
Abstract
Pancreas ductal adenocarcinoma belongs to the most common cancers, but also to the tumors with the poorest prognosis. Here, we pharmacologically targeted a mitochondrial potassium channel, namely mitochondrial Kv1.3, and investigated the role of sphingolipids and mutated Kirsten Rat Sarcoma Virus (KRAS) in Kv1.3-mediated cell death. We demonstrate that inhibition of Kv1.3 using the Kv1.3-inhibitor PAPTP results in an increase of sphingosine and superoxide in membranes and/or membranes associated with mitochondria, which is enhanced by KRAS mutation. The effect of PAPTP on sphingosine and mitochondrial superoxide formation as well as cell death is prevented by sh-RNA-mediated downregulation of Kv1.3. Induction of sphingosine in human pancreas cancer cells by PAPTP is mediated by activation of sphingosine-1-phosphate phosphatase and prevented by an inhibitor of sphingosine-1-phosphate phosphatase. A rapid depolarization of isolated mitochondria is triggered by binding of sphingosine to cardiolipin, which is neutralized by addition of exogenous cardiolipin. The significance of these findings is indicated by treatment of mutated KRAS-harboring metastasized pancreas cancer with PAPTP in combination with ABC294640, a blocker of sphingosine kinases. This treatment results in increased formation of sphingosine and death of pancreas cancer cells in vitro and, most importantly, prolongs in vivo survival of mice challenged with metastatic pancreas cancer.
Key messages
-
Pancreatic ductal adenocarcinoma (PDAC) is a common tumor with poor prognosis.
-
The mitochondrial Kv1.3 ion channel blocker induced mitochondrial sphingosine.
-
Sphingosine binds to cardiolipin thereby mediating mitochondrial depolarization.
-
Sphingosine is formed by a PAPTP-mediated activation of S1P-Phosphatase.
-
Inhibition of sphingosine-consumption amplifies PAPTP effects on PDAC in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer-related death in the United States. Unfortunately, the majority of patients with pancreatic cancer are metastatic at the time of diagnosis or already developed early micrometastatic disease with seemingly localized disease. This is seen with systemic recurrences despite treatment with surgery alone. The cornerstone of treatment to reduce recurrences remains systemic chemotherapy. Despite incremental improvements in treatment outcomes, results remain abysmal with 5-year survival just recently surpassing 10% [1, 2]. Attempts to improve treatment efficacy, for instance by introducing check-point inhibitors to trigger an immune response against the tumor, have largely been studied with negative results [1, 2]. These failures highlight our lack of knowledge and understanding of the molecular underpinnings of the aggressive nature of this malignancy. Here, we focused on the role of a mitochondrial ion channel as a potential target for the treatment of PDAC.
Ion channels are transmembrane proteins that form aqueous pores driving ions through the plasma membrane in favor of an electrochemical gradient. We have previously demonstrated that Kv1.3 is not only expressed on the cell membrane, but also in the inner mitochondrial membrane. The voltage-gated Kv1.3 channel of the Kv1 subfamily is mostly expressed in immune cells, including T and B lymphocytes, macrophages, neutrophils, microglia, and dendritic cells and in neurons [3, 4]. However, we and others have previously shown the expression of Kv1.3 in numerous types of cancers [5,6,7]. The mitochondrial Kv1.3 channel has been shown to be very important for the induction of cell death in a variety of cells, including cancer cells [8, 9].
In detail, inhibition of mitochondrial Kv1.3 results in changes in mitochondrial membrane potential, formation of reactive oxygen species (ROS), opening of the permeability transition pore (PTP) [10], release of cytochrome c, and cell death [8, 9, 11, 12]. All of these events are abrogated in Kv1.3-deficient cells. The formation of ROS upon inhibition of mitochondrial Kv1.3 was shown to be a crucial step in the induction of cell death [9]. We developed two novel mitochondria-targeted specific inhibitors of Kv1.3, i.e., PAPTP and PCARBTP, and demonstrated that the basal level of ROS production and expression levels of mitochondrial Kv1.3 determine whether these inhibitors kill cells or not [9]. The new drugs allowed us to target exclusively malignant tumor cells, because their basal levels of ROS and Kv1.3 expression level are higher than those of non-malignant cells [13, 14]. However, many details of the mechanisms of how Kv1.3 inhibitors induce cell death are still unknown.
Here, we tested a role of sphingolipids in killing of pancreas cancer cells by inhibitors of mitochondrial Kv1.3. and established their importance in the context of cancer therapy. Sphingolipids are a class of lipids that share an amino alcohol (sphingoid base) backbone. Ceramides are a subfamily of sphingolipids in which the amino group on sphingosine is linked to a fatty acid [15]. We have previously shown that ceramide alters the organization and the distribution of receptors and associated signalling molecules within membranes, and this activity allows the generation of intracellular signals [16]. Thus, ceramide may act via a change in membrane biophysics that results in the formation of ceramide-enriched membrane domains and the reorganization of receptors and signalling molecules within these domains [16], but it may also act by direct interaction with certain proteins [17,18,19,20,21,22]. Sphingosine may regulate certain protein kinase C isoforms [23], but at present it is largely unknown how sphingosine acts on and in cells. It was shown that under stress, ceramide synthase 1 traffics from the endoplasmic reticulum to the cytoplasmic leaflet of the outer mitochondrial membrane and generates C18 ceramide [22]. Mitochondrial C18 ceramide in the cytosolic leaflet of the outer mitochondrial membrane binds to LC3-II and thereby mediates mitophagy [22]. At present, the functions of mitochondrial sphingosine are completely unknown; however, general consensus is that sphingosine is pro-apoptotic, while sphingosine-1 phosphate (S1P) is mitogenic and anti-apoptotic [24].
Here, we demonstrate that inhibition of mitochondrial Kv1.3 using one of the inhibitors we recently developed, i.e., PAPTP, results in the formation of mitochondrial/mitochondria-associated sphingosine. Sphingosine binds to cardiolipin in mitochondrial membranes and induces a rapid depolarization of mitochondria, which may trigger cell death. In addition, we explored the role of sphingosine in KRAS modulation, since sphingomyelin metabolism has been proposed to act as a KRAS regulator [25]. Activating point mutations in RAS genes are associated with approximately 30% of all human cancers. In particular, mutant KRAS is the most oncogenic and frequent, accounting for 85% of all mutated RAS proteins reported, and is predominantly found in PDAC. Interestingly, the mitochondria-targeted anti-oxidant MitoQ has been shown to decrease KRAS-caused formation of PDAC in vivo [26], suggesting that mitochondria are the primary source of KRASG12D-induced ROS. This is in agreement with a translocation of mutant KRAS to mitochondria [27]. We hypothesize that oncogenic KRAS alters the physiology/metabolism of mitochondria resulting in increased sensitivity to mitoKv1.3 inhibition and serves as a paradigm for alterations in tumor cells or pharmacologically induced manipulations that result in increased mitochondrial ROS and sensitize cancer cells to these drugs.
Methods
Mice and in vivo studies
Ethics approval
All animal experiments were approved by the University of Cincinnati Ethics Committee and the Institutional Animal Care and Use Committee.
All experiments were performed according to the FELASA regulations and we also followed the ARRIVE guidelines.
Eight- to 12-week-old, wild-type C57BL/6 J mice were employed. Pancreas cancer was established in the peritoneum by intraperitoneal injection of 50,000 KPC cells resuspended in PBS. Mice were treated i.v. 6 times starting at day 3 and then every 2nd day with 4 nmol/g PAPTP + 50 mg/kg ABC294640. PAPTP was injected i.v. in a total volume of 125 µL DMSO and 0.9% NaCl (1:4, v/v); ABC294640 was injected i.p. Controls were injected with the solvent only. Survival was determined over 65 days.
Tumor-injected mice were randomly divided into experimental groups.
Cell culture and treatment
Human PDAC cell lines BxPc3, Panc-1, and MIA Paca-2 were obtained from the pHionic consortium. KPC tumor cells were originally generated from KRASLSL−G12D/+, Trp53loxP/+, and Pdx1-Cre (KPC) mice in C57BL/6 mice background (Jackson Laboratories). Rodent pathogen status of the tumor cell lines was originally verified by Charles River Laboratory diagnosis services.
All cells were cultured in modified Eagle medium (MEM) supplemented with 10 mM HEPES (pH 7.4; Carl Roth GmbH, Karlsruhe, Germany), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 µM nonessential amino acids, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal calf serum (MEM/K10).
If indicated, cells were treated with 10 µM XY-14 (Echelon, #L9218) for 30 min prior to the addition of PAPTP.
Transfections
Panc-1 and MIA Paca-2 cells expressing wild-type or oncogenic RAS were transfected with shRNA targeting KRAS or sphingosine kinase 2 (all from Santa Cruz Inc., #sc-35731-SH for KRAS, sc-39225-SH for sphingosine kinase 2). Cells were stably transfected by electroporation.
Kv1.3 was downregulated by transient transfection using shRNA targeting Kv1.3 (Santa Cruz Inc., #sc-42712-SH) using a BTX electroporator. Cells were employed 48 h after transient transfection.
For transfections, cells were trypsinized and washed 3 times in HEPES/saline (H/S; 132 mM NaCl, 20 mM HEPES [pH 7.4], 5 mM KCl, 1 mM CaCl2, 0.7 mM MgCl2, 0.8 mM MgSO4). Cells were then resuspended in H/S at 20 × 106 cells/400 µL, aliquoted into electroporation cuvettes, the plasmids were added, and samples were incubated on ice for 15 min and electroporated at 500 V with 5 pulses each for 1 ms. Cells were incubated for 15 min on ice and finally resuspended in MEM/K10. Transfected cells were then selected by incubation with 3.5 µg/mL puromycin for 3–4 weeks. We used bulk cultures in the present experiments to avoid any clonal artifacts.
Reactive oxygen species (ROS)
Cells were grown in MEM/K10 as above, removed from the plates using cell dissociation buffer (ThermoFisher, #13,151,014), washed in PBS, incubated with 2.5 µM mitoSOX Red (ThermoFisher, # M36008) for 20 min at 37 °C, and stimulated with 0.25 or 1 µM PAPTP or left untreated. Cells were then analyzed by flow cytometry employing a FACSCalibur.
Cell death
Cells were treated as indicated, trypsinized, washed in H/S, resuspended in a Ca2+-containing staining buffer as provided by the vendor, and stained for 15 min with FITC-Annexin V (1:1000, Roche). Samples were analyzed by flow cytometry on a FACSCalibur. Positive controls were permeabilized for 5 min with 0.1% Triton X-100 at room temperature before incubation with FITC-Annexin V.
Protein measurements
Protein concentrations were measured employing the BioRad Protein Assay Dye (cat. no. #500,006) and served to normalize the samples.
Sphingosine measurements
Cells were grown in 24-well plates overnight, washed, and stimulated with 1 µM PAPTP for 30 min in H/S or left untreated, the medium was removed, and cells were washed once with cold H/S and shock-frozen in liquid nitrogen. Cells were then lysed in 250 µL 0.5% NP40 in H2O and 200 µL was extracted in 600 µL CHCl3:CH3OH:1N HCl (100:100:1, v/v/v). Phases were separated, and an aliquot of the lower phase was dried in a Speedvac and resuspended in 20 µL of a detergent solution (7.5% [w/v] n-octyl glucopyranoside, 5 mM cardiolipin in 1 mM diethylenetriaminepentaacetic acid [DTPA]). The samples were sonicated in a bath sonicator for 10 min and the kinase reaction was performed by the addition of 80 µL of 0.001 units sphingosine kinase in 50 mM HEPES (pH 7.4), 250 mM NaCl, 30 mM MgCl2 1 mM ATP, and 10 µCi [32P]γATP. The kinase reaction was performed for 30 min and terminated by extraction of the samples in 20 µL 1N HCl followed by the addition of 800 µL CHCl3:CH3OH:1N HCl (100:200:1, v/v/v), and 240 µL each of CHCl3 and 2 M KCl. Samples were vortexed between additions. Phases were separated, and the lower phase was collected, dried, dissolved in 20 µL CHCl3:CH3OH (1:1, v/v), and separated on Silica G60 TLC plates with CHCl3:CH3OH:acetic acid:H2O (90:90:15:5, v/v/v/v) as developing solvent. The TLC plates were analyzed with a phosphorimager. Sphingosine levels were determined with a standard curve of C18-sphingosine.
Ceramide measurements
Cells were treated and extracted as described for sphingosine. The dried samples were resuspended in 20 µL of a detergent solution (7.5% [w/v] n-octyl glucopyranoside and 5 mM cardiolipin in 1 mM diethylenetriamine-pentaacetic acid [DTPA]), and micelles were obtained by bath sonication for 10 min. The kinase reaction was initiated by the addition of 70 µL of a reaction mixture containing 10 µL diacylglycerol (DAG) kinase (GE Healthcare Europe, Munich, Germany), 0.1 M imidazole/HCl (pH 6.6), 0.2 mM DTPA, 70 mM NaCl, 17 mM MgCl2, 1.4 mM ethylene glycol tetraacetic acid, 2 mM dithiothreitol, 1 µM adenosine triphosphate (ATP), and 5 µCi [32P]γATP. The kinase reaction was performed for 30 min at room temperature under shaking at 300 rpm, terminated by the addition of 1 mL CHCl3:CH3OH:1N HCl (100:100:1, v/v/v), 170 µL buffered saline solution (135 mM NaCl, 1.5 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 10 mM HEPES; pH 7.2), and 30 µL of a 100 mM ethylenediaminetetraacetic acid (EDTA) solution. The samples were vortexed, phases were separated, and the lower phase was collected, dried, dissolved in 20 µL CHCl3:CH3OH (1/1, v/v), separated on Silica G60 thin-layer chromatography (TLC) plates using chloroform/acetone/methanol/acetic acid/H2O (50:20:15:10:5, v/v/v/v/v) as solvent, and developed employing a Fuji phosphorimager. Ceramide levels were determined by comparison with a standard curve; C16 to C24 ceramides were used as substrates.
Sphingosine-1-phosphate-phosphatase activity
Cells were stimulated with 1 µM PAPTP for 30 min or left untreated, and mitochondria were prepared as above and incubated with 10 µM [3H]S1P complexed with fatty acid–free BSA for 30 min at 37 °C. The samples were organically extracted as described above for sphingosine measurements, separated by TLC, and consumption of S1P to sphingosine was determined.
Sphingosine kinase activity
Cells were stimulated with 1 µM PAPTP for 30 min or left untreated; mitochondria were prepared as above and resuspended after purification in a buffer consisting of 50 mM HEPES, pH 7.4, 150 mM NaCl, 30 mM MgCl2 in the presence of 500 pmol sphingosine (i.e., excess substrate), 10 µM ATP, and 10 µCi[32P]γATP. The kinase reaction was allowed to proceed for 60 min at 30 °C and stopped by the addition of 20 µL 1N HCl followed by the addition of 800 µL CHCl3:CH3OH:1N HCl (100:200:1, v/v/v), and 240 µL each of CHCl3 and 2 M KCl. Samples were then processed as described above for sphingosine measurements.
Measurement of cardiolipin binding to sphingosine
Cells were treated with 1 µM PAPTP for 30 min and lysed in 125 mM NaCl, 25 mM Tris HCl (pH 7.4), 10 mM EDTA, 10 mM sodium pyrophosphate, 2% Nonidet P40, and 10 µg/mL aprotinin and leupeptin for 10 min on ice. Sphingosine was immunoprecipitated from the lysates using a monoclonal anti-sphingosine antibody (clone NHSPH, Alfresa Pharma Corporation, Japan) for 60 min at 4 °C. Immunocomplexes were immobilized by incubation with protein A/G-coupled agarose for 45 min at 4 °C. The precipitates were extensively washed 6 times in H/S and extracted in CHCl3/CH3OH/1N HCl (100:100:1, v/v/v). Samples were dried and then resuspended in 10 µM of a cardiolipin probe (Abcam, ab241036), a fluorescent probe for the detection and quantification of cardiolipin. The samples were incubated for 5 min, 250 rpm shaking and the fluorescence was measured using a fluorescence reader. Cardiolipin concentration was determined using a standard curve of cardiolipin.
Incubation of isolated mitochondria with sphingosine and measurement of mitochondrial membrane potential in vitro
Mitochondria were isolated as above, incubated with 2.5 nM TMRM (ThermoFisher M20036) for 5 min at 37 °C and incubated with 100 nM sphingosine (Avanti Polar Lipids, #860,490) or 100 nM PAPTP. If indicated, we added 10 µM Cardiolipin (Merck, #C0563). Samples were analyzed by flow cytometry using a FACSCalibur. The results were analyzed for the mean fluorescence using the BD software.
Flow cytometry
Cells were treated with sphingosine, resuspended in buffer A consisting of 0.3 M sucrose, 10 mM TES (pH 7.4), and 0.5 mM EGTA, and incubated for 30 min on ice. Cells were then pottered in a tight glass potter with 40 strokes, centrifuged at 600 × g for 5 min at 4 °C, and the supernatants were collected. The supernatants were centrifuged at 6000 × g for 10 min at 4 °C, the pellets were resuspended in buffer B consisting of 50 mM Pipes-KOH (pH 7.4), 50 mM KCl, 2 mM MgCl2, 2 mM EGTA, 10 µg/mL A/L, 2 mM ATP, 10 mM phosphocreatine, 5 mM succinate, and 50 µg/mL creatine kinase, and samples were stained for 30 min with anti-TIM23 (1:250 dilution; BD, #611,222) and anti-sphingosine antibodies (1:1000 dilution, Alfresa, clone NHSPH) or anti-Kv1.3 antibodies (1:250, Alamone, #APC-101), respectively. Samples were washed and stained with Cy3-coupled donkey anti-mouse IgG F(ab)2 fragments (1:500 dilution; Jackson Immunoresearch, #715–166-150) for the mouse anti-TIM23 IgG + Alexa Fluor 647-coupled donkey anti-mouse IgM F(ab)2 fragments (1:500 dilution; Jackson Immunoresearch, #715–606-020) for the IgM anti-sphingosine or APC-coupled donkey anti-rabbit IgG F(ab)2 fragments (1:500 dilution; Jackson Immunoresearch, #711–136-152) for the rabbit anti-Kv1.3 IgG. Samples were washed again and analyzed by flow cytometry using a FACSCalibur employing FL2 vs. FL4. The mean fluorescence was analyzed using BD software.
Statistics
Data are expressed as arithmetic means ± SD. For the comparison of continuous variables from independent groups with one variable, we used one-way ANOVA followed by post hoc Tukey test for all pairwise comparisons, applying the Bonferroni correction for multiple testing. The P values for the pairwise comparisons were calculated after the Bonferroni correction. All values were tested for normal distribution using the Kolmogorov–Smirnov test. Statistical significance was set at a P value of 0.05 or lower (two-tailed). Outliers were not removed and all data are provided. The sample size planning was based on the results of two-sided Wilcoxon-Mann–Whitney tests (free software: G*Power, Version 3.1.7, University of Duesseldorf, Germany). Investigators were blinded to the samples in microscopic studies and to animal identity. Animals were randomly assigned to cages by a technician who was not involved in the experiments; thus, the mice were purely randomly assigned for every experiment. Cages were then randomly assigned to the various experimental groups.
Results
KRAS influences PAPTP-induced death of pancreas ductal adenocarcinoma cells
To define mechanisms that determine the mitochondrial effects of PAPTP, we tested whether oncogenic KRAS, which drives tumorigenesis in pancreas cancer, influences the effects of PAPTP on tumor cells. We treated human BxPc-3 pancreas cancer cells, which express wild-type (wt) RAS, and Panc-1 and MiaPaCa-2 cells, which both express oncogenic Ras (Gly12Asp-RAS, Gly12Cys-RAS) [28], with 250 nM and 1 µM PAPTP for 48 h. To test whether oncogenic KRAS alters death by PAPTP, we down-modulated KRAS in Panc-1 and MiaPaCa-2 cells by transfection of shRNA targeting KRAS. Downregulation was confirmed by western blotting (not shown). The results show that 75–90% of the pancreas cancer cells expressing oncogenic KRAS died after treatment with PAPTP, while cells expressing wild-type KRAS or cells transfected with shRNA targeting KRAS were less sensitive to PAPTP (Fig. 1A).

Oncogenic RAS sensitizes pancreas cancer to PAPTP-induced cell death. A PDAC cells expressing wild-type or oncogenic RAS or cells that were transfected with shRNA targeting RAS or control shRNA were treated for 48 h with PAPTP (0.25 or 1 µM) and cell death was determined by FITC-Annexin-V staining and flow cytometry analysis. Displayed are the mean ± SD, n = 8, ***p < 0.001, ANOVA and post hoc Tukey test. B Reactive oxygen species were determined by loading the cells with mitoSox and flow cytometry analysis upon treatment with PAPTP. Expression of oncogenic Ras increased mitochondrial ROS, and this sensitization was reversed by shRNA-mediated suppression of RAS expression in pancreas carcinoma cells (B). PAPTP induces a release of ROS. Shown are the mean ± SD, n = 5, ***p < 0.001, ANOVA and post hoc Tukey test. C–E Kv1.3 was downregulated in Panc-1 or MIA Paca-2 cells by transfection of siRNA targeting Kv1.3. Controls (Ctrl) were transfected with irrelevant siRNA constructs. Si-RNA-mediated downregulation of Kv1.3 prevented PAPTP-induced cell death (C) and mitochondrial superoxide release (D). E Control flow cytometry studies confirm the downregulation of Kv1.3 in mitochondria upon siRNA transfection. Given are the mean ± SD, n = 5, ***p < 0.001, ANOVA and post hoc Tukey test
Expression of oncogenic KRAS resulted in an increase of mitochondrial ROS in accordance with, e.g., [26]. The increased release of ROS in cells expressing oncogenic KRAS was reduced by shRNA-mediated suppression of KRAS expression in pancreas carcinoma cells (Fig. 1B). PAPTP treatment drastically enhanced the release of ROS, which was much higher in PDAC-expressing oncogenic KRAS than in those lacking oncogenic RAS (Fig. 1B). We hypothesize that oncogenic KRAS mediates increased sensitivity to PAPTP treatment that results in increased mitochondrial ROS and sensitize cancer cells.
Downregulation of Kv1.3 in Panc-1 or MIA Paca-2 cells, respectively, prevented the effects of PAPTP on mitochondrial superoxide release and cell death, confirming Kv1.3 as target for PAPTP (Fig. 1C,D). It is important to note that a downregulation of Kv1.3, which allows the cells to compensate and maintain the mitochondrial membrane potential, differs from the acute inhibition of the channel by an inhibitor resulting in membrane potential changes as a signal as previously described [9, 11, 12]. Downregulation of Kv1.3 in mitochondria was confirmed by staining isolated mitochondria from transfected cells with APC-anti-Kv1.3 and Cy3-anti-TIM23 followed by flow cytometry analysis and determination of the mean fluorescence signal for APC (Fig. 1E) in the Cy3-positive population.
PAPTP induces sphingosine in mitochondria
To identify targets of PAPTP-induced ROS, we tested the effects of PAPTP on sphingolipid metabolism, since the sphingolipid metabolism is redox-sensitive [29, 30]; we tested whether PAPTP induces the formation of intracellular ceramide and sphingosine. Our data indicate a marked increase of mitochondria-associated sphingosine concentrations upon treatment of pancreas cancer cells with PAPTP (Fig. 2A–C). We also observed a decrease of ceramide upon treatment with PAPTP (Fig. 2D). The formation of sphingosine was greatly enhanced in cells expressing oncogenic RAS, such as MIA Paca-2 cells, compared to cells not expressing oncogenic RAS, such as BxPc3 (Fig. 2A). Downregulation of KRAS by shRNA also decreased the formation of sphingosine in pancreas cancer cells (Fig. 2A). Furthermore, neutralization of ROS by incubating the cells with the anti-oxidant Tiron also reduced the release of sphingosine from pancreas cancer cells upon treatment with PAPTP (Fig. 2B). Si-RNA-mediated downregulation of Kv1.3 in MIA Paca-2 cells prevented the induction of sphingosine by PAPTP (Fig. 2C).

PAPTP induces the formation of sphingosine in mitochondria/mitochondria-associated membranes of pancreas cancer cells expressing oncogenic RAS, while cells lacking oncogenic RAS release much less sphingosine. A, B MiaPaca-2 PDAC cells expressing oncogenic RAS were transiently transfected with shRNA targeting RAS, control shRNA, or left untransfected. BxPc-3 lacking oncogenic RAS were used as controls. Cells were stimulated with 250 nM PAPTP in the presence or absence of the 10 µM anti-oxidant Tiron for 30 min and lysed, mitochondria were isolated and organically extracted, and sphingosine was quantified using kinase assays. C Downregulation of Kv1.3 in MIA Paca-2 cells using siRNA transfections prevented the formation of sphingosine by PAPTP. Shown is the quantitative analysis of the mean fluorescence of the sphingosine signal (FL4 channel) in the Cy3-positive population (FL2 channel) that represents mitochondria. Displayed are the mean ± SD, n = 5 or 6; *p < 0.001 ANOVA and post hoc Tukey’s multiple comparison test. D Ceramide was determined in cells treated with PAPTP or left untreated by a ceramide kinase assay. Shown are the mean ± SD, n = 5; *p < 0.001 ANOVA and post hoc Tukey’s multiple comparison test
To identify mechanisms of how PAPTP induces the formation of sphingosine, we tested the activity of the sphingosine 1-phosphate-(S1P-)phosphatase in mitochondria/mitochondria-associated membranes of oncogenic KRAS-expressing or lacking cells (Fig. 3A). Sphingosine can be generated from sphingosine 1-phosphate (S1P) by the activity of a S1P-phosphatase. The results show that PAPTP induces an activation of S1P-phosphatase, which is enhanced in cells expressing oncogenic KRAS, compared to pancreas cancer cells lacking oncogenic KRAS (Fig. 3A). We did not observe a change of sphingosine kinase activity upon treatment with PAPTP in pancreas cancer cells (not shown).

PAPTP induces the activity of sphingosine-1 phosphate-phosphatase. A Cells were disrupted and mitochondria isolated and incubated with 10 µM [3H]S1P complexed with fatty acid–free BSA for 30 min at 37 °C, organically extracted, and separated by TLC, and consumption of S1P to sphingosine was determined. B MiaPaca-2 PDAC cells were treated with PAPTP for 30 min in the presence or absence of the S1P-phosphatase inhibitor XY-14, swollen, and pottered, and mitochondria were isolated. The samples were then stained with Cy3-coupled anti-TIM23 antibodies and Alexa Fluor 647-coupled anti-sphingosine antibodies and analyzed by flow cytometry. Shown are a typical flow cytometry result and the quantitative analysis of the mean fluorescence of the sphingosine signal (FL4 channel) in the Cy3-positive population (FL2 channel) that represents mitochondria. This population is indicated by the square. Shown are the mean ± SD, n = 6, or representative stainings from 6 independent experiments; ***p < 0.001 ANOVA and post hoc Tukey test
Next, to confirm the formation of sphingosine in mitochondria and the role of S1P-phosphateses, we treated MIA Paca-2 cells with PAPTP in the presence or absence of the S1P inhibitor XY-14 [31]. Cells were then disintegrated, mitochondria isolated and stained with Cy3-anti-TIM23 and Alexa Fluor 647-anti-sphingosine. Samples were then analyzed by flow cytometry and the fluorescence signal intensity of the TIM23 positive population was determined. The results indicate a formation of sphingosine in mitochondria upon treatment with PAPTP, which was abrogated by treatment with the S1P-inhibitor XY-14 (Fig. 3B).
Sphingosine binds to cardiolipin in mitochondrial membranes
We have previously shown that sphingosine binds to cardiolipin and thereby mediates rapid permeabilization of bacterial membranes and bacterial cell death [32]. Bacterial cell death was mediated by a massive, sphingosine-mediated clustering of cardiolipin resulting in the generation of very rigid membrane domains and finally in membrane leakiness, depolarization, loss of ATP, and bacterial cell death [32]. The endosymbiont hypothesis indicates that mitochondria and bacteria are similar, and in fact in mammalian cells only the inner mitochondrial membrane contains cardiolipin. Here, we show that PAPTP induced binding of sphingosine to cardiolipin in mitochondria after stimulation (Fig. 4A). Sphingosine and PAPTP induced rapid depolarization of isolated mitochondria (Fig. 4B), which was prevented by co-incubation of the mitochondria with exogenous cardiolipin (Fig. 4B).

Sphingosine binds to cardiolipin upon treatment of cells with PAPTP. A MiaPaca-2 cells were treated with 1 µM PAPTP for 30 min and lysed and sphingosine was immunoprecipitated from the lysates to detect associating cardiolipin. Shown is the mean ± SD of the ratio of cardiolipin, n = 4, ***p < 0.001 ANOVA and post hoc test. B Isolated mitochondria were loaded with TMRM and incubated with 100 nM sphingosine or 100 nM PAPTP, and the mitochondrial membrane potential was determined by flow cytometry 10 min and 20 min after the addition of compounds. Addition of cardiolipin (10 µM) greatly reduced the effects of sphingosine and PAPTP on mitochondrial membrane potential determined after 10 and 20 min using the same laser settings for all samples. Shown is the mean ± SD of the fluorescence determined by flow cytometry, n = 6, ***p < 0.001 ANOVA and post hoc test
Pharmacological or genetic manipulation of sphingosine formation determines PAPTP-induced tumor cell death in vitro and in vivo
If PAPTP-induced sphingosine is involved in cell death, inhibition of S1P-phosphatase should prevent cell death induced by PAPTP. We therefore treated BxPc-3 PDAC (wt RAS) and MiaPaCa-2 cells (Gly12Cys KRAS) with 250 nM PAPTP in the presence or absence of 10 µM of the S1P phosphatase inhibitor XY-14. The results show that PAPTP-induced sphingosine formation and death of pancreas cancer cells were greatly reduced by S1P-phosphatase inhibition (Fig. 5A).

Pharmacological inhibition of S1P-phosphatase prevents the formation of sphingosine in and cell death of pancreas cancer cells. MIA PaCa-2 cells were treated with 10 µM S1P-inhibitor XY-14 or 10 µM of the sphingosine kinase 2 (SK2) inhibitor ABC294640 or transfected with siRNA targeting RAS or sphingosine kinase 2. Untransfected or control vector-transfected cells (Ctrl) served as controls. Cells were treated with 1 µM PAPTP for 30 min to determine sphingosine A or 48 h to measure cell death B. Cells were lysed, mitochondria isolated, and sphingosine quantified by kinase assays; cell death was measured by FITC-Annexin V staining and flow cytometry. Given are the mean ± SD, n = 6; *p < 0.001 ANOVA and post hoc Tukey’s multiple comparison test
Although PAPTP does not regulate sphingosine kinase 2, sphingosine kinase 2 consumes sphingosine by its constitutive activity and counteracts S1P-phosphatase. Thus, we also inhibited sphingosine kinase 2 using ABC294640 [33]. The results show that PAPTP-induced formation of sphingosine (Fig. 5A) and cell death (Fig. 5B) of pancreas cancer cells were increased upon pharmacological inhibition of sphingosine kinase 2.
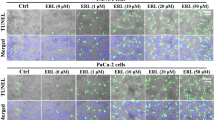
To test the significance of these findings, we established a metastatic pancreas cancer model by intraperitoneal injection of 50,000 PDAC cells. Treatment with PAPTP and ABC294640 markedly delayed tumor growth in vivo (Fig. 6), while ABC294640 alone had no effect on pancreas cancer growth (not shown).

The combination of PAPTP and carmofur allows long-term survival of mice with metastasized pancreas cancer. Metastasized pancreas cancer was established in the peritoneum by injection of 50,000 KPC cells; mice were treated i.v. 6 times starting at day 3 with 4 nmol/g PAPTP + every 2nd day with one i.p. injection/day of 50 mg/kg ABC294640. Controls were injected with solvent. Survival was determined over 60 days. Six mice were investigated per group. ***p < 0.001 compared to the survival of untreated or PAPTP-treated mice
Discussion
The present data suggest that ROS-triggered sphingosine significantly contributes to PAPTP-induced cell death. The levels of sphingosine are controlled (1) by hydrolysis of sphingosine-1-phosphate by a S1P-phosphatase associated with mitochondrial fractions and (2) by sphingosine kinases, which are not affected by PAPTP, but constitutively consume sphingosine. It is certainly possible that ceramidases by their activity also contribute to mitochondrial sphingosine, but the exact definition of the function and regulation of these enzymes in mitochondria seems to be beyond the present manuscript. Our data suggest that sphingosine binds to cardiolipin and induces rapid depolarization of isolated mitochondria, which is neutralized by the addition of exogenous cardiolipin (Fig. 7). Thus, a change of mitochondrial membrane biophysics, in particular fluidity, might be at least in part a mechanism how sphingosine mediates cell death.

Scheme of the proposed PAPTP-induced mechanisms
We have previously shown that sphingosine binds via the protonated NH2 group to cardiolipin in bacterial and model membranes and thereby mediates a marked change in the fluidity and also the permeability of bacterial and model membranes [32]. Here, we suggest that sphingosine synthesized in mitochondria or associated mitochondrial membranes has a similar effect and induces rapid induction of mitochondrial membrane potential loss, possibly thereby mediating cell death. This might be one additional mechanism how PAPTP-mediated inhibition of Kv1.3 induces cell death. Other mechanisms include opening of the permeability transition pore and the recruitment of Bax/Bak to the outer mitochondrial membrane [11]. Mitochondrial depolarization has been known for a long time to be a key step in the induction of cell death however, molecular details are unknown. Here, we suggest that sphingosine, via its biophysical interaction with cardiolipin, constitutes one of the molecules triggering mitochondrial depolarization during cell death.
Cardiolipin might be simply a binding molecule for sphingosine followed by clustering of lipids, altered membrane fluidity, and increased membrane permeabilization. It might be also possible that the binding of sphingosine to cardiolipin interferes with the function of cardiolipin in the respiratory chain. Preliminary studies suggested that the binding of sphingosine does not displace cytochrome c from mitochondria, at least early after incubation. However, cardiolipin also functions to organize proteins of the respiratory chain and thereby binding of sphingosine to cardiolipin might interfere with the organization of the respiratory chain, followed by impaired energy generation and mitochondrial depolarization.
Our data also show that the expression of oncogenic KRAS, which is one of the key oncogenes in pancreas cancer, sensitizes pancreas cancer cells to the treatment with PAPTP and promotes the formation of sphingosine in mitochondria/mitochondria-associated membranes after treatment with PAPTP. Oncogenic KRAS has been previously shown to promote the formation of reactive oxygen species in mitochondria [26]. We have previously demonstrated that the basal level of ROS production and expression levels of mitoKv1.3 determine whether these inhibitors kill cells or not [9, 11]. Since the formation of ROS upon inhibition of Kv1.3 was shown to be a crucial step in the induction of cell death [9, 11], it might be possible that oncogenic KRAS facilitates death induced by PAPTP, consistent with the present data. It is presently unknown how the formation of sphingosine is linked to the release of ROS. The acid sphingomyelinase is regulated by reactive oxygen species and redox mechanisms [29, 30]. Thus, it might be possible that reactive oxygen species also influence the activity of S1P-phosphatase.
Our in vitro and in vivo data employing ABC294640 as an inhibitor of the SK2 [33] suggests the significance of sphingosine for PAPTP-induced cell death. ABC294640 enhanced PAPTP-induced death of pancreas cancer cells in vitro and greatly delayed, in combination with PAPTP, the growth of metastatic pancreas cancer.
Kv1.3 is not only expressed in malignant cancer cells, but also in lymphocytes and macrophages. It is therefore possible that the in vivo effect observed after the application of PAPTP is not only mediated by a direct effect on malignant pancreas cancer cells, but also by killing activated immunosuppressive regulatory T lymphocytes and/or myeloid-derived suppressor cells. Such an elimination of immunosuppressive cells may then allow the immune system to become activated and contribute to the elimination of the tumor.
Our studies employed a density-based method to purify mitochondria. However, it is impossible to obtain absolutely pure mitochondria and our preparations will contain some mitochondria-associated membranes and very likely also other cellular vesicle structures. The flow cytometry studies indicate a co-localization of sphingosine with Tim23, expressed in the inner mitochondrial membrane. However, also these studies do not prove a mitochondrial formation of sphingosine and it might be possible that sphingosine is mainly formed in mitochondria-associated membranes and then transferred to mitochondria. Our studies also do not exclude that PAPTP induces the formation of sphingosine in other organelles than mitochondria (although the drug is accumulating in mitochondria by virtue of the positively charged triphenyl-phosphonium group), for instance lysosomes, and then promotes an exchange of sphingosine with mitochondria.
In summary, our data indicate that inhibition of mitochondrial Kv1.3 by PAPTP induces the formation of sphingosine, which seems to contribute to PAPTP-induced death of pancreas cancer cells in vitro and in vivo.
Availability of data and material
All data are presented in the manuscript. All material is freely available.
Abbreviations
- KRAS:
-
Kirsten Rat Sarcoma Virus
- PDAC:
-
Pancreatic ductal adenocarcinoma
- S1P:
-
Sphingosine-1 phosphate
- PTP:
-
Permeability transition pore
- Gly/Asp:
-
Glycine/aspartate
- Gly/Cys:
-
Glycine/cysteine
- si-RNA:
-
Small inhibitory RNA
- Tim23:
-
Translocase of the inner membrane 23
- H/S:
-
HEPES/saline
- ROS:
-
Reactive oxygen species
- TMRM:
-
Tetramethylrhodamine
- Cy3:
-
Cyanine 3
- APC:
-
Allophycocyanine
- FITC:
-
Fluorescein isothiocyanate
References
EER Cancer Stat Facts: Pancreatic Cancer. National Cancer Institute. Bethesda MD. https://seer.cancer.gov/statfacts/html/pancreas.html
Siegel RL, Miller KD, Fuchs HE, Jemal A (2021) Cancer statistics. CA Cancer J Clin 71:7–33. https://doi.org/10.3322/caac.21654
Cahalan MD, Chandy KG (2009) The functional network of ion channels in T lymphocytes. Immunol Rev 231:59–87. https://doi.org/10.1111/j.1600-065X.2009.00816
Feske S, Wulff H, Skolnik EY (2015) Ion channels in innate and adaptive immunity. Annu Rev Immunol 33:291–353. https://doi.org/10.1146/annurev-immunol-032414-112212
Pardo LA, Stuhmer W (2014) The roles of K(+) channels in cancer. Nat Rev Cancer 14:39–48. https://doi.org/10.1038/nrc3635
Bielanska J, Hernandez-Losa J, Perez-Verdaguer M, Moline T, Somoza R, Ramon YCS, Condom E, Ferreres JC, Felipe A (2009) Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr Cancer Drug Targets 9:904–914
Arcangeli A, Becchetti A (2015) Novel perspectives in cancer therapy: targeting ion channels. Drug resistance updates : Reviews and Commentaries in antimicrobial and anticancer Chemotherapy 21–22:11–19. https://doi.org/10.1016/j.drup.2015.06.002
Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabo I (2012) Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med 4:577–593. https://doi.org/10.1002/emmm.201200235
Leanza L, Romio M, Becker KA, Azzolini M, Trentin L, Manago A, Venturini E, Zaccagnino A, Mattarei A, Carraretto L et al (2017) Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell 31:516-531.e510. https://doi.org/10.1016/j.ccell.2017.03.003
Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa-Itoh K, Bogers JFM et al (2019) Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat Commun 10:4341. https://doi.org/10.1038/s41467-019-12331-1
Szabo I, Bock J, Grassmé H, Soddemann M, Wilker B, Lang F, Zoratti M, Gulbins E (2008) Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc Natl Acad Sci U S A 105:14861–14866. https://doi.org/10.1073/pnas.0804236105
Szabò I, Soddemann M, Leanza L, Zoratti M, Gulbins E (2011) Single-point mutations of a lysine residue change function of Bax and Bcl-x(L) expressed in Bax- and Bak-less mouse embryonic fibroblasts: novel insights into the molecular mechanisms of Bax-induced apoptosis. Cell Death Differ 18:427–438. https://doi.org/10.1038/cdd.2010.112
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discovery 12:931–947. https://doi.org/10.1038/nrd4002
Wahl DR, Byersdorfer CA, Ferrara JL, Opipari AW Jr, Glick GD (2012) Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol Rev 249:104–115. https://doi.org/10.1111/j.1600-065X.2012.01148.x
Hannun YA, Obeid LM (2018) Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 19:175–191. https://doi.org/10.1038/nrm.2017.107
Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, Kolesnick R, Gulbins E (2001) CD95 signaling via ceramide-rich membrane rafts. J Biol Chem 276:20589–20596. https://doi.org/10.1074/jbc.M101207200
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J et al (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J 18:5252–5263. https://doi.org/10.1093/emboj/18.19.5252
Huwiler A, Johansen B, Skarstad A, Pfeilschifter J (2001) Ceramide binds to the CaLB domain of cytosolic phospholipase A2 and facilitates its membrane docking and arachidonic acid release. FASEB J 15:7–9. https://doi.org/10.1096/fj.00-0370fje
Dobrowsky RT, Hannun YA (1993) Ceramide-activated protein phosphatase: partial purification and relationship to protein phosphatase 2A. Adv Lipid Res 25:91–104
Muller G, Ayoub M, Storz P, Rennecke J, Fabbro D, Pfizenmaier K (1995) PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J 14:1961–1969
Huwiler A, Fabbro D, Pfeilschifter J (1998) Selective ceramide binding to protein kinase C-alpha and -delta isoenzymes in renal mesangial cells. Biochemistry 37:14556–14562. https://doi.org/10.1021/bi981401i
Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, Szulc ZM, Ruvolo P, Kornblau S, Andreeff M et al (2016) Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128:1944–1958. https://doi.org/10.1182/blood-2016-04-708750
Hannun YA, Loomis CR, Merrill AH Jr, Bell RM (1986) Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J Biol Chem 261:12604–12609
Newton J, Lima S, Maceyka M, Spiegel S (2015) Revisiting the sphingolipid rheostat: evolving concepts in cancer therapy. Exp Cell Res 333:195–200. https://doi.org/10.1016/j.yexcr.2015.02.025
van der Hoeven D, Cho KJ, Zhou Y, Ma X, Chen W, Naji A, Montufar-Solis D, Zuo Y, Kovar SE, Levental KR et al (2018) Sphingomyelin metabolism is a regulator of K-Ras function. Mol Cell Biol 38. https://doi.org/10.1128/mcb.00373-17
Liou GY, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P (2016) Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep 14:2325–2336. https://doi.org/10.1016/j.celrep.2016.02.029
Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H et al (2012) K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res 22:399–412. https://doi.org/10.1038/cr.2011.145
Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, Lu W, Liu J, Pang X, Liu M (2016) Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun 7:11363. https://doi.org/10.1038/ncomms11363
Qiu H, Edmunds T, Baker-Malcolm J, Karey KP, Estes S, Schwarz C, Hughes H, van Patten SM (2003) Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J Biol Chem 278:32744–32752. https://doi.org/10.1074/jbc.M303022200
Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, Li PL (2007) Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal 9:817–828. https://doi.org/10.1089/ars.2007.1509
Zhao Y, Kalari SK, Usatyuk PV, Gorshkova I, He D, Watkins T, Brindley DN, Sun C, Bittman R, Garcia JG, Berdyshev EV, Natarajan V (2007) Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J Biol Chem 282:14165–14177. https://doi.org/10.1074/jbc.M701279200
Verhaegh R, Becker KA, Edwards MJ, Gulbins E (2020) Sphingosine kills bacteria by binding to cardiolipin. J Biol Chem 295:7686–7696. https://doi.org/10.1074/jbc.RA119.012325
French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD (2010) Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther 333:129–139. https://doi.org/10.1124/jpet.109.163444
Acknowledgements
The authors thank pHionic consortium for providing the human PDAC cell lines. I.S is grateful also to the Italian Association for Cancer Research. Publication costs were supported by the University of Duisburg-Essen.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was supported by grant WWCR22-0348 to IS and EG.
Author information
Authors and Affiliations
Contributions
SHP, GCW, SAA, IS, and EG planned the studies. SHP, GCW, and EG performed in vivo studies. SK, BW, and YW did fluorescence microscopy studies. SK and EG transfected cells. SK did selection and cell culture. EG performed measurements of sphingosine, ceramide, enzyme activities, flow cytometry, transfections, and the studies on isolated mitochondria. AM synthesized PAPTP. All authors read the manuscript and commented on it.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were approved by the University of Cincinnati Ethics Committee and the Institutional Animal Care and Use Committee.
No human studies were performed.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, S.H., Wilson, G.C., Wu, Y. et al. Sphingosine is involved in PAPTP-induced death of pancreas cancer cells by interfering with mitochondrial functions. J Mol Med 102, 947–959 (2024). https://doi.org/10.1007/s00109-024-02456-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-024-02456-2