
Abstract
Cystinuria is a rare genetic disorder characterized by defective l-cystine reabsorption from the renal proximal tubule, resulting in abnormally high concentrations of L-cystine and subsequent l-cystine crystallization and stone formation in urine. l-Cystine diamides have shown great promise as inhibitors of l-cystine crystallization. The free α-amino groups in l-cystine diamides have previously been shown to be necessary for l-cystine crystallization inhibitory activity. In this study, three additional series of l-cystine diamide analogs were designed to explore further the structure-activity relationships for l-cystine crystallization inhibition. It has been demonstrated that the middle disulfide bond is required for optimal l-cystine crystallization inhibitory activity, and the only regions that can be modified are the two terminal amides. The presence of another basic amine 2–3 atoms away from the amide nitrogen is also critical for optimal activity. Disulfide exchange was found to be the main metabolic pathway resulting in the formation of two molecules of the active mixed disulfide metabolite from a single l-cystine diamide. l-Cystine diamides have the potential to be developed into a much-needed therapy for cystinuria.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cystinuria, as an autosomal recessive disorder, results from the dysfunction in the transport of l-cystine amino acid in the renal proximal tubule [1,2,3,4,5,6,7]. It is classified into Type A or Type B based on the mutations found on the two heterodimeric transporter subunits. Mutations in SLC3A1, encoding rBAT subunit, are classified as Type A, whereas mutations in SLC7A9, encoding b0,+AT subunit, are classified as Type B. Although the two genotypes are distinctly different, their clinical phenotypes are similar [5].
Globally, the prevalence of cystinuria varies from 1 in 2500 in Israeli Jews of Libyan origin to 1 in 100,000 in Sweden, averaging worldwide around 1 in 7000 [2, 3]. The prevalence in the US population is estimated to be 1 in 15,000. In patients with cystinuria, the excretion of l-cystine in the urine is estimated to be >400 mg/day, as compared to <30 mg/day in normal subjects. The clinical consequence of high levels of the poorly soluble l-cystine in these patients’ urine is the precipitation of l-cystine to form urinary tract stones, primarily in the kidneys and less often in the bladder [8, 9]. While the incidence is equal between the sexes, males generally experience more severe effects [5].
Current clinical management of cystinuria is limited. Patients are advised to drink 3–4 liters of fluid to help reduce the concentration of free l-cystine in urine. They can also take citrate or bicarbonate to increase their urinary pH to help make l-cystine more soluble [10, 11]. Two most common medications used for the treatment of cystinuria are tiopronin and D-penicillamine, which act through a disulfide exchange between l-cystine and the free thiol present in these drugs forming the more soluble mixed disulfides [10]. However, these therapies are known to have limited efficacy and are associated with frequent side effects that lead to patient compliance issues [11, 12]. We, through molecular mimicry, are attempting to develop new, safer, and more efficacious drugs for cystinuria without the issues associated with current therapies [13,14,15,16,17].
In the present study, we explored further the structure-activity relationships (SAR) of our previously reported l-cystine diamide, l-cystine bis(N′-methylpiperazide) (LH708, 2) [13, 14]. These SAR studies provided critical information on the structural components required for potency as l-cystine crystallization inhibitors and led to the discovery of 8-l-cystinyl bis(1,8-diazaspiro[4.5]decane) (LH1753, 3i), as a promising orally bioavailable inhibitor of l-cystine crystallization for cystinuria [16].
Results and discussion
Analog design principles
As we reported previously [13, 14], l-cystine diamides like l-cystine bis(N′-methylpiperazide) (LH708, 2) have been shown to be chemically and metabolically more stable than l-CDME (1). LH708 has been shown to be active in preventing l-cystine stone formation in a Slc3a1-knockout mouse model of cystinuria after oral gavage. In our earlier SAR study, we demonstrated that the smallest possible modification like methylation of the α-amino groups in l-cystine diamides would lead to their inactivation as shown by absence of any significant effects on the apparent aqueous solubility of l-cystine as compared to water control [14]. Therefore, in our current SAR study, we avoided any modification of α-amino groups in l-cystine diamides but focused on optimization of the amide terminal groups and the disulfide bond in the middle as shown in Fig. 1.

The regions of LH708 (2) for structural optimization and the various analogs designed to explore the structure-activity relationships
We designed the first series of compounds (3a-3j) focusing on modifying the N4-methyl substituent on the piperazine terminal groups of LH708 in addition to methylation on the piperazine ring and replacement of the piperazine ring with a 4-aminopiperidine or a spiro bicyclic diamine. A second series of analogs (4a-4h) was designed with the replacement of cyclic piperazine with linear ethylene or propylene diamines with the exception of 4e, where the terminal nitrogen is part of a lactam instead of an amine to evaluate the requirement of terminal basic nitrogen for the inhibition of l-cystine crystallization. A third series (5a-5i) was designed to focus on replacing the disulfide bond with a sulfide or amide bioisostere or homologation of the disulfide to investigate the importance of the disulfide bond on the inhibition of l-cystine crystallization.
Chemical synthesis
Synthesis of l-cystine diamides 3a-3j and 4a-4h. As illustrated in Scheme 1, a straightforward two-step synthesis was carried out to synthesize l-cystine diamides 3a-3j and 4a-4h: amide coupling between various amines and Nα,Nα’-di-Boc-l-cystine (6) facilitated by PyAOP, followed by Boc deprotection using 4 N HCl in 1,4-dioxane, with overall yields ranging from 33 to 94%.

Synthesis of L-cystine diamides 3a-3j and 4a-4h with modification of the terminal amidesa. aReagents and conditions: (i) Amines, PyAOP, DIPEA, DCM, r.t., 0.5–3 h, 33–100%; (ii) 4 N HCl in 1,4-Dioxane, MeOH, 0 °C, 1–2 h; r.t., 1 h, 45–100%
Synthesis of compounds 5a-5b and 5f-5g by replacing [S-S] with sulfide bioisosteres [CH2S] or [CH2SCH2]. As shown in Scheme 2, the synthesis of compounds 5a-5b and 5f-5g started with l-homocystine (9) and either Boc-Ser-OMe (13a) or homoserine (12b). 9 was converted to Boc-Hcy-OMe (11) in three steps with 90% overall yield: methyl esterification with thionyl chloride and methanol, followed by Boc protection, and finally reduction of the disulfide bridge using triphenylphosphine. Tosylation of 13a led to intermediate 14a in 98% yield. Intermediate 14b was obtained by first Boc protection of the α-amino group of homoserine (12b), followed by methylation of the α-carboxylate group with methyl iodide, and finally tosylation of the side chain hydroxyl group of Boc-Hse-OMe (13b), with an overall yield of 29%. Nucleophilic substitution of the tosylate group of 14a-14b with the thiol intermediate 11 in the presence of sodium hydride afforded intermediates 15a-15b (yield = 48–67%), which were successively subjected to lithium hydroxide-mediated methyl ester hydrolysis, amide coupling with either N-methylpiperazine or N,N-dimethylethylenediamine facilitated by HATU (35–65% yield over two steps), and Boc deprotection to yield compounds 5a-5b and 5f-5g.

Synthesis of compounds 5a-5b and 5f-5g by replacing [S-S] with sulfide bioisosteres [CH2S] or [CH2SCH2]a. aReagents and conditions: (i) SOCl2, MeOH, 0 °C to r.t., overnight; (ii) Boc2O, Na2CO3, 1,4-Dioxane/H2O, 0 °C to r.t., 6 h; (iii) PPh3, NaOAc,AcOH, MeOH, H2O, 60 °C, 75 min, 90% over three steps; (iv) Boc2O, NaOH, MeCN/H2O (1:1), 0 °C to r.t., 1.5 h; (v) MeI, DMF, 0 °C to r.t.,5 h; (vi) TsCl, Pyridine, DCM, 0 °C, 15 min; r.t., 45 min, 98% for 14a and 29% over three steps for 14b; (vii) NaH, DMF, 0 °C, 0.5 h; r.t., 3–4 h, 48–67%; (viii) LiOH, THF/H2O (1:1), 0 °C to r.t., 1–2 h; (ix) N-Methylpiperazine or N,N-Dimethylethylenediamine, HATU, DIPEA, DMF,r.t., 15 min to 3 h, 37–65% over two steps; (x) 4 N HCl in 1,4-Dioxane, MeOH, 0 °C, 1–2 h; r.t., 5 h, 29–87%
Synthesis of compounds 5c, 5d, 5h and 5i by replacing [S-S] with amide bioisosteres [CONH] and [CH2CONH]. Scheme 3 presents the synthesis of compounds 5c, 5d, 5h and 5i. Boc-Ser(OTs)-OMe (14a) was reacted with sodium azide to replace the tosylate functionality with an azide group forming 18, which was then catalytically hydrogenated in the presence of palladium on carbon to give the primary amine intermediate 19. 19 was coupled with the side chain carboxylic acid of either Boc-Asp-OMe using HATU or Boc-Glu-OMe using PyAOP, leading to the dimethyl esters 15c-15d (overall yield = 18% over three steps). The following three steps were similar to those shown in Scheme 2: dimethyl esters were hydrolyzed with lithium hydroxide, and the resulting dicarboxylic acids 16c-16d were activated using PyAOP and coupled with either N-methylpiperazine or N,N-dimethylethylenediamine to give the diamides 17c, 17d, 17h, and 17i with yields ranging 43–88% over the two steps. These diamides were finally Boc deprotected to give the target compounds 5c, 5d, 5h, and 5i.

Synthesis of compounds 5c, 5d, 5h and 5i by replacing [S-S] with amide bioisosteres [CONH] and [CH2CONH]a. aReagents and conditions: (i) NaN3, DMF, 50 °C, 3 h, 43%; (ii) H2 (1 atm), 10% Pd/C, MeOH, r.t., 3 h; (iii) Boc-Asp-OMe, HATU, DIPEA, DMF, r.t.,130 min, 41% over two steps for 15c, or Boc-Glu-OMe, PyAOP, DIPEA, DCM, r.t., 1.5 h, 42% over two steps for 15d; (iv) LiOH, THF/H2O(1:1), 0 °C to r.t., 1–3 h; (v) N-Methylpiperazine or N,N-Dimethylethylenediamine,PyAOP, DIPEA, DCM, r.t., 30 min to 2 h, 43–88% over two steps; (vi) 4 N HCl in1,4-Dioxane, MeOH, 0 °C, 1–2 h; r.t., 1 h, 55–98%.
Synthesis of l-homocystine diamide 5e. Starting with Boc-protected l-homocystine, 5e was made using the same two step sequence as shown in Scheme 1: amide coupling with N-methylpiperazine facilitated by PyAOP, and Boc deprotection using 4 N HCl in 1,4-dioxane with an overall yield of about 50%.
Synthesis of mixed disulfide 25 as the major metabolite of LH708. The synthesis of the mixed disulfide 25 is described in Scheme 4. Di-Boc-protected LH708 (20), reported previously [13], was reduced using triphenylphosphine to give Boc-l-cysteine N-methylpiperazide (21). The fully protected mixed disulfide was synthesized via activation of the thiol in 23 using 1-chlorobenzotriazole (BtCl), followed by addition of thiourea to quench excess BtCl, and finally addition of Boc-Cys-OtBu (21), which was obtained in 53% yield from di-Boc-l-cystine (6) in two steps: treatment with t-butyl 2,2,2-trichloroacetimidate and then reduction with triphenylphosphine. Final Boc deprotection and t-Bu removal of 24 using 4 N HCl in 1,4-dioxane afforded 25 as the major metabolite of LH708 with an overall yield of 31%.

Synthesis of the mixed disulfide 25 as the major metabolite of LH708a. aReagents and conditions: (i) PPh3, NaOAc, AcOH, MeOH, H2O, 60 °C, 75 min, 84–87%; (ii) TBTA, DCM, r.t., 5 h, 61%; (iii) BtCl, BtaH, DCM, ‒78 °C, 40 min; thiourea, THF, ‒78 °C, 15 min; 23, DCM, ‒78 °C to r.t., overnight, 58%; (iv) 4 N HCl in 1,4-Dioxane, 0 °C, 1 h; r.t.,overnight, 100%.
Effect on the inhibition of l-cystine crystallization and structure-activity relationships
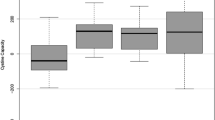
The effect of the various structural modifications on their ability to inhibit l-cystine crystallization was evaluated using a convenient crystallization inhibition assay we recently published by incubating a supersaturated solution of l-cystine in the presence of varying concentrations of tested inhibitors for 72 h and then measuring the concentrations of l-cystine remaining in the supernatant after centrifugation [18]. A preliminary screening of potential inhibitors at two concentrations (2 and 10 µM) is usually performed as shown in Fig. 2A before a full dose response curve study is conducted to derive EC50 for the more potent inhibitors as we have reported previously and shown in Fig. 2B [14]. EC50 is calculated as the inhibitor concentration required to achieve 50% of the maximal increase in apparent aqueous solubility of l-cystine. EC50 is the preferred activity metric after we had standardized the conditions used in our crystallization inhibition assays. The EC50 is similar in value to the EC2x, which we were using in earlier studies to represent the concentration required to double the l-cystine concentration in solution due to inconsistent maximum of plateau reached with different inhibitors because of the inherent instability of supersaturated solutions prepared [13].

l-Cystine crystallization inhibition assays: (A) initial screening of inhibitors at two concentrations (2 and 10 μM) relative to water control and the concentration of supersaturated solution (SSS) of l-cystine and (B) representative dose-response curves with LH1753 and LH1754 as compared to LH708 and l-CDME
Effect of modification of terminal amide groups on the aqueous solubility of l-cystine. As shown in Table 1, the terminal amide groups in l-cystine diamides have shown the greatest effect on the aqueous solubility of l-cystine. We varied the substituent on the N4-piperazine in compounds 3a to 3e, and found that N4-phenyl substitution decreased the l-cystine crystallization inhibition activity by >10-fold and N4-cyclopropyl substitution decreased the crystallization inhibition activity by about 38% while the other alkyl and cycloalkyl groups on the N4-piperazine had much less significant effect with EC50 values ranging between 32 and 53 nM. The significant decrease in l-cystine crystallization inhibition activity caused by a cyclopropyl substitution in 3c and the phenyl substitution in 3e could be attributed to the significantly reduced basicity of N4-nitrogen by 1–2 pKa units when it is substituted by a cyclopropyl group [19] or by 4–5 pKa units when it is conjugated to a phenyl ring. Dimethyl substitution next to N1-nitrogen on the piperazine ring as in 3 f also adversely affected its crystallization inhibition activity by about 50%, increasing its EC50 to 108 nM. Moving the N4-nitrogen exocyclic as in 4-amino-piperidine analogs 3g-3i maintained the strong crystallization inhibition activity, leading to the discovery of LH1753 (3i), which is the most active inhibitor of l-cystine crystallization inhibition we discovered so far and which we recently highlighted in a letter of communication [16]. Compound 3j as a structure isomer of 3i with 8-l-cystinyl bis(1,8-diazaspiro[4.5]decane) piperidine diamide changed to the 1-l-cystinyl bis(1,8-diazaspiro[4.5]decane) pyrrolidine diamide, however, had significantly reduced l-cystine crystallization inhibition by almost 5-fold, suggesting that a specific conformation of the basic amine-containing terminal amide group is needed for optimal crystallization inhibition.
We next focused on replacing the cyclic piperazine terminal groups with linear ethylene or propylene diamines as in 4a-4h. As shown in Table 1, all of the linear ethylene or propylene diamines retained most of the l-cystine crystallization inhibition activity with EC50 ranging between 80 and 160 nM. When the terminal amine is dimethyl amine, there seems to be a slightly improved inhibition activity for the longer propylene diamine with EC50 of 163 vs 114 nM for 4a and 4 f, respectively. When the terminal amine is a cyclic pyrrolidine or morpholine, there seems to be an improved inhibition activity for the shorter ethylene diamines. When the pyrrolidine is replaced by a non-basic pyrrolidine-2-one, the resulting analog 4e lost almost all of its crystallization inhibition activity, further supporting the earlier conclusion that another basic center in the terminal amides is required to maintain l-cystine crystallization inhibition activity.
Effect of bioisosteric replacement of the disulfide bond on the aqueous solubility of l-cystine. Despite its plasma stability, the disulfide bond is known to be susceptible to intracellular reduction due to the overall intracellular reducing environment [20]. We wanted to explore the possibility of replacing the disulfide bond with other bioisosteres such as sulfides and amides that cannot be easily reduced. Two series of the analogs were used for this purpose: one cyclic piperazine series 5a-5e and another acyclic N,N-dimethyl ethylenediamine series 5f-5i. In the cyclic piperazine series, one analog has one of the two sulfur atoms in the disulfide bond replaced with a methylene as in 5a. A second analog has another methylene added to the other side of sulfur to make up for the reduced sulfide bond length as in 5b. The third and fourth analogs have the disulfide bond replaced by an amide as in 5c or by a methylene amide as in 5d. In a fifth analog, the disulfide bond is further extended by a methylene on each side to give 5e. As shown in Fig. 2A and Table 2, any such replacement of the disulfide bond led to a complete loss of l-cystine crystallization inhibition activity. Similar complete loss of activity was also observed in the acyclic N,N-dimethyl ethylenediamine series 5f-5i. These results suggest that the disulfide bond plays an important role in the nucleation process of l-cystine and its subsequent crystal growth. Likewise, for an effective inhibitor of l-cystine crystallization, the disulfide bond remains a critical structural component needed to maintain the interactions with l-cystine molecules in the nuclei or on the crystal surfaces to interfere with the l-cystine nucleation or crystal growth. This is consistent with the crystal growth inhibition studies using AFM as reported previously [17].
In vivo metabolism and pharmacokinetics of l-cystine diamides
Based on the structure of LH708 (2), disulfide exchange and demethylation were predicted to be its main metabolic pathways as shown in Fig. 3. We already synthesized two of these metabolites, 25 (M1, LH1727, LH708M1) as a mixed disulfide between LH708 and l-cystine and 3a (M2, LH1726) as a demethylated LH708. Our crystallization inhibition assays demonstrated that both 25 and 3a are active with EC50 within about twofold of their parent.

Expected metabolites of l-cystine diamide LH708 and their activity in inhibiting the crystallization of l-cystine. EC50 refers to the inhibitor concentration required to achieve 50% of the maximal apparent aqueous solubility of l-cystine
In our in vivo PK studies, we found that disulfide exchange was the main metabolic pathway observed leading to the formation of LH1727 as the major metabolite detected. As shown in Fig. 4A, after a single iv or oral dose of LH708 to Slc3a1 knockout mice, we detected significant amounts of the mixed disulfide LH1727 as the major metabolite in both plasma and urine samples collected. This has been shown to be similar for several other l-cystine diamides including LH1753 [16].

Pharmacokinetic analysis of LH708 in 3-months-old Slc3a1 KO male mice. A Changes in plasma concentration of LH708 and its major metabolite LH1727 over time after p.o. (oral) and i.v. administration of LH708 at a single dose of 150 µmol/kg. B Concentration of LH708 and its major metabolite LH1727 in urine samples collected 24 h after an oral and i.v. administration of LH708 at a single dose of 150 µmol/kg
Figure 4A shows the time course of plasma concentration of LH708 and its major metabolite LH1727 after a single dose of LH708, and Fig. 4B shows their concentrations in urine samples collected 24 h after dosing. The oral bioavailability calculated for the parent LH708 was 2.3% while that for the major active metabolite was 25% based on the plasma levels of LH708 and LH1727 with a combined oral bioavailability of 18%. Micromolar concentrations of LH708 and its metabolite LH1727 were found in urine samples collected at 24 h after administration. Interestingly, LH708 concentration was higher than that of LH1727 in urine samples collected 24 h after administration even though LH708 has a short plasma half-life of ~10 min and the metabolite LH1727 has a plasma half-life of 1 h. The finding of disulfide exchange as the major route of metabolism of l-cystine diamides makes it possible to produce two molecules of the active mixed disulfide metabolite from one molecule of the parent l-cystine diamide, thus providing an advantage for the disulfide-containing l-cystine crystallization inhibitors.
Crystal growth inhibition using atomic force microscopy (AFM)
For the AFM studies, we tested three inhibitors, LH1726, LH1729, and LH1753 selected based on their EC50 values. Figure 5 shows the growth of the l-cystine {0001} face growing in the absence of inhibitors and in the presence of 45 µM of LH1726, LH1729, and LH1753, while Fig. 6 shows in situ AFM images of {0001} l-cystine crystal faces growing in the absence of inhibitor and in the presence of 15, 30, and 45 µM of LH1729. AFM measurements were performed in areas containing active growth hillocks. Step roughening of the growth hillocks indicates the effectiveness of inhibitors. The effects of the various cystine diamides on l-cystine crystallization are summarized in Table 3.

In situ AFM images of {0001} L-cystine crystal faces (2 mM L-cystine) growing in the absence of inhibitors (A), and in the presence of 45 µM (B) LH1726, (C) LH 1729, and (D) LH 1753. The area of all images is 6 µm × 6 µm. Close inspection of the images in (B–D) reveals step roughening not observed in (A), signaling binding of the inhibitor to sites on the step edges

In situ AFM images of {0001} L-cystine crystal faces (2 mM L-cystine) growing in the absence of inhibitor (A), and in the presence of 15 µM (B), 30 µM (C), and 45 µM (D) LH1729. The area of each image is 6 µm × 6 µm. The images reveal changes in morphology and an increase in step edge roughness as the concentration is increased
All three compounds inhibited l-cystine crystal growth, as observed from step roughening and decrease of {1010} step velocity from in situ AFM measurements on {0001} growth hillocks. LH1726 was less effective than the other two compounds (LH1729 and LH1753), however. The V/Vo ratios corresponded somewhat with the EC50 values measured for the respective inhibitors (Table 3). Some of this data has been presented in a recent communication [16]. The inhibition of {1010} step advancement on the {0001} is a consequence of the Cabrera−Vermilyea mechanism of impurity action [21, 22], in which inhibitor molecules at the step edge act as stoppers that block step propagation through pinning [15, 23, 24]. Pinning induces curvature of the step edge as it advances between the adsorbates. This curvature increases the step edge energy, tantamount to a decrease in supersaturation, as described by the Gibbs−Thomson law. Increasing the inhibitor concentration decreases the average distance between pinning sites, further increasing the step edge curvature. When the separation between posts is smaller than the diameter of the critical nucleus, then step curvature is sufficiently high to stop growth. It should be noted, however, that while step growth inhibition on the {0001} face is diagnostic of inhibitor effectiveness, L-cystine growth also is inhibited by inhibitor binding to steps and kinks on the {10\(\bar{1}\)0} faces as well. This mechanism may actually be the dominant one, but high-quality AFM imaging of the {10\(\bar{1}\)0} faces was precluded by the small size and roughness. Instead, the inhibition on the{10\(\bar{1}\)0} faces needed to be measured by measurements of growth on bulk crystals in the presence of inhibitors [17].
Conclusion
In this study, we explored further the structure-activity relationships of l-cystine diamides as l-cystine crystallization inhibitors. Previously we have shown that the α-amino groups in l-cystine diamides need to remain unsubstituted to possess any l-cystine crystallization inhibitory activity. Now through this study, we further demonstrated that the middle disulfide bond needs to be retained for optimal l-cystine crystallization inhibitory activity. The only regions that can be modified are the two terminal amides. It was also clearly demonstrated that another basic amine 2–3 atoms away from the amide nitrogen is needed for optimal activity. In vivo metabolism and pharmacokinetic studies indicate that the major metabolic pathway for the l-cystine diamides is the disulfide exchange which provides the ability to produce two molecules of the active mixed disulfide metabolite from a single l-cystine diamide. Our l-cystine diamides are designed to be highly water soluble so that they, upon absorption after oral administration, would filter through the kidney tubules and reach the urine, where the site of action is. l-Cystine diamides are showing great promise as a potential therapy to prevent l-cystine crystallization and thus kidney stone formation in Slc3a1-knockout mouse models of cystinuria.
Experimental
General chemistry
All reagents and solvents were purchased as ACS/HPLC grade. All reactions were monitored by TLC and/or by LC/MS using a Shimadzu 2010 series HPLC and/or an Agilent 1200 series HPLC coupled with an Agilent G6140A Quadrupole MS system (Santa Clara, CA). LC/MS analyses were achieved using 0.1% formic acid in water and 0.1% formic acid in methanol as mobile phase A and B, respectively. The column temperature was maintained at 40 °C, and the UV wavelength for detection was set at 280 nm. The gradient was set from 10 to 90% B over 5 min at a flow rate of 0.8 mL/min. Compound purification was carried out using flash column chromatography on a Teledyne ISCO CombiFlash system (Thousand Oaks, CA). Lyophilization of final compounds was performed on a VirTis freezemobile freeze dryer (SP Scientific, Warminster, PA). 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded at ambient temperature on a Bruker UltrashieldTM 400 MHz Multinuclear NMR spectrometer (Billerica, MA) and calibrated using residual undeuterated solvents (chloroform, DMSO, water) as the internal reference. All chemical shifts are reported in parts per million (ppm). All coupling constants are reported in Hz. Spin multiplicities were abbreviated as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), m (multiplet), and br (broad). High-resolution mass spectrometry (HRMS) experiments were conducted by the Center for Integrative Proteomics Research (CIPR) at Rutgers University using a Thermo LTQ Orbitrap Velos ETD with Dionex UltiMate 3000 nano-flow 2D LC system (Thermo Fischer Scientific Inc., Waltham, MA).
General amide coupling procedure for the synthesis of intermediates 7a-j and 8a-h
To a suspended mixture of Boc-L-cystine-OH in anhydrous dichloromethane was added DIPEA (6 equiv.), leading to a clear solution. Subsequently, the amine (3 equiv.) and PyAOP (2 equiv.) were added to the solution, and the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with water and extracted with dichloromethane three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography using pre-packed normal phase silica gel column.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bispiperazide (7a)
The compound was eluted with 2% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a light yellow solid (74 mg, yield: 60%). 1H NMR (400 MHz, CDCl3): δ 5.44 (d, 2H, J = 8 Hz), 4.93 (t, 2H, J = 8 Hz), 3.67-3.40 (m, 16H), 3.01 (br, 4H), 1.47 (s, 18H), 1.43 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 169.2, 155.0, 154.4, 80.4, 49.1, 45.8, 42.2, 41.5, 28.3; LC-MS (ESI+): m/z 777.1 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-tert-butylpiperazide) (7b)
The compound was eluted with 5% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a light yellow solid (80 mg, yield: 73%). 1H NMR (400 MHz, CDCl3): δ 5.46 (d, 2H, J = 8 Hz), 4.93 (br, 2H), 3.64 (br, 8H), 3.02-2.94 (m, 4H), 2.56 (br, 8H), 1.42 (s, 18H), 1.06 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 168.6, 155.0, 80.0, 49.1, 46.4, 45.5, 42.9, 41.8, 28.4, 25.8; LC-MS (ESI+): m/z 689.2 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-cyclopropylpiperazide) (7c)
The compound was eluted with 1.2% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a yellow solid (70 mg, yield: 67%). 1H NMR (400 MHz, CDCl3): δ 5.47 (d, 2H, J = 8 Hz), 4.93 (t, 2H, J = 8 Hz), 3.56 (br, 8H), 3.03-2.96 (m, 4H), 2.62-2.57 (m, 8H), 1.60 (br, 2H), 1.41 (s, 18H), 0.46-0.40 (m, 8H); 13C NMR (100 MHz, CDCl3): δ 168.8, 155.0, 80.1, 53.1, 49.1, 45.8, 41.8, 38.2, 28.4, 5.9; LC-MS (ESI+): m/z 657.1 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-cyclopentylpiperazide) (7d)
The compound was eluted with 5% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a light yellow solid (85 mg, yield: 75%). 1H NMR (400 MHz, CDCl3): δ 5.44 (d, 2H, J = 8 Hz), 4.91 (br, 2H), 3.75 (br, 8H), 3.15-3.01 (m, 2H), 2.98 (br, 4H), 2.68 (br, 8H), 1.89 (br, 4H), 1.74 (br, 4H), 1.58–1.48 (m, 8H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 168.8, 155.1, 80.3, 67.6, 52.1, 51.6, 49.0, 45.8, 41.5, 29.7, 28.3, 23.9; LC-MS (ESI+): m/z 713.2 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-phenylpiperazide) (7e)
The compound was eluted with 1.7% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a dark yellow solid (84 mg, yield: 73%). 1H NMR (400 MHz, CDCl3): δ 7.27-7.23 (m, 4H), 6.89 (d, 6H, J = 8 Hz), 5.48 (d, 2H, J = 8 Hz), 5.00 (br, 2H), 3.77 (br, 8H), 3.17 (br, 8H), 3.02 (br, 4H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 169.0, 155.0, 150.8, 129.3, 120.6, 116.6, 80.2, 55.9, 49.8, 45.8, 42.3, 41.7, 28.4; LC-MS (ESI+): m/z 729.1 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(4′-tert-butoxycarbonyl-2’,2’-dimethylpiperazide) (7f)
The compound was eluted with 2.2% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a dark yellow solid (80 mg, yield: 63%). 1H NMR (400 MHz, CDCl3): δ 5.32 (br, 2H), 4.88 (br, 2H), 3.88–3.48 (m, 12H), 2.99 (br, 4H), 1.46 (s, 18H), 1.41 (s, 18H), 1.36 (s, 12H); 13C NMR (100 MHz, CDCl3): δ 170.9, 155.1, 154.8, 80.3, 56.1, 54.1, 50.6, 49.3, 45.3, 44.0, 39.8, 28.4, 25.0; LC-MS (ESI+): m/z 833.0 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(4’-(dimethylamino)piperidineamide) (7g)
The compound was eluted with 7% methanol in dichloromethane with 0.3 M ammonia and concentrated under reduced pressure to give a colorless oil (68 mg, yield: 94%). 1H NMR (400 MHz, CDCl3): δ 5.46 (br, 2H), 4.93 (br, 2H), 4.54 (br, 2H), 4.07 (br, 2H), 3.09-2.92 (m, 6H), 2.67-2.58 (m, 2H), 2.34 (br, 2H), 2.24 (s, 12H), 1.85 (br, 4H), 1.40 (s, 22H); 13C NMR (100 MHz, CDCl3): δ 168.6, 155.0, 80.0, 74.8, 61.8, 53.4, 49.1, 44.8, 41.6, 29.0, 28.3; LC-MS (ESI+): m/z 661.4 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(4’-(1”-pyrrolidinyl)piperidine) (7h)
The compound was eluted with 6% methanol in dichloromethane with 0.3 M ammonia and concentrated under reduced pressure to give a colorless oil (70 mg, yield: 90%). 1H NMR (400 MHz, CDCl3): δ 5.50 (br, 2H), 4.94 (br, 2H), 4.40 (br, 2H), 4.01 (br, 2H), 3.15-2.76 (m, 8H), 2.55 (br, 8H), 2.25 (br, 4H), 1.92 (br, 4H), 1.76 (br, 8H), 1.40 (s, 20H); 13C NMR (100 MHz, CDCl3): δ 168.6, 155.1, 80.0, 74.8, 61.4, 51.4, 49.1, 44.2, 41.1, 31.9, 31.0, 28.3, 23.2.
N,N′-Bis(tert-butoxycarbonyl)-1-l-cystinyl bis(1,8-diazaspiro[4.5]decane)-8-carboxylate tert-butylester (7j)
The compound was eluted with 5% methanol in dichloromethane with 0.3 M ammonia and concentrated under reduced pressure to give a colorless oil (80 mg, yield: 33%). 1H NMR (400 MHz, CDCl3): δ 5.34 (br, 2H), 4.65 (br, 2H), 4.10 (br, 2H), 4.01 (br, 2H), 3.70 (br, 2H), 3.61 (br, 2H), 2.98–2.70 (m, 12H), 2.05-1.86 (m, 8H), 1.41 (d, 36H, J = 8 Hz), 1.25-1.21 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 168.9, 155.2, 154.5, 80.0, 79.4, 65.2, 52.4, 48.7, 41.8, 41.0, 35.0, 32.4, 28.5, 28.3, 22.7.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′,N’-dimethylethyleneamide) (8a)
The compound was eluted with 14% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a yellowish-white solid (250 mg, yield: 95%). 1H NMR (400 MHz, DMSO-d6): δ 8.01 (br, 2H), 7.05 (d, 2H, J = 8 Hz), 4.14 (br, 2H), 3.24 (br, 4H), 3.08 (dd, 2H, J = 12, 4 Hz), 2.83 (dd, 2H, J = 12, 8 Hz), 2.64 (t, 4H, J = 4 Hz), 2.40 (s, 12H), 1.36 (s, 18H); 13C NMR (100 MHz, DMSO-d6): δ 170.4, 155.2, 78.4, 56.8, 53.6, 43.8, 40.5, 35.6, 28.1; LC-MS (ESI+): m/z 581.2 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-pyrrolidinoethyleneamide) (8b)
The compound was eluted with 6% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a white solid (82 mg, yield: 81%). 1H NMR (400 MHz, CDCl3): δ 7.64 (br, 2H), 5.65 (br, 2H), 4.47 (br, 2H), 3.30 (br, 4H), 3.10-2.86 (m, 4H), 2.60 (br, 12H), 1.68 (br, 8H), 1.26 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 170.5, 155.5, 79.9, 54.7, 54.2, 54.0, 44.2, 37.9, 28.3, 23.3; LC-MS (ESI+): m/z 633.3 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-pyrrolidin-2-oneethyleneamide) (8c)
The compound was eluted with 6% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a white solid (82 mg, yield: 81%). 1H NMR (400 MHz, CDCl3): δ 7.94 (br, 2H), 5.58 (br, 2H), 4.60 (br, 2H), 3.52–3.34 (m, 12H), 2.98 (br, 4H), 2.34 (t, 4H, J = 8 Hz), 2.01–1.95 (m, 4H), 1.43 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 175.9, 170.7, 155.6, 80.2, 54.5, 47.3, 44.8, 42.1, 37.1, 31.0, 28.4, 18.0 ppm. LC-MS (ESI+): m/z 661.4 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-piperidinoethyleneamide) (8d)
The compound was eluted with 4% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a yellowish-white solid (95 mg, yield: 90%). 1H NMR (400 MHz, CDCl3): δ 7.65 (br, 2H), 5.65 (d, 2H, J = 8 Hz), 4.53 (br, 2H), 3.38 (br, 4H), 3.10-2.96 (m, 4H), 2.54 (br, 12H), 1.59 (br, 8H), 1.37 (s, 22H) ppm. 13C NMR (100 MHz, CDCl3): δ 170.4, 155.5, 80.0, 57.2, 55.2, 54.2, 43.9, 36.1, 28.3, 25.1, 23.6; LC-MS (ESI+): m/z 661.3 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(N′-morpholinoethyleneamide) (8e)
The compound was eluted with 7% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a white solid (85 mg, yield: 80%). 1H NMR (400 MHz, CDCl3): δ 7.51 (br, 2H), 5.63 (d, 2H, J = 12 Hz), 4.66 (br, 2H), 3.67 (t, 8H, J = 4 Hz), 3.45–3.29 (m, 4H), 3.01-2.97 (m, 4H), 2.50-2.44 (m, 12H), 1.44 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 170.2, 155.7, 80.2, 66.9, 57.5, 54.5, 53.5, 45.3, 36.4, 28.4; LC-MS (ESI+): m/z 665.3 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine-bis(N′,N’-dimethyl-1’,3’-diaminopropaneamide) (8f)
The compound was eluted with 15% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a light yellow solid (55 mg, yield: 58%). 1H NMR (400 MHz, CDCl3): δ 7.90 (br, 2H), 5.61 (d, 2H, J = 8 Hz), 4.69 (br, 2H), 3.33-3.25 (m, 4H), 3.02-2.94 (m, 4H), 2.31 (t, 4H, J = 8 Hz), 2.20 (s, 12H), 1.72-1.64 (m, 4H), 1.45 (s, 18H); 13C NMR (100 MHz, DMSO-d6): δ 170.1, 155.8, 80.1, 57.7, 54.5, 45.9, 45.4, 38.5, 28.4, 27.2; LC-MS (ESI+): m/z 609.2 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(1’-pyrrolidine-3’-aminopropaneamide) (8g)
The compound was eluted with 16% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a yellow solid (85 mg, yield: 81%). 1H NMR (400 MHz, CDCl3): δ 8.35 (br, 2H), 5.78 (br, 2H), 4.40 (br, 2H), 3.60 (br, 4H), 3.30 (br, 4H), 3.18 (br, 4H), 2.93 (br, 4H), 2.02 (br, 12H), 1.31 (s, 18H); 13C NMR (100 MHz, DMSO-d6): δ 171.2, 155.3, 79.7, 54.1, 53.6, 52.9, 42.7, 36.6, 28.3, 25.6, 23.2; LC-MS (ESI+): m/z 661.3 [M + H]+.
N,N′-Bis(tert-butoxycarbonyl)-L-cystine bis(1’-morpholine-3’-aminopropaneamide) (8h)
The compound was eluted with 16% methanol in dichloromethane with 0.1% triethylamine and concentrated under reduced pressure to give a white solid (85 mg, yield: 80%). 1H NMR (400 MHz, CDCl3): δ 7.76 (br, 2H), 5.60 (br, 2H), 4.49 (br, 2H), 3.61 (br, 8H), 3.20-3.15 (m, 4H), 2.88 (br, 4H), 2.40 (br, 12H), 1.68-1.60 (m, 4H), 1.30 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 170.3, 155.6, 80.0, 66.3, 56.5, 54.5, 53.5, 45.0, 38.1, 28.3, 25. 6; LC-MS (ESI+): m/z 693.3 [M + H]+.
General procedure for the synthesis of L-cystine diamides 3a-3i, 4a-4h, and 5a-5i
Respective intermediates 7a-7i, 8a-8h, and 17a-17i were dissolved in methanol and cooled to 0 °C, to which 4 N HCl in 1,4-dioxane (at least 20 equiv.) was added in portions every half an hour over 1–3 h. The reaction mixture was gradually warmed up to room temperature after the last addition of 4 N HCl and stirred at room temperature for 0.5–5 h. The reaction mixture was concentrated under reduced pressure and then purified by recrystallization.
L-Cystine bis(piperazide) tetrahydrochloride (3a)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (6 mg, yield: 85%). 1H NMR (400 MHz, D2O): δ 3.87-4.14 (m, 7H), 3.80–3.69 (m, 2H), 3.52 − 3.28 (m, 13H); 13C NMR (100 MHz, D2O): δ 166.6, 49.7, 42.9, 42.7, 42.5, 39.4, 37.9; LC/MS (ESI+): m/z 377.2 [M + H]+; HRMS (ESI+): m/z calculated for C14H29N6O2S2+ [M + H]+ 377.1788, found 377.1799.
L-Cystine bis(N′-tert-butylpiperazide) tetrahydrochloride (3b)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (21 mg, yield: 84%). 1H NMR (400 MHz, D2O): δ 4.30-4.24 (m, 2H), 3.98-3.57 (m, 9H), 3.57–3.01 (m, 11H), 1.44 (s, 9H), 1.43 (s, 9H); 13C NMR (100 MHz, D2O): δ 166.3, 64.8, 49.7, 45.7, 45.4, 42.9, 39.9, 23.6; LC/MS (ESI+): m/z 489.2 [M + H]+; HRMS (ESI+): m/z calculated for C22H45N6O2S2+ [M + H]+ 489.3040, found 489.3075.
L-Cystine bis(N′-cyclopropylpiperazide) tetrahydrochloride (3c)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (19 mg, yield: 80%). 1H NMR (400 MHz, D2O): δ 4.69 (b, 2H), 3.30–3.86 (m, 20H), 2.91 (b, 2H), 1.03 (s, 8H); 13C NMR (100 MHz, D2O): δ 166.5, 52.0, 51.9, 49.8, 42.6, 39.5, 37.9, 3.6; LC/MS (ESI+): m/z 457.3 [M + H]+; HRMS (ESI+): m/z calculated for C20H37N6O2S2+ [M + H]+ 457.2414, found 457.2444.
L-Cystine bis(N′-cyclopentylpiperazide) tetrahydrochloride (3d)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (25 mg, yield: 76%). 1H NMR (400 MHz, D2O): δ 5.01 – 4.92 (m, 2H), 4.69 – 4.53 (m, 2H), 4.24 (d, 2H, J = 14.7 Hz), 3.84 – 3.57 (m, 8H), 3.46 – 3.11 (m, 10H), 2.28 – 2.09 (m, 4H), 1.85 – 1.61 (m, 12H); 13C NMR (100 MHz, D2O): δ 166.2, 68.2, 50.2, 50.1, 49.8, 49.7, 42.8, 39.7, 27.9, 23.3; LC/MS (ESI+): m/z 513.3 [M + H]+; HRMS (ESI+): m/z calculated for C24H45N6O2S2+ [M + H]+ 513.3040, found 513.3069.
L-Cystine bis(N′-phenylpiperazide) tetrahydrochloride (3e)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (23 mg, yield: 88%). 1H NMR (400 MHz, D2O): δ 7.55 – 7.45 (m, 4H), 7.45–7.28 (m, 6H), 4.94 (t, 2H, J = 6.0 Hz), 4.12–3.94 (m, 6H), 3.94 – 3.83 (m, 2H), 3.69–3.49 (m, 8H), 3.46 – 3.27 (m, 4H); 13C NMR (100 MHz, D2O): δ 166.4, 144.7, 130.3, 127.1, 119.5, 52.5, 50.0, 49.8, 44.2, 41.1, 37.9; LC/MS (ESI+): m/z 529.3 [M + H]+; HRMS (ESI+): m/z calculated for C26H37N6O2S2+ [M + H]+ 529.2414, found 529.2446.
L-Cystine bis(2′,2′-dimethylpiperazide) tetrahydrochloride (3f)
The compound was recrystallized three times with diethyl ether/methanol to give a white solid (4 mg, yield: 80%). 1H NMR (400 MHz, D2O): δ 4.35–4.19 (m, 1H), 4.06 – 3.89 (m, 3H), 3.88–3.74 (m, 2H), 3.72 – 3.61 (m, 1H), 3.57–3.27 (m, 11H), 1.55 – 1.37 (m, 12H); 13C NMR (100 MHz, D2O): δ 167.1, 55.1, 54.5, 52.5, 49.6, 42.3, 39.1, 38.9, 38.3, 37.8, 22.8, 22.5, 20.7, 20.1; LC/MS (ESI+): m/z 433.3 [M + H]+; HRMS (ESI+): m/z calculated for C18H37N6O2S2+ [M + H]+ 433.2414, found 433.2444.
L-Cystine bis(4′-(dimethylamino)piperidineamide) tetrahydrochloride (3g)
The compound was recrystallized with diethyl ether/methanol to give a white solid (22 mg, yield: 100%). 1H NMR (400 MHz, D2O): δ 4.99 – 4.89 (m, 2H), 4.70 – 4.55 (m, 2H), 4.17 (t, 2H, J = 12.2 Hz), 3.70 – 3.54 (m, 2H), 3.48 – 3.18 (m, 6H), 3.05 – 2.80 (m, 14H), 2.40 – 2.17 (m, 4H), 1.96 – 1.62 (m, 4H); 13C NMR (100 MHz, D2O): δ 166.2, 62.6, 49.9, 44.1, 41.3, 39.8, 26.3, 25.5; LC/MS (ESI+): m/z 461.3 [M + H]+; HRMS (ESI+): m/z calculated for C20H41N6O2S2+ [M + H]+ 461.2727, found 461.2757.
L-Cystine bis(4′-(1′′-pyrrolidinyl)piperidineamide) tetrahydrochloride (3h)
The compound was recrystallized with diethyl ether/methanol to give a white solid (20 mg, yield: 100%). 1H NMR (400 MHz, D2O): δ 5.00–4.89 (m, 2H), 4.65 – 4.53 (m, 2H), 4.21–4.05 (m, 2H), 3.79 – 3.66 (m, 4H), 3.60 – 3.50 (m, 2H), 3.47 – 3.15 (m, 10H), 2.98–2.83 (m, 2H), 2.45–2.30 (m, 4H), 2.26 – 2.14 (m, 4H), 2.11 – 1.95 (m, 4H), 1.95 – 1.61 (m, 4H); 13C NMR (100 MHz, D2O): δ 165.9, 60.9, 51.8, 49.9, 43.9, 41.2, 28.5, 27.7, 22.5; LC/MS (ESI+): m/z 513.3 [M + H]+; HRMS (ESI+): m/z calculated for C24H45N6O2S2+ [M + H]+ 513.3040, found 513.3070.
L-Cystine bis(1,8-diazaspiro[4.5]decan-1-yl) tetrahydrochloride (3j)
The compound was recrystallized three times with diethyl ether/methanol to give a white solid (6 mg, yield: 100%). 1H NMR (400 MHz, D2O): δ 4.59 (dd, 2H, J = 8.4, 4.3 Hz), 3.79 (dt, 2H, J = 9.8, 6.2 Hz), 3.69 (dt, 2H, J = 9.8, 7.0 Hz), 3.55 (q, 2H, J = 7.1 Hz), 3.51–3.36 (m, 6H), 3.21–3.04 (m, 6H), 3.00 – 2.87 (m, 4H), 2.19 (dt, 2H, J = 12.5, 6.2 Hz), 2.07 (q, 2H, J = 6.9 Hz), 1.99 (p, 4H, J = 5.7 Hz), 1.75 (d, 4H, J = 13.9 Hz); 13C NMR (100 MHz, D2O): δ 165.9, 64.1, 51.9, 49.0, 42.2, 36.8, 34.6, 28.7, 28.1, 22.3; LC/MS (ESI+): m/z 485.3 [M + H]+; HRMS (ESI+): m/z calculated for C22H41N6O2S2+ [M + H]+ 485.2727, found 485.2761.
L-Cystine bis(N′,N’-dimethylethyleneamide) tetrahydrochloride (4a)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (160 mg, yield: 73%). 1H NMR (400 MHz, D2O): δ 4.44 (dd, 2H, J = 8.0, 5.2 Hz), 3.80 (m, 2H), 3.69–3.58 (m, 2H), 3.48 (m, 2H), 3.45–3.38 (m, 4H), 3.26 (m, 2H), 2.99 (s, 12H); 13C NMR (100 MHz, D2O): δ 168.8, 56.1, 52.0, 43.3, 43.1, 37.2, 35.0; LC/MS (ESI+): m/z 381.2 [M + H]+; HRMS (ESI+): m/z calculated for C14H33N6O2S2+ [M + H]+ 381.2101, found 381.2125.
L-Cystine bis(N′-pyrrolidinoethyleneamide) tetrahydrochloride (4b)
The compound was recrystallized six times with diethyl ether/methanol to give a yellow solid (25 mg, yield: 86%). 1H NMR (400 MHz, D2O): δ 4.38 (dd, 2H, J = 8.4, 4.7 Hz), 3.72 – 3.57 (m, 6H), 3.55–3.46 (m, 2H), 3.42–3.27 (m, 6H), 3.20–2.96 (m, 6H), 2.14–2.01 (m, 4H), 2.00 – 1.90 (m, 4H); 13C NMR (100 MHz, D2O): δ 168.7, 54.7, 54.5, 53.4, 52.0, 37.1, 36.1, 22.7; LC/MS (ESI+): m/z 433.3 [M + H]+; HRMS (ESI+): m/z calculated for C18H37N6O2S2+ [M + H]+ 433.2414, found 433.2439.
L-Cystine bis(N′-piperidinoethyleneamide) tetrahydrochloride (4c)
The compound was recrystallized six times with diethyl ether/methanol to give a yellow solid (31 mg, yield: 86%). 1H NMR (400 MHz, D2O): δ 4.45 – 4.36 (m, 2H), 3.80–3.69 (m, 2H), 3.66–3.53 (m, 6H), 3.50 – 3.40 (m, 2H), 3.37–3.28 (m, 4H), 3.27–3.18 (m, 2H), 3.06 – 2.94 (m, 4H), 2.01–1.90 (m, 4H), 1.86 – 1.69 (m, 6H), 1.57 – 1.41 (m, 2H); 13C NMR (100 MHz, D2O): δ 168.6, 55.1, 53.7, 53.6, 52.0, 37.1, 34.5, 22.8, 21.0; LC/MS (ESI + ): m/z 461.4 [M + H]+; HRMS (ESI+): m/z calculated for C20H41N6O2S2+ [M + H]+ 461.2000, found 461.2028.
L-Cystine bis(N′-morpholinoethyleneamide) tetrahydrochloride (4d)
The compound was recrystallized four times with diethyl ether/methanol to give a yellow solid (49 mg, yield: 100%). 1H NMR (400 MHz, D2O): δ 4.47 – 4.40 (m, 2H), 4.22 – 4.04 (m, 4H), 3.93 – 3.76 (m, 6H), 3.71–3.56 (m, 6H), 3.52–3.40 (m, 6H), 3.36–3.18 (m, 6H); 13C NMR (100 MHz, D2O): δ 168.7, 63.7, 55.6, 52.1, 52.0, 37.0, 34.1; LC/MS (ESI+): m/z 465.4 [M + H]+; HRMS (ESI+): m/z calculated for C18H37N6O4S2+ [M + H]+ 465.2313, found 465.2340.
L-Cystine bis(N′-pyrrolidin-2-one ethyleneamide) dihydrochloride (4e)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (67 mg, yield: 99%). 1H NMR (400 MHz, D2O): δ 4.32 (b, 2H), 3.28-3.61 (m, 14H), 3.12–3.19 (m, 2H), 2.44 (b, 4H), 2.04 (b, 4H); 13C NMR (100 MHz, D2O): δ 178.9, 167.9, 51.9, 47.9, 41.9, 37.2, 36.9, 31.1, 17.4; LC/MS (ESI+): m/z 461.4 [M + H]+; HRMS (ESI+): m/z calculated for C18H33N6O4S2+ [M + H]+ 461.2000, found 461.2028.
L-Cystine bis(N′,N′-dimethyl-1′,3′-diaminopropaneamide) tetrahydrochloride (4f)
The compound was recrystallized three times with diethyl ether/methanol to give a pale yellow solid (11 mg, yield: 68%). 1H NMR (400 MHz, D2O): δ 4.36 (dd, 2H, J = 7.5, 5.7 Hz), 3.46 – 3.30 (m, 6H), 3.30–3.15 (m, 6H), 2.91 (s, 12H), 2.21 – 1.85 (m, 4H) ppm. 13 C NMR (100 MHz, D2O): δ 168.11, 55.34, 52.07, 42.90, 42.85, 37.40, 36.68, 23.91; LC/MS (ESI+): m/z 409.3 [M + H]+; HRMS (ESI+): m/z calculated for C16H37N6O2S2+ [M + H]+ 409.2414, found 409.2438.
L-Cystine bis(1′-pyrrolidine-3′-aminopropaneamide) tetrahydrochloride (4g)
The compound was recrystallized six times with diethyl ether/methanol to give a yellow solid (16 mg, yield: 45%). 1H NMR (400 MHz, D2O): δ 4.40 (t, 2H, J = 6.6 Hz), 3.80–3.65 (m, 4H), 3.50 – 3.35 (m, 6H), 3.30 (ddd, 6H, J = 8.1, 6.4, 3.3 Hz), 3.14 (dt, 4H, J = 10.3, 7.5 Hz), 2.26 – 2.14 (m, 4H), 2.06 (ttd, 8H, J = 9.6, 7.6, 2.4 Hz); 13C NMR (100 MHz, D2O): δ 168.1, 54.3, 52.6, 52.2, 37.5, 36.9, 25.3, 22.7; LC/MS (ESI+): m/z 461.5 [M + H]+; HRMS (ESI+): m/z calculated for C20H41N6O2S2+ [M + H]+ 461.2727, found 461.2757.
L-Cystine bis(1′-morpholine-3′-aminopropaneamide) tetrahydrochloride (4h)
The compound was recrystallized six times with diethyl ether/methanol to give a yellow solid (40 mg, yield: 85%). 1H NMR (400 MHz, D2O): δ 4.41 (t, 2H, J = 6.7 Hz), 4.19 (d, 4H, J = 13.5 Hz), 3.89 (t, 4H, J = 12.8 Hz), 3.70 – 3.55 (m, 4H), 3.50 – 3.37 (m, 6H), 3.37–3.19 (m, 10H), 2.17–2.01 (m, 4H); 13C NMR (100 MHz, D2O): δ 168.2, 63.9, 54.9, 51.9, 37.5, 36.9, 23.1; LC/MS (ESI+): m/z 493.5 [M + H]+; HRMS (ESI+): m/z calculated for C20H41N6O4S2+ [M + H]+ 493.2625, found 493.2662.
Methyl N-(tert-butoxycarbonyl)-O-tosyl-L-serinate (14a)
Boc-Ser-OMe (13a, 2.5 g, 11.4 mmol) was dissolved in 50 mL of dry dichloromethane and cooled to 0 °C. Pyridine (2 mL, 23 mmol) was added to the chilled solution followed by para-toluenesulfonyl chloride (6.6 g, 34.2 mmol) and the reaction mixture was left to stir at 0 °C for 15 min, then warmed to room temperature, and left to stir for 45 min. The solvent was evaporated under reduced pressure and the white residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 15%. The solvent was evaporated under reduced pressure to give a colorless oil (4180 mg, 98% yield). 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H), 2.43 (s, 3H), 3.67 (s, 3H), 4.25-4.39 (m, 2H), 4.49 (b, 1H), 5.29 (b, 1H), 7.33 (d, 2H, J = 8 Hz), 7.74 (d, 2H, J = 8 Hz); 13C NMR (100 MHz, CDCl3) δ 169.0, 155.0, 145.2, 132.4, 129.9, 128.0, 80.5, 69.5, 53.0, 52.9, 28.2, 21.0; LC-MS (ESI+) m/z 374.49 [M + H]+.
Methyl N-(tert-butoxycarbonyl)-S-((R)-2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxo-propyl)-L-homocysteinate (15a)
Boc-L-Hcy-OMe was prepared from the L-Hcy in three steps of esterification, Boc protection, and reduction. Boc-L-Hcy-OMe (11, 255 mg, 0.7 mmol) and Boc-Ser(OTs)-OMe (14a, 209.4 mg, 0.84 mmol) were dissolved in 9 mL of degassed anhydrous N,N-dimethylformamide and cooled to 0 °C. NaH (28.6 mg, 1.19 mmol) and the reaction mixture was left to stir at 0 °C for 30 m then was brought to room temperature and left to stir under argon for 4 h. The mixture was diluted with 10 mL of water. The organic layer was washed with (3 ×40 mL) of dichloromethane. The combined organic layers were washed with (20 mL) of brine and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the brown residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 25%. The solvent was evaporated under reduced pressure to give a yellow solid (150 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 18H), 1.88-2.12 (m, 2H), 2.54-2.58 (m, 2H), 2.94 (b, 2H), 3.75 (s, 6H), 4.38 (b, 1H), 4.51 (b, 1H), 5.13 (b, 1H), 5.36 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 172.6, 171.5, 155.1, 80.2, 53.2, 52.6, 52.4, 34.6, 32.6, 28.3; LC-MS (ESI+) m/z 451.33 [M + H]+.
N-(tert-Butoxycarbonyl)-S-((R)-2-((tert-butoxycarbonyl)amino)-2-carboxyethyl)-L-homocysteine (16a)
Compound 15a (60 mg, 0.13 mmol) was dissolved in 900 µL of tetrahydrofuran and cooled to 0 °C. Lithium hydroxide (7 mg, 0.28 mmol) was dissolved in 900 µL of water then was added to the chilled solution and the reaction mixture was left to stir at room temperature for 1 h. The reaction mixture was diluted with 5 mL of water and was acidified by 0.5 N potassium hydrogen sulfate to pH 2 then was extracted with ethyl acetate (3 ×10 mL). The combined organic layers were washed with brine (5 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure to give a yellow solid (56 mg, 100% yield). 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 18H), 1.86-2.10 (m, 2H), 2.63 (b, 2H), 3.03 (b, 2H), 4.42 (b, 1H), 4.57 (b, 1H), 5.45 (b, 1H), 5.56 (b, 1H), 9.20 (b, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.7, 172.5, 155.3, 78.16, 53.8, 52.5, 32.7, 30.9, 28.2; LC-MS (ESI+) m/z 423.17 [M + H]+.
tert-Butyl ((R)-3-(((S)-3-((tert-butoxycarbonyl)amino)-4-(4-methylpiperazin-1-yl)-4-oxobutyl)-thio)-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)carbamate (17a)
To a solution of compound 16a (55 mg, 0.13 mmol) in 300 µL of anhydrous N,N-dimethylformamide, HATU (104 mg, 0.273 mmol) and DIPEA (140 µL, 0.8 mmol) were added. Then 1-methyl piperazine (36 µL, 0.32 mmol) was added to the mixture and the reaction mixture was left to stir at room temperature for 3 h. The mixture was diluted with ethyl acetate (20 mL) and washed with water (3 ×10 mL), brine (5 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 55%. The solvent was evaporated under reduced pressure to give a yellow oil (28 mg, 37% yield). 1H NMR (400 MHz, CDCl3) δ 1.43 (s, 18H), 1.74-1.81 (m, 2H), 2.38 (s, 6H), 2.57 (b, 10H), 2.73-2.88 (m, 2H), 3.48 (b, 8H), 4.72 (b, 2H), 5.40 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 170.1, 155.6, 79.9, 54.9, 54.4, 49.5, 48.9, 45.6, 41.7, 35.3, 33.6, 28.3; LC-MS (ESI+) m/z 587.18 [M + H]+.
(S)-2-Amino-4-(((R)-2-amino-3-(4-methylpiperazin-1-yl)-3-oxopropyl)thio)-1-(4-methyl-piperazin-1-yl)butan-1-one tetrahydrochloride (5a)
Compound 17a (23 mg, 0.04 mmol) was dissolved in 200 µL of methanol and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (90 µL, 0.36 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. There was still some mono-deprotected product so additional 4 N HCl in dioxane (45 µL, 0.18 mmol) was added in two portions to the solution at 0 °C and the solution was warmed to room temperature and left to stir for 5 h then in fridge for overnight. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated three times. The residue was dried to give a white solid (18 mg, 85% yield). 1H NMR (400 MHz, D2O) δ 2.19 (b, 2H), 2.78 (b, 2H), 2.97 (s, 6H), 3.10-3.25 (m, 10H), 3.66 (b, 8H), 4.19 (b, 2H); 13C NMR (100 MHz, D2O) δ 167.8, 166.9, 52.7, 49.7, 43.1, 42.8, 39.7, 31.9, 29.8, 27.1; LC-MS (ESI+) m/z 387.19 [M + H]+; HRMS (ESI+) m/z calculated for C17H35N6O2S+ [M + H]+ 387.2537, found 387.2562.
Di-tert-butyl ((7 R,12 S)-2,17-dimethyl-6,13-dioxo-9-thia-2,5,14,17-tetraazaocta-decane-7,12-di-yl)dicarbamate (17f)
To a solution of 16a (250 mg, 0.59 mmol) in 2.5 mL of anhydrous N,N-dimethylformamide, HATU (718 mg, 1.89 mmol) and DIPEA (924 µL, 5.31 mmol) were added. Then N,N-dimethylethylenediamine (216 µL, 2.1 mmol) was added to the mixture and the reaction mixture was left to stir at room temperature for 3 h. The mixture was diluted with ethyl acetate (20 mL), washed with water (3 ×10 mL) and brine (5 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the light brown oil was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 80%. The solvent was evaporated under reduced pressure to give white crystals (189 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 18H), 1.93-2.03 (m, 2H), 2.34 (s, 12H), 2.56 (b, 4H), 2.67-2.95 (m, 2H), 3.02-3.08 (m, 2H), 3.40 (b, 4H), 4.26 (b, 2H), 5.61 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 171.8, 170.8, 155.6, 79.9, 57.7, 53.4, 50.3, 44.8, 36.7, 34.9, 34.2, 33.1, 28.5; LC-MS (ESI+) m/z 563.19 [M + H]+.
(S)-2-Amino-4-(((R)-2-amino-3-((2-(dimethylamino)ethyl)amino)-3-oxopropyl)thio)-N-(2-(di-methylamino)ethyl)butanamide tetrahydrochloride (5f)
Compound 17 f (165 mg, 0.3 mmol) was dissolved in 1.5 mL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (900 µL, 3.6 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated five times. The residue was dried to give a white solid (133 mg, 87% yield). 1H NMR (400 MHz, D2O) δ 2.18-2.26 (m, 2H), 2.75 (t, 2H, J = 8 Hz), 2.97 (s, 12H), 3.05-3.22 (m, 2H), 3.38 (t, 4H, J = 8 Hz), 3.55–3.85 (m, 4H), 4.15 (t, 1H, J = 8 Hz), 4.24 (t, 1H, J = 8 Hz); 13C NMR (100 MHz, D2O) δ 170.8, 56.3, 51.9, 43.7, 34.5, 32.8, 29.9, 26.6; LC-MS (ESI+) m/z 363.32 [M + H]+; HRMS (ESI+) m/z calculated for C15H35N6O2S+ [M + H]+ 363.2537, found 363.2567.
Methyl (tert-butoxycarbonyl)-L-homocysteinate (11)
Compound 10 (838 mg, 1.69 mmol), triphenyl phosphine (499 mg, 1.9 mmol), sodium acetate (58 mg, 0.7 mmol) were suspended in a mixture of 8 mL of methanol, 3.5 mL of water, and 58 µL of glacial acetic acid and heated to 60 °C for 75 min. The mixture was diluted with ethyl acetate (80 mL), washed with water (2 ×40 mL) and brine (10 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the colorless oil was purified via ISCO (ethyl acetate/ hexane) and the product was eluted at 15%. The solvent was evaporated under reduced pressure to give a colorless oil (778 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 9H), 1.55 (t, 1H, J = 8 Hz), 1.84-2.08 (m, 2H), 2.55 (t, 2H, J = 8 Hz), 3.72 (s, 3H), 4.42 (b, 1H), 5.12 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 172.8, 155.4, 80.1, 52.4, 52.2, 37.2, 28.3, 20.7; LC-MS (ESI+) m/z 250.21 [M + H]+.
Methyl (tert-butoxycarbonyl)-L-homoserinate (13b)
A flask was charged with L-homoserine (12b, 600 mg, 5.04 mmol) and 5 mL of water and 5 mL of MeCN were added then the solution was cooled to 0 °C. Sodium hydroxide (202 mg, 5.04 mmol) was added followed by di-tert-butyl carbonate (1430 mg, 6.55 mmol) and the reaction mixture was brought to room temperature and left to stir for 1.5 h. The reaction mixture was concentrated as possible under reduced pressure and then the water was lyophilized to give a white foam (1121 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.37 (s, 9H), 1.64-1.71 (m, 2H), 3.64 (b, 4H), 5.98 (b, 1H); 13C NMR (100 MHz, DMSO-d6) δ 173.6, 154.6, 79.1, 59.7, 54.6, 36.5, 28.2.
The Boc-homoserine intermediate obtained above (1121 mg, 4.65 mmol) was dissolved in 10 mL of anhydrous N,N-dimethylformamide. Iodomethane (350 µL, 5.6 mmol) was added at 0 °C then the reaction mixture was brought to room temperature and left to stir for 5 h. The reaction mixture was diluted with ethyl acetate (150 mL), washed with water (2 ×25 mL), sodium bicarbonate (2 ×25 mL), 0.5 N potassium hydrogen sulfate (25 mL), and brine (15 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure to give a colorless oil (487 mg, 45% yield). 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 9H), 1.59-1.65 (m, 1H), 1.99 (b, 1H), 3.42 (b, 1H), 3.59 (b, 2H), 3.64 (s, 3H), 4.34 (b, 1H), 5.51 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 173.3, 156.2, 80.1, 58.3, 52.3, 50.8, 35.4, 28.2; LC-MS (ESI+) m/z 234.72 [M + H]+.
Methyl N-(tert-butoxycarbonyl)-O-tosyl-L-homoserinate (14b)
Compound 13b (487 mg, 2.09 mmol) was dissolved in 12 mL of dry dichloromethane and cooled to 0 °C. Pyridine (337 µL, 4.18 mmol) was added to the chilled solution followed by para-toluenesulfonyl chloride (1196 mg, 6.27 mmol) and the reaction mixture was left to stir at 0 °C for 15 min, then was warmed to room temperature and left to stir for 1 h. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 30%. The solvent was evaporated under reduced pressure to give a colorless oil (561 mg, 69% yield). 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 9H), 2.08-2.24 (m, 2H), 2.43 (s, 3H), 3.70 (s, 3H), 4.07 (b, 2H), 4.31 (b, 1H), 5.12 (b, 1H), 7.34 (d, 2H, J = 8 Hz), 7.77 (d, 2H, J = 8 Hz); 13C NMR (100 MHz, CDCl3) δ 172.0, 155.5, 144.9, 132.7, 129.9, 128.0, 80.2, 66.2, 52.6, 50.5, 31.6, 28.2, 21.6; LC-MS (ESI+) m/z 388.57 [M + H]+.
Dimethyl 4,4’-thio(2 S,2’S)-bis(2-((tert-butoxycarbonyl)amino)butanoate) (15b)
Compound 14b (550 mg, 1.42 mmol) was dissolved in 10 mL of degassed anhydrous N,N-dimethylformamide and cooled to 0 °C. Compound 11 (354 mg, 1.42 mmol) was added to the chilled solution followed by NaH (58 mg, 2.4 mmol) and the reaction mixture was left to stir at 0 °C for 30 m then was brought to room temperature and left to stir under argon for 3 h. The mixture was quenched with water (2 mL) and diluted with ethyl acetate (150 mL). The organic layer was washed with water (2 ×50 mL) and brine (20 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the brown residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 30%. The solvent was evaporated under reduced pressure to give a light yellow oil (443 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 1.84-1.91 (m, 2H), 2.03 (b, 2H), 2.52 (t, 4H, J = 8 Hz), 3.71 (s, 6H), 4.36 (b, 2H), 5.16 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 172.7, 155.3, 80.0, 60.3, 52.5, 52.4, 32.6, 28.3; LC-MS (ESI+) m/z 465.29 [M + H]+.
(2 S,2’S)-4,4’-Thiobis(2-((tert-butoxycarbonyl)amino)butanoic acid) (16b)
Compound 15b (443 mg, 0.95 mmol) was dissolved in 6 mL of tetrahydrofuran and cooled to 0 °C. Lithium hydroxide (48 mg, 2 mmol) was dissolved in 6 mL of water then was added to the chilled solution and the reaction mixture was left to stir at room temperature for 2 h. The reaction mixture was diluted with water (20 mL) and washed with dichloromethane (3 ×20 mL). Then the aqueous layer was acidified by 0.5 N potassium hydrogen sulfate to pH 2 and then was extracted with dichloromethane (3 ×40 mL). The combined organic layers were washed with brine (10 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure to give a pale yellow solid (361 mg, 87% yield). 1H NMR (400 MHz, CDCl3) δ 1.45 (s, 18H), 2.11 (b, 4H), 2.45–2.69 (m, 4H), 4.31 (b, 2H), 5.43 (b, 1H), 7.59 (b, 1H), 10.81 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 174.2, 157.1, 82.0, 54.1, 32.7, 32.2, 28.3; LC-MS (ESI+) m/z 437.13 [M + H]+.
Di-tert-butyl ((2 S,2’S)-thiobis(1-(4-methylpiperazin-1-yl)-1-oxobutane-4,2-diyl))di-carbamate (17b)
To a solution of compound 16b (150 mg, 0.344 mmol) in 1 mL of anhydrous N,N-dimethylformamide, HATU (274 mg, 0.72 mmol) and DIPEA (360 µL, 2.1 mmol) were added. Then 1-methylpiperazine (96 µL, 0.86 mmol) was added to the mixture and the reaction mixture was left to stir at room temperature for 3 h. The mixture was diluted with ethyl acetate (25 mL), washed with water (3 ×10 mL) and brine (5 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the light brown oil was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 65%. The solvent was evaporated under reduced pressure to give white crystals (155 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 1.75-1.93 (m, 4H), 2.55 (b, 4H), 2.63 (s, 6H), 2.90 (b, 8H), 3.86 (b, 8H), 4.70 (b, 2H), 5.36 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 170.5, 155.6, 80.1, 54.1, 48.9, 45.9, 44.5, 43.8, 40.3, 33.3, 28.3; LC-MS (ESI+) m/z 601.16 [M + H]+.
(2 S,2’S)-4,4’-Thiobis(2-amino-1-(4-methylpiperazin-1-yl)butan-1-one) tetrahydro-chloride (5b)
Compound 17b (23 mg, 0.04 mmol) was dissolved in 250 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (42 µL, 0.17 mmol) in two portions every half an hour and the solution was warmed to room temperature after each addition. There was still starting material and mono-deprotected product so additional 4 N HCl in dioxane (42 µL, 0.17 mmol) was added in four portions to the solution at 0 °C and the solution was warmed to room temperature and left to stir for 5 h then in fridge for overnight. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated three times. The residue was dried to give a white solid (6 mg, 29% yield). 1H NMR (400 MHz, D2O) δ 2.18 (b, 4H), 2.72 (b, 4H), 2.96 (s, 6H), 3.14-3.26 (m, 8H), 3.64 (b, 8H), 4.19 (b, 2H); 13C NMR (100 MHz, D2O) δ 167.9, 52.6, 49.7, 43.1, 42.6, 39.6, 29.7, 26.1; LC-MS (ESI+) m/z 401.33 [M + H]+; HRMS (ESI+) m/z calculated for C18H37N6O2S+ [M + H]+ 401.2693, found 401.2722.
Di-tert-butyl-((7 S,13 S)-2,18-dimethyl-6,14-dioxo-10-thia-2,5,15,18-tetraazanona-decane-7,13-diyl)dicarbamate (17g)
To a solution of compound 16b (361 mg, 0.83 mmol) in 1.5 mL of anhydrous N,N-dimethylformamide, HATU (663 mg, 1.74 mmol) and DIPEA (868 µL, 4.98 mmol) were added. The reaction mixture was left to stir at room temperature for 15 min. N,N-Dimethylethylenediamine (271 µL, 2.48 mmol) was added to the mixture and the reaction mixture was left to stir at room temperature for 2 h. The mixture was diluted with ethyl acetate (120 mL), washed with water (2 ×20 mL) and brine (20 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the dark yellow oil was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 65%. The solvent was evaporated under reduced pressure to give a light yellow oil (208 mg, 44% yield). 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 18H), 1.84-1.89 (m, 2H), 1.99-2.03 (m, 2H), 2.20 (s, 12H), 2.40 (t, 4H, J = 8 Hz), 2.53 (b, 4H), 3.29 (b, 4H), 4.22 (b, 2H), 5.46 (d, 2H, J = 4 Hz), 6.87 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 171.5, 155.5, 79.9, 57.8, 53.5, 46.0, 45.1, 36.9, 32.7, 28.3, 27.8; LC-MS (ESI+) m/z 577.24 [M + H]+.
(2 S,2’S)-4,4’-Thiobis(2-amino-N-(2-(dimethylamino)ethyl)butanamide) tetrahydro-chloride (5g)
Compound 17 g (190 mg, 0.33 mmol) was dissolved in 400 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (989 µL, 3.96 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated three times. The residue was dried to give a white solid (104 mg, 84% yield). 1H NMR (400 MHz, D2O) δ 2.23-230 (m, 4H), 2.77 (t, 4H, J = 8 Hz), 3.02 (s, 12H), 3.40-3.45 (m, 4H), 3.61-3.66 (m, 2H), 3.81-3.89 (m, 2H), 4.21–4.24 (m, 2H); 13C NMR (100 MHz, D2O) δ 170.2, 56.3, 52.7, 43.4, 35.0, 30.4, 26.3; LC-MS (ESI+) m/z 377.33 [M + H]+; HRMS (ESI+) m/z calculated for C16H37N6O2S+ [M + H]+ 377.2693, found 377.2724.
Methyl (S)-3-azido-2-((tert-butoxycarbonyl)amino)propanoate (18)
Compound 14a (4100 mg, 10.98 mmol) was dissolved in 6.5 mL of anhydrous N,N-dimethylformamide. Sodium azide (1785 mg, 27.5 mmol) was added slowly to the solution and the reaction mixture was shaken at 50 °C for 3 h. The reaction mixture was diluted with (100 mL) of cold water and extracted with dichloromethane (2 ×50 mL). The combined organic layers were washed with brine (5 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the colorless residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 10%. The solvent was evaporated under reduced pressure to give a colorless oil (1150 mg, 43% yield). 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H), 3.69 (b, 2H), 3.76 (s, 3H), 4.45 (b, 1H), 5.37 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 170.2, 155.1, 80.5, 53.5, 52.8, 52.7, 28.3; LC-MS (ESI+) m/z 245.05 [M + H]+.
Methyl (S)-3-amino-2-((tert-butoxycarbonyl)amino)propanoate (19)
Compound 63 (1150 mg, 4.7 mmol) was dissolved in 15 mL of methanol and 10% palladium on carbon (64 mg) was added and the reaction mixture was deoxygenated and stirred at room temperature under H2 at balloon pressure for 3 h. The mixture was filtered through a celite layer and the filtrate was dried under reduced pressure to get a pale yellow oil (1024 mg, 100% yield). 1H NMR (400 MHz, CDCl3) δ 1.24 (b, 2H), 1.41 (s, 9H), 3.01 (b, 2H), 3.73 (s, 3H), 4.27 (b, 1H), 5.42 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 172.1, 155.5, 80.0, 55.9, 52.4, 43.9, 28.3; LC-MS (ESI+) m/z 219.1 [M + H]+.
Methyl-N2-(tert-butoxycarbonyl)-N4-((S)-2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxo-propyl)-L-asparaginate (15c)
To a solution of Boc-Asp-OMe (866 mg, 3.5 mmol) in 3 mL of anhydrous N,N-dimethylformamide, HATU (1472 mg, 3.87 mmol) and DIPEA (1.9 mL, 10.62 mmol) were added. The reaction mixture was left to stir at room temperature for 10 min. Compound 19 (769 mg, 3.5 mmol) was dissolved in 2 mL of anhydrous N,N-dimethylformamide and then was added to the mixture and the reaction mixture was left to stir at room temperature for 2 h. The mixture was diluted with ethyl acetate (100 mL), washed with water (3 ×50 mL) and brine (5 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 40%. The solvent was evaporated under reduced pressure to give a colorless oil (630 mg, 41% yield). 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 18H), 2.60 (b, 1H), 3.02-3.09 (m, 1H), 3.75 (s, 6H), 3.85-3.93 (m, 2H), 4.33 (b, 1H), 4.60 (b, 1H), 5.45 (b, 2H), 7.22 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 175.8, 174.5, 170.1, 155.6, 80.3, 52.9, 51.7, 49.8, 40.8, 36.5, 28.3; LC-MS (ESI+) m/z 448.18 [M + H]+.
N2-(tert-Butoxycarbonyl)-N4-((S)-2-((tert-butoxycarbonyl)amino)-2-carboxyethyl)-L-asparagine (16c)
Compound 15c (600 mg, 1.34 mmol) was dissolved in 9 mL of tetrahydrofuran and cooled to 0 °C. Lithium hydroxide (81 mg, 3.35 mmol) was dissolved in 9 mL of water then was added to the chilled solution and the reaction mixture was left to stir at room temperature for 1 h. The reaction mixture was diluted with water (60 mL) and washed with dichloromethane (3 ×20 mL). Then the aqueous layer was acidified by 0.5 N potassium hydrogen sulfate to pH 2 and then was extracted with ethyl acetate (3 ×120 mL). The combined organic layers were washed with brine (20 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure to give a pale yellow solid (439 mg, 79% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.37 (s, 18H), 2.45–2.49 (m, 2H), 3.21–3.58 (m, 2H), 4.00 (b, 1H), 4.22 (b, 1H), 6.80 (b, 1H), 6.90 (b, 1H), 7.92 (b, 1H), 12.46 (b, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.2, 172.1, 169.8, 155.3, 78.2, 53.4, 50.1, 48.6, 36.9, 28.1; LC-MS (ESI+) m/z 420.42 [M + H]+.
tert-Butyl ((S)-3-((S)-3-((tert-butoxycarbonyl)amino)-4-(4-methylpiperazin-1-yl)-4-oxobutan-amido)-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)carbamate (17c)
Compound 16c (206 mg, 0.5 mmol) was suspended in 6 mL of dry dichloromethane. DIPEA (523 µL, 3 mmol) was added and gave a clear solution. Then 1-methyl piperazine (164 µL, 1.5 mmol) and PyAOP (548 mg, 1 mmol) were added to the solution and the reaction mixture was left to stir at room temperature for 1.5 h. The mixture was diluted with (40 mL) of water and extracted with dichloromethane (7 ×100 mL). The combined organic layers were washed with brine (20 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 60%. The solvent was evaporated under reduced pressure to give a pale yellow solid (160 mg, 55% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.35 (s, 18H), 2.18 (s, 6H), 2.26 (b, 10H), 3.02-3.52 (m, 10H), 4.48 (b, 1H), 4.69 (b, 1H), 6.84 (b, 1H), 7.03 (b, 1H), 7.88 (b, 1H); 13C NMR (100 MHz, DMSO-d6) δ 169.9, 169.2, 168.3, 155.0, 79.1, 54.6, 54.2, 49.6, 46.9, 45.5, 44.5, 41.4, 37.6, 28.1; LC-MS (ESI+) m/z 584.22 [M + H]+.
(S)-3-Amino-N-((S)-2-amino-3-(4-methylpiperazin-1-yl)-3-oxopropyl)-4-(4-methylpiperazin-1-yl)-4-oxobutanamide tetrahydrochloride (5c)
Compound 17c (140 mg, 0.24 mmol) was dissolved in 550 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (725 µL, 2.9 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. There was still some mono-deprotected product so additional 4 N HCl in dioxane (363 µL, 1.45 mmol) was added in two portions to the solution at 0 °C and the solution was warmed to room temperature and left to stir for 1 h. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated five times. The residue was dried to give a yellowish white solid (125 mg, 98% yield). 1H NMR (400 MHz, D2O) δ 2.97 (s, 6H), 3.19-3.43 (m, 8H), 3.66 (b, 12H), 4.30 (b, 2H); 13C NMR (100 MHz, D2O) δ 173.0, 166.1, 52.6, 51.1, 50.1, 43.3, 42.5, 40.9, 39.6, 35.1; LC-MS (ESI+) m/z 384.29 [M + H]+; HRMS (ESI+) m/z calculated for C17H34N7O3+ [M + H]+ 384.2718, found 384.2755.
Di-tert-butyl ((7 S,12 S)-2,17-dimethyl-6,10,13-trioxo-2,5,9,14,17-pentaazaoctadecane-7,12-di-yl)dicarbamate (17h)
Compound 16c (208 mg, 0.5 mmol) was suspended in 6 mL of dry dichloromethane. DIPEA (523 µL, 3 mmol) was added and gave a clear solution. Then N,N-dimethylethylenediamine (164 µL, 1.5 mmol) and PyAOP (548 mg, 1 mmol) were added to the solution and the reaction mixture was left to stir at room temperature for 45 min. The mixture was diluted with water (40 mL) and extracted with dichloromethane (12 ×80 mL). The combined organic layers were washed with brine (20 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 75%. The solvent was evaporated under reduced pressure to give a pale yellow solid (156 mg, 56% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.36 (s, 18H), 2.23 (b, 2H), 2.81 (b, 4H), 3.11 (b, 16H), 3.71 (b, 1H), 3.99 (b, 1H), 6.55 (b, 2H), 8.03 (b, 3H); 13C NMR (100 MHz, CDCl3) δ 171.8, 170.8, 155.1, 79.2, 55.7, 5.17, 51.5, 45.3, 42.4, 37.5, 34.1, 28.1; LC-MS (ESI+) m/z 560.29 [M + H]+.
(S)-2-Amino-N4-((S)-2-amino-3-((2-(dimethylamino)ethyl)amino)-3-oxopropyl)-N1-(2-(di-methylamino)ethyl)succinamide tetrahydrochloride (5h)
Compound 17 h (130 mg, 0.23 mmol) was dissolved in 550 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (700 µL, 2.8 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. There was still some mono-deprotected product so additional 4 N HCl in dioxane (175 µL, 0.7 mmol) was added to the solution at 0 °C and the solution was warmed to room temperature and left to stir for 1 h. The solvent was evaporated under reduced pressure and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated three times. The residue was dried to give a yellowish white solid (75 mg, 65% yield). 1H NMR (400 MHz, D2O) δ 2.97 (s, 6H), 3.19-3.43 (m, 8H), 3.66 (b, 12H), 4.30 (b, 2H); 13C NMR (100 MHz, D2O) δ 171.6, 169.5, 168.5, 56.3, 53.1, 50.0, 43.2, 39.9, 35.3, 34.9; LC-MS (ESI+) m/z 360.29 [M + H]+; HRMS (ESI+) m/z calculated for C15H34N7O3+ [M + H]+ 360.2718, found 360.2748.
Methyl-N2-(tert-butoxycarbonyl)-N5-((S)-2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxo-propyl)-L-glutaminate (15d)
Compound 19 (200 mg, 0.92 mmol) was dissolved in 6 mL of dry dichloromethane. DIPEA (481 µL, 2.76 mmol) was added to give a clear solution. Then Boc-Glu-OMe (240 mg, 0.92 mmol) and PyAOP (528 mg, 1 mmol) were added to the solution and the reaction mixture was left to stir at room temperature for 1.5 h. The mixture was diluted with water (40 mL) and extracted with dichloromethane (3 ×80 mL). The combined organic layers were washed with brine (10 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the residue was purified via ISCO (ethyl acetate/hexane) and the product was eluted at 50%. The solvent was evaporated under reduced pressure to give a white solid (176 mg, 42% yield). 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 1.81 (b, 1H), 2.12-2.26 (m, 3H), 3.51 (b, 1H), 3.72 (s, 6H), 3.82 (b, 1H), 4.29 (b, 1H), 4.40 (b, 1H), 5.35 (b, 1H), 5.78 (b, 1H), 6.73 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 172.7, 171.2, 155.1, 80.4, 53.8, 52.6, 41.7, 32.6, 29.7, 28.3; LC-MS (ESI+) m/z 462.31 [M + H]+.
N2-(tert-Butoxycarbonyl)-N5-((S)-2-((tert-butoxycarbonyl)amino)-2-carboxyethyl)-L-glutamine (16d)
Compound 15d (170 mg, 0.37 mmol) was dissolved in 3 mL of tetrahydrofuran and cooled to 0 °C. Lithium hydroxide (23 mg, 0.93 mmol) was dissolved in 3 mL of water, which was then added to the chilled solution and the reaction mixture was left to stir at room temperature for 3 h. The reaction mixture was concentrated under reduced pressure and then diluted with water (30 mL) and washed with dichloromethane (3 ×10 mL). Then the aqueous layer was acidified by 0.5 N potassium hydrogen sulfate to pH 2 and then was extracted with ethyl acetate (6 ×60 mL). The combined organic layers were washed with (20 mL) of brine and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the residue was purified via ISCO (ethyl acetate/hexane + 0.1% AcOH) and the product was eluted at 60%. The solvent was evaporated under reduced pressure to give a colorless oil (146 mg, 91% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.36 (s, 18H), 1.49-1.79 (m, 2H), 2.20 (t, 2H, J = 8 Hz), 3.16–3.48 (m, 2H), 3.95 (b, 2H), 6.87 (b, 2H), 7.84 (b, 1H), 10.81 (b, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.9, 172.1, 155.3, 89.9, 78.2, 53.6, 36.2, 28.1, 27.2; LC-MS (ESI+) m/z 434.6 [M + H]+.
tert-Butyl ((S)-5-(((S)-2-((tert-butoxycarbonyl)amino)-3-(4-methylpiperazin-1-yl)-3-oxopropyl)-amino)-1-(4-methylpiperazin-1-yl)-1,5-dioxopentan-2-yl)carbamate (17d)
Compound 16d (50 mg, 0.12 mmol) was suspended in 1.5 mL of dry dichloromethane. DIPEA (126 µL, 0.72 mmol) was added to give a clear solution. Then 1-methyl piperazine (40 µL, 0.36 mmol) and PyAOP (132 mg, 0.25 mmol) were added to the solution and the reaction mixture was left to stir at room temperature for 30 min. The mixture was diluted with water (10 mL) and extracted with dichloromethane (3 ×20 mL). The combined organic layers were washed with brine (5 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 60%. The solvent was evaporated under reduced pressure to give a pale yellow solid (69 mg, 97% yield). 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 1.64-1.93 (m, 2H), 2.23 (b, 2H), 2.53 (d, 6H, J = 12 Hz), 2.84 (b, 8H), 3.37–3.59 (m, 2H), 3.83 (b, 8H), 4.66 (b, 2H), 5.62 (b, 1H), 6.02 (b, 1H), 6.94 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 172.9, 170.2, 168.3, 155.6, 79.9, 54.5, 50.1, 49.4, 45.1, 44.5, 41.1, 31.9, 29.8, 28.3; LC-MS (ESI+) m/z 598.31 [M + H]+.
(S)-4-Amino-N-((S)-2-amino-3-(4-methylpiperazin-1-yl)-3-oxopropyl)-5-(4-methylpiperazin-1-yl)-5-oxopentanamide tetrahydrochloride (5d)
Compound 17d (16 mg, 0.027 mmol) was dissolved in 40 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 M HCl in dioxane (80 µL, 0.32 mmol) in four portions every half an hour and the solution was warmed to room temperature after each addition. The solvent was evaporated via N2 and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated five times. The residue was dried to give a yellowish white solid (6 mg, 55% yield). 1H NMR (400 MHz, D2O) δ 2.97 (s, 6H), 3.19-3.43 (m, 8H), 3.66 (b, 12H), 4.30 (b, 2H); 13C NMR (100 MHz, D2O) δ 175.1, 166.2, 52.7, 51.0, 50.1, 43.1, 42.5, 39.6, 29.9, 25.3; LC-MS (ESI+) m/z 398.28 [M + H]+; HRMS (ESI+) m/z calculated for C18H36N7O3+ [M + H]+ 398.2874, found 398.2906.
Di-tert-butyl ((7 S,13 S)-2,18-dimethyl-6,10,14-trioxo-2,5,9,15,18-pentaazanona-decane-7,13-di-yl)dicarbamate (17i)
Compound 16d (66 mg, 0.15 mmol) was suspended in 2 mL of dry dichloromethane. DIPEA (160 µL, 0.9 mmol) was added to give a clear solution. Then N,N-dimethylethylenediamine (55 µL, 0.5 mmol) and PyAOP (167 mg, 0.32 mmol) were added to the solution and the reaction mixture was left to stir at room temperature for 2 h. The mixture was diluted with water (15 mL) and extracted with dichloromethane (6 ×40 mL). The combined organic layers were washed with brine (10 mL) and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the residue was purified via ISCO (20% MeOH:DCM + 0.1% TEA/DCM) and the product was eluted at 70%. The solvent was evaporated under reduced pressure to give a pale yellow solid (51 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.39 (s, 18H), 1.72 (b, 2H), 2.10 (b, 2H), 2.51 (s, 12H), 2.71 (b, 4H), 3.17–3.59 (m, 6H), 3.85 (b, 2H), 6.84 (b, 2H), 8.17 (b, 3H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 171.9, 170.5, 155.2, 79.7, 56.9, 54.5, 54.0, 51.4, 43.7, 35.3, 31.9, 28.1, 27.9; LC-MS (ESI+) m/z 574.3 [M + H]+.
(S)-2-Amino-N5-((S)-2-amino-3-((2-(dimethylamino)ethyl)amino)-3-oxopropyl)-N1-(2-(dimethylamino)ethyl)pentanediamide tetrahydrochloride (5i)
Compound 17i (41 mg, 0.071 mmol) was dissolved in 185 µL of MeOH and cooled to 0 °C. To the chilled solution was added 4 N HCl in dioxane (215 µL, 0.86 mmol) in two portions every half an hour and the solution was warmed to room temperature after each addition. There was still some mono-deprotected product so additional 4 N HCl in dioxane (215 µL, 0.86 mmol) was added to the solution at 0 °C and the solution was warmed to room temperature and left to stir for 1 h. The solvent was evaporated via N2 and the residue was dried under vacuum. The residue then was dissolved in a minimal amount of methanol and recrystallized with an excess of diethyl ether then the vial was centrifuged and the solvent was decanted; this purification step was repeated five times. The residue was dried to give a yellowish white solid (30 mg, 83% yield). 1H NMR (400 MHz, D2O) δ 2.20–2.28 (m, 2H), 2.54 (t, 2H, J = 8 Hz), 3.02 (s, 12H), 3.37–3.44 (m, 4H), 3.64-3.69 (m, 2H), 3.77-3.91 (m, 4H), 4.14–4.29 (m, 2H); 13C NMR (100 MHz, D2O) δ 175.2, 170.1, 168.6, 56.3, 53.2, 52.8, 43.3, 40.1, 34.9, 30.6, 26.2; LC-MS (ESI+) m/z 374.28 [M + H]+. HRMS (ESI+) m/z calculated for C16H36N7O3+ [M + H]+ 374.2874, found 374.2903.
tert-Butyl (R)-(3-mercapto-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)carbamate (21)
Compound 20 (1071 mg, 1.77 mmol), triphenyl phosphine (604 mg, 2.3 mmol), sodium acetate (58 mg, 0.7 mmol) were suspended in a mixture of 10 mL of methanol, 5 mL of water, and 42 µL of glacial acetic acid and heated to 60 °C for 75 min. The mixture was concentrated and diluted with (300 mL) of dichloromethane, washed with water (2 ×100 mL) and brine, and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the light yellow oil was purified via ISCO (20% MeOH:DCM + 0.3 N NH3/DCM) and the product was eluted at 35%. The solvent was evaporated under reduced pressure to give a colorless oil (900 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H), 1.50 (t, 1H, J = 8 Hz), 2.25 (s, 3H), 2.33-2.38 (m, 4H), 2.62-2.86 (m, 2H), 3.54-3.61 (m, 4H), 4.70 (b, 2H), 5.59 (d, 2H, J = 12 Hz); 13C NMR (100 MHz, CDCl3) δ 168.6, 155.1, 80.0, 54.6, 51.6, 45.7, 42.2, 28.3, 27.4.
Di-tert-butyl-3,3’-disulfanediyl(2 R,2’R)-bis(2-((tert-butoxycarbonyl)amino)-propanoate) (22)
Boc-L-cystine-OH (1 g, 2.27 mmol) was suspended in 10 mL of anhydrous dichloromethane. Tert-butyl-2,2,2-trichloroacetimidate (1.2 mL, 6.81 mmol) was added to the mixture and the reaction mixture was stirred at room temperature for 4 h. Additional tert-butyl-2,2,2-trichloroacetimidate (0.6 mL, 3.4 mmol) was added to the mixture and the reaction mixture was stirred at room temperature for 1 h. The reaction mixture was filtered and washed with dichloromethane and 3% ethyl acetate/hexane. The filtrate was evaporated under reduced pressure and the residue was purified via ISCO (50% ethyl acetate in hexane/hexane) and the product was eluted at 20%. The solvent was evaporated under reduced pressure to give a colorless oil (760 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 1.45 (d, 36H, J = 12 Hz), 3.09-3.22 (m, 4H), 4.45 (b, 2H), 5.33 (b, 2H). 13C NMR (100 MHz, CDCl3) δ 169.6, 155.1, 82.7, 80.0, 53.8, 42.1, 28.3, 28.0.
tert-Butyl (tert-butoxycarbonyl)-L-cysteinate (23)
Compound 22 (693 mg, 1.25 mmol), triphenyl phosphine (428 mg, 1.63 mmol), sodium acetate (41 mg, 0.7 mmol) were suspended in a mixture of 6 mL of methanol, 3 mL of water, and 30 µL of glacial acetic acid and heated to 60 °C for 75 min. The mixture was concentrated and diluted with dichloromethane (200 mL), washed with water (2 ×100 mL) and brine, and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the yellow residue was purified via ISCO (50% ethyl acetate in hexane/hexane) and the product was eluted at 10%. The solvent was evaporated under reduced pressure to give a white solid (600 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 1.45 (d, 19H, J = 12 Hz), 2.90-2.95 (m, 2H), 4.45 (b, 1H), 5.40 (b, 1H); 13C NMR (100 MHz, CDCl3) δ 169.3, 155.1, 82.7, 80.0, 55.1, 28.3, 28.0, 27.5.
tert-Butyl-N-(tert-butoxycarbonyl)-S-(((R)-2-((tert-butoxycarbonyl)amino)-3-(4-methyl-piperazin-1-yl)-3-oxopropyl)thio)-L-cysteinate (24)
To a stirred solution of 1-chlorobenzotriazole (74 mg, 0.48 mmol) and benzotriazole (29 mg, 0.24 mmol) in 8 mL of anhydrous dichloromethane under argon at ‒78 °C was added dropwise a solution of compound 21 (73 mg, 0.24 mmol) in 2 mL of anhydrous dichloromethane. After 30 min, a solution of thiourea (54 mg, 0.72 mmol) in 6 mL of anhydrous tetrahydrofuran was added and the reaction mixture was left to stir at ‒78 °C for further 15 min. Compound 23 (100 mg, 0.36 mmol) in 2 mL of anhydrous dichloromethane was added slowly at ‒78 °C and the reaction mixture was warmed slowly to room temperature and left to stir for overnight. The reaction mixture was filtered with additional dichloromethane washes, and the filtrate was evaporated under reduced pressure. The residue was purified via ISCO (20% MeOH:DCM + 0.3 N NH3 & DCM) and the product was eluted at 25%. The solvent was evaporated under reduced pressure to give a colorless oil (80 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 1.40 (d, 27H, J = 12 Hz), 2.26 (s, 3H), 2.34-2.41 (m, 4H), 2.83–3.16 (m, 4H), 3.60 (b, 4H), 4.39 (b, 1H), 4.87-4.93 (m, 1H), 5.43 (b, 2H); 13C NMR (100 MHz, CDCl3) δ 169.6, 168.9, 155.1, 82.7, 80.1, 55.0, 54.5, 53.9, 49.0, 45.8, 42.1, 28.4, 28.0.
S-(((R)-2-Amino-3-(4-methylpiperazin-1-yl)-3-oxopropyl)thio)-L-cysteine trihydrochloride (25)
The compound was recrystallized three times with diethyl ether/methanol to give a white solid (11 mg, yield: 100%). 1H NMR (400 MHz, D2O): δ 4.37 (b, 1H), 4.23 (b, 1H), 3.67-3.80 (m, 4H), 3.18-3.48 (m, 8H), 2.83 (d, 3H, J = 8 Hz); 13C NMR (100 MHz, D2O): δ 171.6, 166.7, 52.5, 49.5, 43.0, 42.5, 39.6, 37.1, 36.6; LC/MS (ESI+): m/z 323.2 [M + H]+; HRMS (ESI+): m/z calculated for C11H23N4O3S2+ [M + H]+ 323.1206, found 323.1233.
L-cystine crystallization inhibition assay
The detailed procedure for the L-cystine crystallization inhibition assay has recently been published [18]. Briefly, supersaturated L-cystine solutions were incubated with inhibitors at varying concentrations for 72 h in an incubator set at 20 °C. After the incubation period, the incubation mixtures were centrifuged at maximum centrifuge speed (i.e., 14,000 rpm) for 10 min at room temperature. 50 µL supernatant was diluted 10-fold with Millipore deionized water. The diluted L-cystine solutions were plated in triplicates (40 µL) into a 96-well plate (Corning 3631 black, clear bottom). 80 µL aqueous solution of potassium cyanide (30% w/v) in phosphate-buffered saline (pH = 7.4) (1:7 ratio) was added to each well. The plate was shaken at 550 rpm for 25 min, followed immediately by addition of 10 µL aqueous solution of sodium nitroprusside (20% w/v) using an automated dispenser (Flexdrop IV Precision reagent dispenser, Perkin Elmer, Waltham, MA). The plate was centrifuged at 1,500 rpm for 20 s. The absorbance of the wells were measured at 530 nm using a Wallac Victor 3 V multi-label microplate reader (Perkin Elmer, Waltham, MA). A standard curve of absorbance vs. L-cystine concentration was used to derive the L-cystine concentration in each test compound solution. Nonlinear regression analysis of the data (OriginPro, Northampton, MA) enabled the calculation of EC50 values for the inhibitors.
In vivo metabolism and pharmacokinetic studies
A method based on size exclusion chromatography (SEC) coupled with ultra-performance liquid chromatography (UPLC) and triple stage mass spectrometry (MS3) was developed for the analysis of LH708 and its metabolites, including LH1727, from mouse plasma and urine samples. Following size exclusion, urine and plasma samples were separated on a Thermo Finnigan UPLC system coupled with a Thermo Finnigan LTQ XL Ion Trap Mass Spectrometer. The separation was carried out on a Waters HSPgel AQ 2.5 6.0x150mm column. An isocratic elution of 40% methanol with 0.15% formic acid in water was at a constant flow of 200 µL/min and the injection volume was 20 μl. LH708 and LH1727 had retention times of 10.5 and 11.2 min, respectively. Electrospray was operated in multiple MS/MS/MS for simultaneous determination of LH708 and LH1727 in positive ion mode. Data acquisition was performed in MS3 mode of 202 m/z after MSMS of 405 m/z for LH708 and MS3 of m/z 202 after MSMS of 323 m/z for LH1727. Integration was performed using Xcalibur software with Genesis peak integration method.
LH708 (100 µl of 16.4 mg/ml, equivalent to 150 µmol/kg) was delivered by tail vein injection or by stomach tube to 3-month-old Slc3a1 KO male mice (3 mice per group). Blood samples were collected from the saphenous vein at 5, 20 min and 1, 4, 8, and 24 hr after administration. Urine was also collected after 24 h. Blood and urine samples were collected from untreated mice as controls (one mouse each).
Atomic force microscopy studies
For the AFM studies, hexagonal L-cystine crystals were crystallized from aqueous supersaturated solutions at pH 6.3 and single crystals were collected by vacuum filtration and air-dried. Cystine crystals were mounted on an AFM specimen disk and real-time in situ AFM was performed with a Bruker Multimode AFM using a Bruker MTFML-V2 cell designed for fluids. The mounted crystals were grown for 20 min prior to measurements in order to regenerate the crystal surface by continually flowing a supersaturated solution (2 mM cystine) through the fluid cell at a rate of 20 mL/h using a syringe pump. Measurements of the velocities of the {1010} steps on the {0001} basal plane were acquired under the same conditions in the presence and absence of crystal growth inhibitors. Step velocities were measured in the absence of the inhibitor (Vo) and then in the presence of the inhibitor at three concentrations (15, 30, and 45 µM).
Abbreviations
- AcOH:
-
acetic acid
- AFM:
-
atomic force microscopy
- Asp:
-
L-aspartic acid
- BtaH:
-
1H-benzotriazole
- BtCl:
-
1-chlorobenzotriazole
- Boc2O:
-
di-tert-butyl carbonate
- L-CDME:
-
L-cystine dimethyl ester
- Cys:
-
L-cysteine
- DCM:
-
dichloromethane
- DIPEA:
-
diisopropylethylamine
- DMF:
-
dimethyl formamide
- DMSO:
-
dimethyl sulfoxide
- Glu:
-
L-glutamic acid
- HATU:
-
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- Hcy:
-
L-homocysteine
- Hse:
-
L-homoserine
- LiOH:
-
lithium hydroxide
- MeCN:
-
acetonitrile
- MeI:
-
methyl iodide
- MeOH:
-
methanol
- Na2CO3 :
-
sodium carbonate
- NaH:
-
sodium hydride
- NaN3 :
-
sodium azide
- NaOAc:
-
sodium acetate
- NaOH:
-
sodium hydroxide
- NH3 :
-
ammonia
- OTs:
-
tosylate
- Pd/C:
-
palladium on carbon
- PPh3 :
-
triphenylphosphine
- PyAOP:
-
(7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- SAR:
-
structure-activity relationship
- SEC:
-
size exclusion chromatography
- Ser:
-
L-serine
- SLC3A1 :
-
solute carrier family 3 member 1
- SLC7A9 :
-
solute carrier family 7 member 9
- SOCl2 :
-
thionyl chloride
- TBTA:
-
tert-butyl-2,2,2-trichloroacetimidate
- TEA:
-
triethylamine
- THF:
-
tetrahydrofuran
- TsCl:
-
para-toluenesulfonyl chloride
- UPLC:
-
ultra-performance liquid chromatography
References
Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJS. How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis. 2016;44:65–76. https://doi.org/10.1007/s00240-015-0841-x
Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28:1923–42. https://doi.org/10.1007/s00467-012-2329-z
Eggermann T, Venghaus A, Zerres K. Cystinuria: an inborn cause of urolithiasis. Orphanet J Rare Dis. 2012;7:11. https://doi.org/10.1186/1750-1172-7-19
Pereira DJC, Schoolwerth AC, Pais VM. Cystinuria: current concepts and future directions. Clin Nephrol. 2015;83:138–46. https://doi.org/10.5414/cn108514
Sahota A, Tischfield JA, Goldfarb DS, Ward MD, Hu L. Cystinuria: genetic aspects, mouse models, and a new approach to therapy. Urolithiasis. 2019;47:57–66. https://doi.org/10.1007/s00240-018-1101-7
Sumorok N, Goldfarb DS. Update on cystinuria. Curr Opin Nephrol Hypertens. 2013;22:427–31. https://doi.org/10.1097/MNH.0b013e3283621c5d
Thomas K, Wong K, Withington J, Bultitude M, Doherty A. Cystinuria-a urologist’s perspective. Nat Rev Urol. 2014;11:270–7. https://doi.org/10.1038/nrurol.2014.51
Schwartz BF, Stoller ML. The vesical calculus. Urol Clin North Am. 2000;27:333. https://doi.org/10.1016/s0094-0143(05)70262-7
Schwentner C, Oswald J, Lunacek A, Bartsch G, Radmayr C. Giant cystine stone in an infant bladder with no evidence of cystinuria - Valence of possible pathomechanisms. Urol Int. 2005;75:285–7. https://doi.org/10.1159/000087810
D’Ambrosio V, Capolongo G, Goldfarb D, Gambaro G, Ferraro PM. Cystinuria: an update on pathophysiology, genetics, and clinical management. Ped Nephrol. 2022;37:1705–11. https://doi.org/10.1007/s00467-021-05342-y
Bhatt NP, Deshpande AV, Starkey MR. Pharmacological interventions for the management of cystinuria: a systematic review. J Nephrol. 2023. https://doi.org/10.1007/s40620-023-01795-6
Zisman AL. Effectiveness of treatment modalities on kidney stone recurrence. Clin J Am Soc Nephrol. 2017;12:1699–708. https://doi.org/10.2215/cjn.11201016
Hu L, Yang Y, Aloysius H, Albanyan H, Yang M, Liang J-J, et al. L-cystine diamides as L-cystine crystallization inhibitors for cystinuria. J Med Chem. 2016;59:7293–8. https://doi.org/10.1021/acs.jmedchem.6b00647
Yang Y, Albanyan H, Lee S, Aloysius H, Liang J-J, Kholodovych V, et al. Design, synthesis, and evaluation of L-cystine diamides as L-cystine crystallization inhibitors for cystinuria. Bioorg Med Chem Lett. 2018;28:1303–8. https://doi.org/10.1016/j.bmcl.2018.03.024
Shtukenberg AG, Hu L, Sahota A, Kahr B, Ward MD. Disrupting Crystal Growth through Molecular Recognition: Designer Therapies for Kidney Stone Prevention. Acc Chem Res. 2022;55:516–25. https://doi.org/10.1021/acs.accounts.1c00631
Hu L, Albanyan H, Yang J, Wang Y, Yang M, Tan X, et al. 8-L-Cystinyl bis (1,8-diazaspiro[4.5]decane) as an orally bioavailable L-cystine crystallization inhibitor for cystinuria. ACS Med Chem Lett. 2024;15:in press. https://doi.org/10.1021/acsmedchemlett.4c00066
Poloni LN, Zhu Z, Garcia-Vázquez N, Yu AC, Connors DM, Hu L, et al. Role of molecular recognition in L-cystine crystal growth inhibition. Cryst Growth Des. 2017;17:2767–81. https://doi.org/10.1021/acs.cgd.7b00236
Yang J, Albanyan H, Wang Y, Yang Y, Sahota A, Hu L. Development of convenient crystallization inhibition assays for structure-activity relationship studies in the discovery of crystallization inhibitors. Med Chem Res. 2023;32:1391–9. https://doi.org/10.1007/s00044-023-03061-7
Gillaspy ML, Lefker BA, Hada WA, Hoover DJ. A simple method for the formation of cyclopropylamines: the first synthesis of tri-cyclopropylamine. Tetrahedron Lett. 1995;36:7399–402. https://www.sciencedirect.com/science/article/pii/0040403995015604
Go Y-M, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta, Gen Subj. 2008;1780:1273–90. https://www.sciencedirect.com/science/article/pii/S0304416508000263
Cabrera N, Vermilyea D, Doremus R, Roberts B, Turnbull D, editors. Growth and Perfection of Crystals: Proceedings of the International Conference. International Conference on Crystal Growth; 1958 August 1958; Cooperstown, N.Y. New York: Wiley; 1958.
De Yoreo JJ, Vekilov PG Principles of crystal nucleation and growth. In: Dove PM, De Yoreo JJ, Weiner S, editors. Biomineralization. Washington, DC: Mineralogical Society of America Geochemical Society; 2003. p. 57–93.
Lee-Thorp JP, Shtukenberg AG, Kohn RV. Crystal growth inhibition by mobile randomly distributed stoppers. Cryst Growth Des. 2020;20:1940–50. https://doi.org/10.1021/acs.cgd.9b01609
Shtukenberg AG, Ward MD, Kahr B. Crystal growth with macromolecular additives. Chem Rev. 2017;117:14042–90. https://doi.org/10.1021/acs.chemrev.7b00285
Acknowledgements
We gratefully thank the National Institutes of Health for their financial support of this work via grant DK112782.
Author contributions
LH conceived of the compound designs and experiments. HA synthesized the compounds and performed the crystallization inhibition assay. YW worked on some of the experimental details. MY performed the animal studies under the direction of LH and AS. XZ performed the AFM studies under the direction of MW. JY wrote an initial draft of the manuscript based on the dissertation authored by HA. All authors discussed the experiments, commented on the manuscript, and approved of the final draft. Note: Some authors (LH, AS, and HA) are inventors of patents on compounds discussed in this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
*Dedicated to Prof. Ron Borchardt on the occasion of his 80th birthday and for his many years of research, teaching, and service at the University of Kansas and in the field of medicinal chemistry and pharmaceutical chemistry
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, L., Albanyan, H., Yang, J. et al. Structure-activity relationships and pharmacokinetic evaluation of L-cystine diamides as L-cystine crystallization inhibitors for cystinuria. Med Chem Res (2024). https://doi.org/10.1007/s00044-024-03228-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00044-024-03228-w