded by G6 and the susceptibility to Ptm pathogen.
The aa substitution D19N associated with disease phenotype is outside the conserved functional domain (CFD) in G6 (Fig. 3), but it is not uncommon that aa substitutions beyond the CFD disrupt protein function (Li et al. 2021, 2016). The specific localization of AtCRPK1 on the plasma membrane indicated the presence of a signal peptide in the protein, although an obvious signal peptide is missing (Liu et al. 2017). The N-terminal sequence of G6 harboring the D19N substitution is homologous to that in AtCRPK1. There may be an uncharacterized signal peptide at the N-terminus, and the aa substitution disturbs protein localization.
In summary, as one of the few genes conferring broad recognition specificity to Ptm, Sptm1 is valuable for variety improvement and fundamental research in barley. The high-resolution mapping in this study provides user-friendly markers and a candidate gene for Sptm1. Cloning of Sptm1 will unravel the genetic mechanism underlying barley susceptibility to this important fungal pathogen, and it will provide a target for gene editing to develop resistant materials. Moreover, it is interesting to investigate if Sptm1 is also involved in cold tolerance. Nevertheless, the candidate of Sptm1 will be functionally validated with genetic transformation in barley.
Data availability
The re-sequencing data in this manuscript have been deposited in NCBI Sequence Read Archive under accession number PRJNA890669.
References
Alhashel AF, Poudel RS, Fiedler J, Carlson CH, Rasmussen J, Baldwin T, Friesen TL, Brueggeman RS, Yang SM (2021) Genetic mapping of host resistance to the Pyrenophora teres f. maculata isolate 13IM8.3. G3-Genes Genom Genet 11
Bayer MM, Rapazote-Flores P, Ganal M, Hedley PE, Macaulay M, Plieske J, Ramsay L, Russell J, Shaw PD, Thomas W, Waugh R (2017) Development and evaluation of a barley 50k iSelect SNP array. Front Plant Sci 8
Brueggeman R, Rostoks N, Kudrna D, Kilian A, Han F, Chen J, Druka A, Steffenson B, Kleinhofs A (2002) The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc Natl Acad Sci USA 99:9328–9333
Bushnell B (2021) BBTools. 38.79 edn. http://sourceforge.net/projects/bbmap/
Cao A, Xing L, Wang X, Yang X, Wang W, Sun Y, Qian C, Ni J, Chen Y, Liu D, Wang X, Chen P (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci USA 108:7727–7732
Chen S, Rouse MN, Zhang W, Zhang X, Guo Y, Briggs J, Dubcovsky J (2020) Wheat gene Sr60 encodes a protein with two putative kinase domains that confers resistance to stem rust. New Phytol 225:948–959
Daba SD, Horsley R, Brueggeman R, Chao SAM, Mohammadi M (2019) Genome-wide association studies and candidate gene identification for leaf scald and net blotch in barley (Hordeum vulgare L.). Plant Dis 103:880–889
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H (2021) Twelve years of SAMtools and BCFtools. Gigascience 10
Duellman K (2015) Characterizing P. teres f. maculata in the northern United States and impact of spot form net blotch on yield of barley. https://doi.org/10.13140/RG.2.1.3045.9925.
Franckowiak JD, Platz GJ (2013) International database for barley genes and barley genetic stocks. Barley Genet Newsl 43:48–223
Frederick RD, Thilmony RL, Sessa G, Martin GB (1998) Recognition specificity for the bacterial avirulence protein AvrPto is determined by Thr-204 in the activation loop of the tomato Pto kinase. Mol Cell 2:241–245
Friesen TL, Faris JD (2021) Characterization of effector-target interactions in necrotrophic pathosystems reveals trends and variation in host manipulation. Annu Rev Phytopathol 59:77–98
Friesen TL, Faris JD, Lai Z, Steffenson BJ (2006) Identification and chromosomal location of major genes for resistance to Pyrenophora teres in a doubled-haploid barley population. Genome 49:855–859
Grewal TS, Rossnagel BG, Pozniak CJ, Scoles GJ (2008) Mapping quantitative trait loci associated with barley net blotch resistance. Theor Appl Genet 116:529–539
Jayakodi M, Padmarasu S, Haberer G, Bonthala VS, Gundlach H, Monat C, Lux T, Kamal N, Lang DI, Himmelbach A, Ens J, Zhang XQ, Angessa TT, Zhou GF, Tan C, Hill C, Wang PH, Schreiber M, Boston LB, Plott C, Jenkins J, Guo Y, Fiebig A, Budak H, Xu DD, Zhang J, Wang CC, Grimwood J, Schmutz J, Guo GG, Zhang GP, Mochida K, Hirayama T, Sato K, Chalmers KJ, Langridge P, Waugh R, Pozniak CJ, Scholz U, Mayer KFX, Spannagl M, Li CD, Mascher M, Stein N (2020) The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588
Klymiuk V, Yaniv E, Huang L, Raats D, Fatiukha A, Chen S, Feng L, Frenkel Z, Krugman T, Lidzbarsky G, Chang W, Jaaskelainen MJ, Schudoma C, Paulin L, Laine P, Bariana H, Sela H, Saleem K, Sorensen CK, Hovmoller MS, Distelfeld A, Chalhoub B, Dubcovsky J, Korol AB, Schulman AH, Fahima T (2018) Cloning of the wheat Yr15 resistance gene sheds light on the plant tandem kinase-pseudokinase family. Nat Commun 9:3735
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10
Leisova L, Minarikova V, Kucera L, Ovesna J (2006) Quantification of Pyrenophora teres in infected barley leaves using real-time PCR. J Microbiol Methods 67:446–455
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li PC, Yu SW, Li K, Huang JG, Wang XJ, Zheng CC (2016) The mutation of Glu at amino Acid 3838 of AtMDN1 provokes peiotropic developmental phenotypes in Arabidopsis. Sci Rep 6:36446
Li M, Hensel G, Melzer M, Junker A, Tschiersch H, Ruwe H, Arend D, Kumlehn J, Borner T, Stein N (2021) Mutation of the ALBOSTRIANS ohnologous gene HvCMF3 impairs chloroplast development and thylakoid architecture in barley. Front Plant Sci 12:732608
Lightfoot DJ, Able AJ (2010) Growth of Pyrenophora teres in planta during barley net blotch disease. Australas Plant Path 39:499–507
Liu Z, Ellwood SR, Oliver RP, Friesen TL (2011) Pyrenophora teres: profile of an increasingly damaging barley pathogen. Mol Plant Pathol 12:1–19
Liu Z, Jia Y, Ding Y, Shi Y, Li Z, Guo Y, Gong Z, Yang S (2017) Plasma membrane CRPK1-mediated phosphorylation of 14-3-3 proteins induces their nuclear import to fine-tune CBF signaling during cold response. Mol Cell 66(117–128):e115
Long YM, Chao WS, Ma GJ, Xu SS, Qi LL (2017) An innovative SNP genotyping method adapting to multiple platforms and throughputs. Theor Appl Genet 130:597–607
Manninen OM, Jalli M, Kalendar R, Schulman A, Afanasenko O, Robinson J (2006) Mapping of major spot-type and net-type net-blotch resistance genes in the Ethiopian barley line CI 9819. Genome 49:1564–1571
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang XQ, Zhang QS, Barrero RA, Li L, Taudien S, Groth M, Felder M, Hastie A, Simkova H, Stankova H, Vrana J, Chan S, Munoz-Amatrian M, Ounit R, Wanamaker S, Bolser D, Colmsee C, Schmutzer T, Aliyeva-Schnorr L, Grasso S, Tanskanen J, Chailyan A, Sampath D, Heavens D, Clissold L, Cao SJ, Chapman B, Dai F, Han Y, Li H, Li X, Lin CY, McCooke JK, Tan C, Wang PH, Wang SB, Yin SY, Zhou GF, Poland JA, Bellgard MI, Borisjuk L, Houben A, Dolezel J, Ayling S, Lonardi S, Kersey P, Lagridge P, Muehlbauer GJ, Clark MD, Caccamo M, Schulman AH, Mayer KFX, Platzer M, Close TJ, Scholz U, Hansson M, Zhang GP, Braumann I, Spannagl M, Li CD, Waugh R, Stein N (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:426–433
Mouchacca J (1999) Compendium of barley diseases, second edition, Mathre D.E. (Ed.). APS Press, The American Phytopathological Society, 139 Pokfulam Road, Hong Kong (1997), 90, ISBN: 0–89054–180–9.
Murray MG, Thompson WF (1980) Rapid isolation of high molecular-weight plant DNA. Nucleic Acids Res 8:4321–4325
Neupane A, Tamang P, Brueggeman RS, Friesen TL (2015) Evaluation of a barley core collection for spot form net blotch reaction reveals distinct genotype-specific pathogen virulence and host susceptibility. Phytopathology 105:509–517
Oh CS, Martin GB (2011) Tomato 14-3-3 protein TFT7 interacts with a MAP kinase kinase to regulate immunity-associated programmed cell death mediated by diverse disease resistance proteins. J Biol Chem 286:14129–14136
Poehlman JM (1987) Breeding barley and oats. Breeding field crops. Springer Netherlands, Dordrecht, pp 378–420
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Raman H, Platz GJ, Chalmers KJ, Raman R, Read BJ, Barr AR, Moody DB (2003) Mapping of genomic regions associated with net form of net blotch resistance in barley. Aust J Agric Res 54:1359–1367
Rapazote-Flores P, Bayer M, Milne L, Mayer CD, Fuller J, Guo WB, Hedley PE, Morris J, Halpin C, Kam J, Mckim SM, Zwirek M, Casao MC, Barakate A, Schreiber M, Stephen G, Zhang RX, Brown JWS, Waugh R, Simpson CG (2019) BaRTv1.0: an improved barley reference transcript dataset to determine accurate changes in the barley transcriptome using RNA-seq. Bmc Genomics 20
Richards J, Chao S, Friesen T, Brueggeman R (2016) Fine mapping of the barley chromosome 6H net form net blotch susceptibility locus. G3 (Bethesda) 6:1809–1818
Sarpeleh A, Wallwork H, Catcheside DE, Tate ME, Able AJ (2007) Proteinaceous metabolites from Pyrenophora teres contribute to symptom development of barley net blotch. Phytopathology 97:907–915
Sarpeleh A, Wallwork H, Tate ME, Catcheside DEA, Able AJ (2008) Initial characterisation of phytotoxic proteins isolated from Pyrenophora teres. Physiol Mol Plant P 72:73–79
Shi Y, Ding Y, Yang S (2018) Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci 23:623–637
Skiba RM, Wyatt NA, Kariyawasam GK, Fiedler JD, Yang S, Brueggeman RS, Friesen TL (2022) Host and pathogen genetics reveal an inverse gene-for-gene association in the P. teres f. maculata-barley pathosystem. Theor Appl Genet 135:3597–3609
Stam P (1993) Construction of integrated genetic-linkage maps by means of a new computer package - JoinMap. Plant J 3:739–744
Tamang P, Richards JK, Alhashal A, Sharma Poudel R, Horsley RD, Friesen TL, Brueggeman RS (2019) Mapping of barley susceptibility/resistance QTL against spot form net blotch caused by Pyrenophora teres f. maculata using RIL populations. Theor Appl Genet 132:1953–1963
Teper D, Salomon D, Sunitha S, Kim JG, Mudgett MB, Sessa G (2014) Xanthomonas euvesicatoria typeIII effector XopQ interacts with tomato and pepper 14-3-3 isoforms to suppress effector-triggered immunity. Plant J 77:297–309
Vatter T, Maurer A, Kopahnke D, Perovic D, Ordon F, Pillen K (2017) A nested association mapping population identifies multiple small effect QTL conferring resistance against net blotch (Pyrenophora teres f. teres) in wild barley. PLoS ONE 12:e0186803
Wang X, Mace ES, Platz GJ, Hunt CH, Hickey LT, Franckowiak JD, Jordan DR (2015) Spot form of net blotch resistance in barley is under complex genetic control. Theor Appl Genet 128:489–499
Whisson SC, Boevink PC, Moleleki L, Avrova AO, Morales JG, Gilroy EM, Armstrong MR, Grouffaud S, van West P, Chapman S, Hein I, Toth IK, Pritchard L, Birch PR (2007) A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature 450:115–118
Williams KJ, Lichon A, Gianquitto P, Kretschmer JM, Karakousis A, Manning S, Langridge P, Wallwork H (1999) Identification and mapping of a gene conferring resistance to the spot form of net blotch (Pyrenophora teres f maculata) in barley. Theor Appl Genet 99:323–327
Williams KJ, Platz GJ, Barr AR, Cheong J, Willsmore K, Cakir M, Wallwork H (2003) A comparison of the genetics of seedling and adult plant resistance to the spot form of net blotch (Pyrenophora teres f. maculata). Aust J Agr Res 54:1387–1394
Wonneberger R, Ficke A, Lillemo M (2017) Mapping of quantitative trait loci associated with resistance to net form net blotch (Pyrenophora teres f. teres) in a doubled haploid Norwegian barley population. PLoS ONE 12:e0175773
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yang X, Wang W, Coleman M, Orgil U, Feng J, Ma X, Ferl R, Turner JG, Xiao S (2009) Arabidopsis 14-3-3 lambda is a positive regulator of RPW8-mediated disease resistance. Plant J 60:539–550
Yun SJ, Gyenis L, Hayes PM, Matus I, Smith KP, Steffenson BJ, Muehlbauer GJM (2005) Quantitative trait loci for multiple disease resistance in wild barley. Crop Sci 45:2563–2572
Zhang Z, Running KLD, Seneviratne S, Peters Haugrud AR, Szabo-Hever A, Shi G, Brueggeman R, Xu SS, Friesen TL, Faris JD (2021) A protein kinase-major sperm protein gene hijacked by a necrotrophic fungal pathogen triggers disease susceptibility in wheat. Plant J 106:720–732
Acknowledgements
This work was supported by North Dakota Barley Council (SY) and the U.S. Department of Agriculture, Agricultural Research Service through project 3060–21000-038-00D (SY). Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
This study was funded by North Dakota Barley Council (to SY) and the U.S. Department of Agriculture, Agricultural Research Service through project 3060–21000-038-00D (to SY).
Author information
Authors and Affiliations
Contributions
SY conceived and designed the project. AFA performed most of the experiments in collaboration with all other authors. JDF and RN contributed new reagents/analytic tools. All authors analyzed the experimental data. AFA and SY wrote the first draft of the manuscript. All authors commented on previous versions and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Communicated by Reem Aboukhaddour.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alhashel, A.F., Fiedler, J.D., Nandety, R.S. et al. Genetic and physical localization of a major susceptibility gene to Pyrenophora teres f. maculata in barley. Theor Appl Genet 136, 118 (2023). https://doi.org/10.1007/s00122-023-04367-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04367-1