
Abstract
Bacterial communities in the phyllosphere are shaped by host genotype and phenotype and spatio-temporal variation of the environment. In turn, bacteria have the potential for altering the plant phenotype. Field experiments can help to estimate bacterial effects on plant functional traits under natural conditions. We used a transplantation approach of culturable bacterial communities to explore how manipulation of leaf-associated microbial communities in two different successional stages within a glacier foreland can influence microbial composition and functional plant traits. Our study documents successional stage-specific variations in the composition of foliar bacterial communities and shifts therein throughout a season and between years. We show that cultured bacteria transferred between plant communities can alter diversity and composition of the microbiome on plant community level as well as species-specific functional plant traits of two selected plant species within one growing season. Furthermore, our results demonstrate a strong resilience of plant-associated bacterial communities and of plants in response to bacterial invaders. Our study illustrates that inoculation experiments in the field with naturally occurring microbial communities of wild plants are suited to investigate complex interactions between microbial communities, the environment, and plant traits.
Similar content being viewed by others


Introduction
Mountain glaciers are retreating rapidly following climate change. As a consequence, unvegetated terrain is exposed in the glacier foreland, characterized by an abundance of highly erodible sediments and high geomorphological process dynamics. The changing biotic and abiotic conditions in a morphodynamic glacier foreland offer unique opportunities for the study of succession and temporal community assembly patterns of whole communities (Matthews 1992). The increasing distance from the glacier is a simple proxy for the time since first colonization by organisms and is correlated with changes in abiotic and biotic properties (Zumsteg et al. 2012). Microbes are the first organisms to colonize the ice-free terrain followed by the establishment of higher plants within 15–35 years after deglaciation—if not disturbed by erosive surface processes such as debris flows, landslides or soil erosion (Kazemi et al. 2016; Junker et al. 2020). Communities subsequently increase in biomass and diversity with a high microbial species turnover, resulting in greater ecosystem stability (Nemergut et al. 2007). Microorganisms are especially important for plant growth in the sparse glacier forelands, as they play a key role in nutrient cycling (Knelman et al. 2018). Plants can exert species-specific effects on microbial communities and, therefore, plant community composition, plant cover, and diversity can predict below-ground microbial diversity, which can be observed in early successional alpine ecosystems (Porazinska et al. 2018; Navrátilová et al. 2019).
Plants are populated by microorganisms both below and above the ground with specific communities on different plant organs. The leaf surface, or phyllosphere, is inhabited by a diverse variety of microorganisms, predominantly bacteria (Stone et al. 2018). Microbial community establishment on plants is not random but undergoes selection depending on host species identity, as well as on temporal and spatial effects (Laforest-Lapointe et al. 2016; Gaube et al. 2020). Leaf-associated microbiota can emerge from the surrounding environment or be transferred across generations of plants via seeds (Mitter et al. 2017; Escobar Rodríguez et al. 2020). Their presence is regulated by the host plant itself as well as through microbe-microbe interactions (Liu et al. 2020; Chaudhry et al. 2021). The phyllosphere is a highly dynamic habitat as it is often subjected to rapid and pronounced dynamic fluctuations of abiotic conditions, such as changes in temperature, UV radiation and humidity (Vorholt 2012). Therefore, the phyllosphere offers a particularly well-suited habitat to study spatio-temporal variability in microbial communities in relation to the environment as well as community assembly processes. Plant–microbe interactions are positive, neutral, or negative, and plant-associated microbiota contribute to essential functions such as seed germination and growth support through the supply of hormones, nutrients, and stress resistance (Berg et al. 2017). Further, phenotypic traits of plants depend not only on the genotype of the plant, but are also shaped by plant–microbe interactions (Friesen et al. 2011). Measuring functional plant traits may be useful for investigating the magnitude of these interactions. This has largely been shown through laboratory studies in controlled and simplified environments, only considering one or a few microbial strains at a time (Beilsmith et al. 2019). Whereas this kind of studies are mostly applied to crop plants or model species, research on natural plant communities is lacking. Therefore, field experiments which manipulate the microbiome of wild plant species are required to estimate the microbial effects on plant functional traits under natural conditions.
By comparing whole phyllosphere communities associated with plant communities in different locations as shown in this study, we can test how spatio-temporal effects and plant communities influence the microbiome composition. The glacier foreland presents a particularly well-suited opportunity to address these questions due to the variable conditions within small distances. We designed an experiment, which allowed us to explore how manipulation of foliar bacterial communities associated with plant communities in two different successional stages within a glacier foreland can influence microbial composition and functional plant traits. Therefore, we used a transplantation approach with cultured leaf-associated bacterial communities within two successional stages and transferred culturable communities from old successional plots to plants on young successional plots and vice versa, as well as back on the same plot age. Next generation amplicon sequencing was applied to samples taken before and after the treatment to determine changes in bacterial community composition. Additionally, we measured leaf traits of two common plant species before and after treatment to track possible alterations in response to the microbiome manipulation. We expected to find (1) temporal (intra- und inter-annual) as well as successional stage-specific differences in the microbiome composition; (2) alterations in the microbiome composition in response to the transfer of culturable bacteria and (3) alterations in leaf traits of two selected alpine plant species in response to the microbiome manipulation.
Our study illustrates that bacterial communities associated with plants can exhibit distinct changes over time as well as site-specific variations and that transferred foliar bacteria between plants of two successional stages can alter the plant microbiome on community level as well as leaf traits on species level.
Materials and methods
Study site and experimental setup
Field work was conducted in the proglacial area of the Gepatschferner glacier in the Kaunertal valley, Tyrol, Austria (46°87′N, 10°74′E). The Gepatschferner is the second largest glacier in Austria and is among the fastest melting glaciers in Austria with a recent annual retreat rate of 52 m between the years 2019 and 2020 (Lieb and Kellerer-Pirklbauer 2020). Since the maximum extent at the Little Ice Age (mid-nineteenth century) the glacier melt uncovered an area of more than 2.5 km2 as glacier foreland. The glacier foreland contains large quantities of poorly consolidated sediment deposited by the retreating glacier, stored in large lateral moraines and thick slope covers. These deposits are subject to high rates of erosion mainly by debris flows, floods, and soil erosion (Hilger et al. 2019). In summer 2018, we selected two sites in the glacier foreland differing in time since deglaciation and, therefore, exhibit plant communities of different successional stages. The young successional site has been deglaciated approximately since 2006 and the old site has been released by the glacier since at least 1971. Terrain ages since deglaciation are based on annual length change measurements published since the early twentieth century (Fischer 2018; Lieb and Kellerer-Pirklbauer 2020). In each of the two sites we established 12 square plots (40 × 40 cm). Eight plots per site were randomly assigned for later treatment with microbiota collected from either the old or young successional site as described below. The remaining four plots in each site were then assigned to serve as controls in which we did not manipulate the microbiome. Therefore, both the young and old successional site ultimately contained four control plots, four plots to be treated with microbes from the old successional stage and four plots to be treated with microbes collected in young successions.
Leaf sampling for cultivation of microbes
For cultivating phyllosphere bacteria for the treatment solution, one intact leaf of every plant individual per plot was collected with sterile forceps and then mixed to form one pooled sample for each of the two successional stages separately, i.e., the pooled sample contained plant material from 12 plots each. The subsequent treatment of the samples in the lab was equivalent for the plant material from young and old sites, however, the two samples were always treated separately. Each sample was split up into three parts and transferred to 20 ml phosphate buffer (PBS) and sonicated in an ultrasonic bath for 7 min to detach the microbes from the leaves’ surface. 1 ml of this solution was transferred in 75 ml 10% tryptic soy broth (TSB) medium including cycloheximide (30 mg/l) as a fungicide and incubated and shaken at room temperature for 48 h (culture 1). 1 ml of the resulting liquid bacterial cultures was taken out and added to 50 ml 10% TSB medium stored at 7 °C for 6 days (culture 2). For the final treatment 150 μl of culture 1 and 150 µl of culture 2 were added to 40 ml 10% TSB medium. After incubation at room temperature in the shaker for 24 h, the final cultures were transported to the field site, mixed with 800 ml PBS and transferred to spray bottles. The two resulting bacterial mixtures were applied to plant communities according to which treatment the plot was assigned to until the plants’ surfaces were saturated. The treatment was applied after sunset to prevent the bacterial cultures from drying up. For control groups only phosphate buffer without microbes was applied. Amplicon sequencing was performed for assessing the OTUs (operational taxonomic units) present in the treatment cultures.
Next generation amplicon sequencing
To identify microbial communities in the phyllosphere, pooled leaf samples of each of the 24 plots were collected with sterile forceps to represent the plant community on each plot. The leaf samples were directly transferred to BashingBeads Lysis tubes from the ZymoBIOMICS DNA Miniprep Kit (Zymo Research) containing lysis solution. This was done at three timepoints, once before the treatment (“pre”), one month after the treatment (“post”) and one year after treatment (“post2”). Lysis tubes were sonicated for 7 min to detach microorganisms from plant surfaces. Lysis tubes were then shaken 5 min at 16 Hz using a ball mill and plant material was subsequently removed from the solution. Extraction and purification of DNA was done following the manufacturer’s guidelines (Zymo Research). Microbiome profiling of isolated DNA samples was performed by Eurofins Genomics (Ebersberg, Germany). Eurofins Genomics amplified and Illumina MiSeq sequenced the V3–V4 region of the 16S rRNA gene to identify bacterial operational taxonomic units (OTUs) following the standard procedure “InView—Microbiome Profiling 3.0 with MiSeq”. Sequences were demultiplexed, the primers were clipped, forward, and reverse reads were merged, and merged reads were quality filtered. Microbiome analysis was performed by Eurofins Genomics using the company’s standard procedure (the following description of analysis is provided by Eurofins Genomics): reads with ambiguous bases (“N”) were removed. Chimeric reads were identified and removed based on the de-novo algorithm of UCHIME (Edgar et al. 2011) as implemented in the VSEARCH package (Rognes et al. 2016). The remaining set of high-quality reads was processed using minimum entropy decomposition (Eren et al. 2015). Entropy Decomposition (MED) provides a computationally efficient means to partition marker gene datasets into OTUs (Operational Taxonomic Units). Each OTU represents a distinct cluster with significant sequence divergence to any other cluster. By employing Shannon entropy, MED uses only the information-rich nucleotide positions across reads and iteratively partitions large datasets while omitting stochastic variation. The MED procedure outperforms classical, identity-based clustering algorithms. Sequences can be partitioned based on relevant single nucleotide differences without being susceptible to random sequencing errors. This allows a decomposition of sequence data sets with a single nucleotide resolution. Furthermore, the MED procedure identifies and filters random “noise” in the dataset, i.e., sequences with a very low abundance (less than 0.02% of the average sample size). To assign taxonomic information to each OTU, DC-MEGABLAST alignments of cluster representative sequences to the sequence database were performed (Reference database: NCBI_nt; Release 2018-07-07). A most specific taxonomic assignment for each OTU was then transferred from the set of best-matching reference sequences (lowest common taxonomic unit of all best hits). Hereby, a sequence identity of 70% across at least 80% of the representative sequence was a minimal requirement for considering reference sequences. Further processing of OTUs and taxonomic assignments was performed using the QIIME software package v1.9.1 (http://qiime.org/; Caporaso et al. 2010). Abundances of bacterial taxonomic units were normalized using lineage-specific copy numbers of the relevant marker genes to improve estimates (Angly et al. 2014).
Plant trait measurements
All vascular plant species were identified on each of the 24 plots during the vegetation peak. The young successional plots contained a mean of 2.5 plant species with a range of 2 to 4 species per plot. The old successional plots consisted of a mean of 9 plant species, ranging from 8 to 12 species per plot. We chose the two most abundant plant species Lotus corniculatus var. alpicola and Achillea moschata, which occurred in all plots, to assess the change in functional leaf traits as a function of plot age, sampling time and treatment. Morphological measurements for sampled leaves were performed at three timepoints (Supplementary Information 1), once before the treatment (pre), one month after the treatment (post) and one year after the treatment (post2). The traits considered for this study have been chosen based on their importance for ecological and physiological processes (Stubbs and Wilson 2004). Young, fully expanded leaves without visible damage were sampled in the field and transported in a cooler to the field station where measurements started within 2 h after picking. For up to n = 5 individuals per plant species and per plot, the following vegetative traits were measured to the nearest 0.01 mm using a calliper: leaf width, leaf length, and thickness. Leaf area was obtained by scanning individual leaves with a standard scanner device (HP Scanjet 200). Numbers of pixels per leaf were compared to number of pixels of a 1 cm2 reference using the freeware imageJ (v1.51). Leaves were subsequently frozen and later weighted to determine the fresh mass. Prior to weighing the dry mass of leaves they were placed in a drying oven for 48 h at 80 °C. Leaf succulence (leaf fresh mass/leaf dry mass) and specific leaf area (SLA = leaf area/leaf dry mass) were calculated from the measured values.
Statistical analyses
Statistical analyses for the bacterial dataset were performed on OTU, genus and family level separately and conducted within R v4.0 software environment (R Core Team, 2020). Statistical significance was assessed at α = 0.05. The numbers of total bacterial reads per sample group are summarized in Supplementary Information 2. The dataset was filtered to exclude OTUs with a relative abundance of < 0.01% (R package RAM v1.2.1.7; Chen et al. 2018) and then collapsed at genus and family level for further analyses. Shannon diversity values were calculated after multiple rarefactions of the count data, averaging the results of 999 iterations (R package rtk v0.2.5.7; Saary et al. 2017) to account for uneven sequencing depth. For assessing beta diversity of bacterial community structure, we performed a cumulative sum scaling (CSS) normalization (R package metagenomeSeq v1.28.2; Paulson et al. 2013) on the count data and calculated a distance matrix based on Bray–Curtis dissimilarity.
For investigating the effect of plot age and sampling time on bacterial community composition, Bray–Curtis distances were used for calculating a constrained analysis of principal coordinates (CAP; R package vegan v2.5-6; Oksanen et al. 2019). Plot age and sampling time were used as explanatory variables for this ordination analysis. The significance of factors used as constraints was tested via permutational analysis of variance (PERMANOVA) with 9999 iterations.
To assess alterations in abundance of relevant taxa after the microbial treatment, the indicator taxa for the treatment were calculated at genus level with random forest and results were ordered by Mean Decrease Gini importance (Supplementary Information 3: SI Fig. S1). A repeated k-fold cross validation method was used to train the random forest model (k-fold = 10, repeats = 10; R package caret v6.0-86; Kuhn 2020) for finding the best mtry value, in the interval between 1 and n−1, where n is the number of genera considered. The random forest analysis was then conducted using the previously calculated mtry, ntree = 10, and nrep = 10 (R package rfPermute, v2.1.81; Archer 2020).
To quantify the effect size of our treatments, we calculated the distances between the measurements at different timepoints on each of the plots separately (Tables 1 and 2). Thereby, we were able to directly quantify the effect of the treatments while excluding the noise introduced by the natural variation in measured parameters between the plots and natural fluctuations between the measurements independent of the treatment. To test the effect of our treatment on bacterial community composition we calculated Bray–Curtis distances between each pair of sampling timepoints before and after treatment for each plot separately. For alpha diversity and plant trait values the differences between sampling timepoints before and after treatment (i.e., difference of post—pre and post2—pre) were calculated for each plot separately. In the case of plant traits, the distances were calculated based on mean values of all measured individual plants of the same species per plot. We used Wilcoxon–Mann–Whitney (WMW) test on the calculated distances to determine the effects of treatment on bacterial diversity, community composition and leaf traits of our two focus plant species. Therefore, we tested each pair of treatment groups against each other, with a significant contrast indicating a detectable treatment effect. Results were plotted using the R packages ggplot2 v3.3.2 (Wickham 2016) and ggpubr v0.4.0 (Kassambara 2020).
Results
Effects of successional stage and sampling timepoint on microbiome
Our results show variable richness of foliar bacterial taxa and community composition between successional stages. Prior to the treatment, we found a lower number of OTUs in young successional plots compared to old plots (Wilcoxon–Mann–Whitney test: n = 24, p = 0.032), whereas Shannon diversity did not differ between the two age groups (p = 0.92). Therefore, older succession plant communities were inhabited by a larger number of bacterial taxa, which were not significantly more diverse than in young succession plants. Bacterial communities also showed distinct compositions between old and young successional stages at genus (PERMANOVA: n = 69, F1,65 = 5.05, p < 0.001; Fig. 1) and family level (PERMANOVA: n = 69, F1,65 = 4.76, p < 0.001; Supplementary Information 3: SI Fig. S2). Similarly, we found divergent community composition for each of the three sampling timepoints at both genus (PERMANOVA: n = 69, F2,65 = 7.70, p < 0.001; Fig. 1) and family level (PERMANOVA: n = 69, F2,65 = 8.93, p < 0.001; Supplementary Information 3: SI Fig. S2), showing distinct clusters in the ordination analysis, which suggests a pronounced shift of taxa over time. Samples collected before and after the treatment in the same year were sequenced in a separate run than the post-treatment samples of the second year and meaningful statistical analyses including all three timepoints on OTU level are, therefore, not possible.

Similarity of bacterial genus composition in relation to plot age and sampling timepoint. Constrained analysis of principal coordinates of bacterial genera composition (Bray–Curtis distances) of leaves sampled in replicated plots within two different successional stages, young (purple) and old (pink). Samples were taken at three different timepoints: before treatment (pre), few weeks after treatment (post) and one year after treatment (post2), which are indicated by the shape of data points (circle, square and triangle, respectively). The position of points in the ordination is determined by bacterial genus composition and distance between the points is a measure for the dissimilarity in the community composition (i.e., the further two points are apart the more they differ). Arrows indicate parameters significantly structuring bacterial community composition and point in the direction of the most rapid increase for the respective parameter, with the length of arrows being proportional to the strength of this relationship. Ellipses are indicating the 95% confidence region of the different factors. The different panels are showing the factors plot age and sampling timepoint together (a) as well as separated for plot age (b) and sampling timepoint (c)
Treatment effects on microbiome
The treatment solution containing bacteria sampled from young succession plants was less diverse than the old succession treatment with 59 OTUs in 2 genera (Pantoea, Pseudomonas) and 168 OTUs in four genera (Bacillus, Pseudomonas, Rhodococcus, Variovorax), respectively. Random forest results based on phyllosphere bacteria samples one month after the treatment revealed the following ten most relevant genera changing in abundance between the different treatment types (Fig. 2): Pseudomonas, Acidiphilium, Sphingomonas, Beijerinckia, Variovorax, Hymenobacter Xylophilus, Methylobacterium, Massilia, Arthrobacter. To investigate the treatment effects on bacterial alpha diversity, we tested whether differences between the Shannon diversity in samples taken before and after the treatments for each plot varied as a function of the treatment. Control samples showed slight variations in diversity between the two measurements, suggesting natural fluctuations over time. Shannon diversity of genus level bacteria increased after inoculation with microbes from old successional plots, in contrast to control plots (WMW: n = 14, p = 0.038; Fig. 3a), whereas there was no significant contrast between young treatment and control (n = 15, p = 0.19; Fig. 3a). The change in Shannon diversity after the application of old succession bacteria was more apparent in young plots than in old plots (Table 1, Supplementary Information 3: SI Fig. S3). There was no apparent variation of Shannon diversity on genus level between different treatments one year after the treatment (Fig. 3b). Alpha diversity of family level bacteria showed no significant alterations in response to the treatment in neither of the two post-treatment samples.

Composition of the ten most abundant bacteria genera on plant leaves sampled in two distinct successional stages (old and young), at two sampling timepoints (pre and post), in relation to different microbial treatments (control, old, young). The relative abundance represents the mean distribution of normalized 16S rDNA gene sequence counts for each genus

Changes of bacterial genus alpha diversity and community composition in relation to microbial treatments. a, b Boxplots showing the change in Shannon diversity of genus level bacterial communities in the phyllosphere in response to treatments with bacteria from old and young successions within the same season (a) and one year after treatment (b). c, d: Changes in community composition (Bray–Curtis distances) of foliar bacterial communities at genus level due to microbial treatment within the same season (c) and one year after treatment (d). The x-axis is showing the different treatments (control, young, old) and the y-axis is indicating the change for each sample before and after treatment. The upper and lower box margins mark the 75th and the 25th percentiles, respectively, with the line in the centre showing the median. The whiskers are indicating the minimum and maximum except for outliers. Values above the boxplots display p values of Wilcoxon–Mann–Whitney tests between each pair of treatment groups
For assessing the effects of treatment on bacterial community composition, we tested whether Bray–Curtis distances between pairs of samples taken before and after the inoculation differed from each other regarding the treatment. Bray–Curtis distances between pre- and post-treatment samples of bacterial community composition increased significantly after old treatment compared to the control samples (n = 14, p < 0.01; Fig. 3c) but did not significantly vary between young treatment and control (n = 15, p < 0.40; Fig. 3c). Hence, the treatment with bacteria isolated from old communities resulted in stronger alterations in the community composition than the treatment with bacteria isolated from young plots and control. Resident bacterial communities on old plots seemed to be more resilient against the experimental introduction of additional bacteria than the young plots, i.e., communities before and after the treatment on young plots tended to have larger distances compared to communities on old successional plots (Table 1, Supplementary Information 3: SI Fig. S4). There was again no significant variation in community composition between treatments one year later (Fig. 3d).
Functional leaf traits
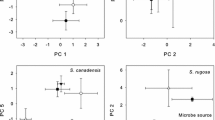
Our study showed alterations of leaf traits after the manipulation of microbial communities on both focus plant species. For A. moschata, we found that three functional leaf traits in old and young successional plots responded differently to the treatment. We found significant alterations in three traits between the plants treated with the two bacterial treatments, but not between the treatments and the control plants: leaf length (WMW: n = 13, p = 0.022; Fig. 4a), leaf dry mass (n = 13, p = 0.035; Fig. 4c), and leaf area (n = 13, p < 0.035, Fig. 4e). After the inoculation with old succession bacteria, length, dry mass, and area of the measured leaves were higher than before the treatment. In contrast, values of all three traits decreased after the inoculation with bacteria from young succession communities. For L. corniculatus we found a significant decrease in leaf length after young treatment (n = 12, p = 0.030, Fig. 5a) and a decreasing trend after old treatment (n = 11, p = 0.052) compared to the control. For both plant species the effect of treatment was not detectable any longer one year later (Figs. 4b, d, f, 5b). Control plants of both plant species showed variations in leaf traits between two measurement timepoints, suggesting natural fluctuations over time independent of the treatment.

Changes in leaf traits of A. moschata in relation to microbiome manipulation. Alterations of leaf length (a, b), leaf dry mass (c, d), and leaf area (e, f) in response to bacterial treatments. The x-axis shows the different treatments (control, young, old), while the y-axis indicates changes in normalized trait values within the same season (a, c, e) and one year after treatment (b, d, f). Values above zero show positive alterations between two measurements and values below zero display negative changes. The upper and lower box margins mark the 75th and the 25th percentiles, respectively, with the line in the centre showing the median. The whiskers are indicating the minimum and maximum except for outliers. Values above the boxplots show p values of Wilcoxon–Mann–Whitney tests between each pair of treatment groups

Alterations in leaf length of L. corniculatus in response to bacterial treatments. The x-axis shows the different treatments (control, young, old), while the y-axis indicates changes in normalized trait values within the same season (a) and one year after treatment (b). Values above zero show positive alterations in traits between two measurements and values below zero display negative changes. The upper and lower box margins mark the 75th and the 25th percentiles, respectively, with the line in the centre showing the median. The whiskers are indicating the minimum and maximum except for outliers. Values above the boxplots show p values of Wilcoxon–Mann–Whitney tests between each pair of treatment groups
Discussion
Our study documents differences in the composition of foliar bacterial communities between plant communities of young and old successional stages as well as shifts throughout a season and between years. Additionally, our data show that cultured bacteria transferred between plant communities of two different successional stages can alter diversity and composition of the microbiome on plant community level as well as species-specific functional plant traits. Furthermore, our results also demonstrate a strong resilience of plant-associated bacterial communities and of plants in response to bacterial invaders.
Community composition of bacteria in the phyllosphere was differing between the three intra- and inter-annual sample events, which is congruent with findings of previous studies showing seasonally dynamic epiphytic microbial communities (Redford and Fierer 2009; Copeland et al. 2015; Vacher et al. 2016). Within microbial communities, there are constant shifts in relative abundance of taxa and this temporal variability likely results from the high exposure of the leaf surface to frequent environmental changes, where microbial epiphytes are influenced by UV radiation, water, nutrients and plant metabolism (Vorholt 2012). In addition to environmental conditions at the leaf surface, changing physiology throughout plant development has a large influence on foliar microbes, leading to fluctuations in bacteria community composition within a plant’s life cycle (Stone et al. 2018; Koskella 2020).
We found that community composition of bacteria was shaped by plant communities of different ages along the succession, which in turn differed in their species composition. Plant species identity is widely considered the predominant factor influencing the composition of foliar bacteria, as plant genetic traits mediate production of secondary metabolites and topology of the leaves (Junker and Keller 2015; Boachon et al. 2019; Gaube et al. 2020). This is specifically the case for the so-called core plant microbiome, which consists of microorganisms that are selected by a certain plant species or genotype, largely independent of environmental conditions (Kumar et al. 2017; Compant et al. 2019). Apart from the core microbiome there is a considerable proportion of non-host-specific but rather site-specific microbial taxa, depending on the environment (Knief et al. 2010; Massoni et al. 2020). Microbes can be transmitted horizontally through the environment or vertically, passed from parent to offspring, resulting in coadaptation with their plant host leading to a more distinct microbiome within a plant population (Mitter et al. 2017; Hassani et al. 2019). Leaf and root microbiome of plants are showing a large overlap, suggesting leaf inoculation from soil (Wagner et al. 2016; Massoni et al. 2021) and this may particularly apply to low-statue alpine plants with aboveground tissues close to the soil. Microbial communities of newly deglaciated unvegetated soils are structured by substrate age and follow predictable composition patterns with increasing biomass and functional diversity along the advancing plant succession where they are directly impacted through litter inputs and root exudates of different species (Knelman et al. 2012; Yuan et al. 2017). Therefore, aboveground plant diversity can predict diversity of soil microbial communities in alpine environments which in turn may affect the assembly of microbiota in aboveground plant tissues through active recruitment of the host plant (Schulz-Bohm et al. 2018). Additionally, species richness in shoots was found to increase with plant species richness, suggesting additional dispersal from neighbouring vegetation (Navrátilová et al. 2019). The contrast between microbes associated with plant communities in different successional stages may, therefore, be the result of a combination of differing local host species composition per se as well as a variation in non-host specific taxa affected by the location and facilitated by vertical and horizontal transmission.
The plant phyllosphere is a highly competitive and challenging habitat for microbes to colonize and the survival of newly arriving microbes is determined by the abiotic environment, the resident microbial composition, and the plant host itself (Koskella 2020). Our experiment illustrates that the transfer of culturable foliar microbial communities from wild plant communities of two different successional stages can alter the plant microbiome on community level. Due to natural variations, there was a large difference in bacterial composition within individual plots before the treatment. We accounted for this difference by quantifying the change of the bacterial community within each plot separately before and after the treatment. We were only able to culture a small part of the highly diverse phyllosphere bacteria for our treatment solutions, which poses a general limitation for approaches trying to transfer entire naturally occurring microbial communities. An alternative approach of transferring a simple leaf slurry proved successful in a study by Zahn and Amend (2017) but was not suited for our research questions due to the additional input of organic material. Nevertheless, the treatment solutions differed in their bacterial community composition between old and young plant communities. The treatment effect was more pronounced after inoculations with the more diverse old succession bacteria especially when applied to young successional plots. This may be facilitated by the lower number of bacterial taxa within young plant communities that are thus more susceptible to invasions. Even at seemingly saturated levels of primary colonizers on leaves, some secondary colonizers arriving in an established community may still be able to reproduce and exert effects on the plant (Remus‐Emsermann et al. 2013). Species-rich communities are considered to be more resistant to invasion due to their larger repertoire of competitive abilities for resources compared to communities with lower diversity (van Elsas et al. 2012; Vila et al. 2019). Plant-associated microbes may produce chemical compounds that directly suppress the growth of other microbes and thereby reducing the success of newly arriving microbiota (Bonanomi et al. 2014; Hassani et al. 2018).
Plant-associated microbial diversity has not just implications for possible invaders but is also important for plant growth and health in general. Phyllosphere-colonizing bacteria produce a range of compounds that directly affect plant metabolism or modulate phytohormones, which are essential for healthy plant growth and can contribute to substantial phenotypic modifications (Lau and Lennon 2011; Vandenkoornhuyse et al. 2015; Hubbard et al. 2019). We found that leaf length, leaf area and leaf mass were affected differently by the microbiome manipulation treatments. The affected traits also differed between the two observed plant species. Although traits of A. moschata after the treatment did not differ significantly compared to the control plants, there is still an apparent effect as the changes in traits after the young and the old succession treatments go in opposite directions, showing an increase or decrease in leaf size, respectively. The species-specific results are possibly due to contrasting resident microbial composition of the host plant as well as distinct topological and chemical conditions of the leaf surface. There are several examples of single bacterial taxa tested to have positive effects on plant traits with promising implications for biotechnological use in agriculture and potential applications for nature-based solutions (Abhilash et al. 2016; Finkel et al. 2017). However, plants in natural systems interact with far more complex microbial communities and studies investigating effects of whole microbe communities on plant traits remain sparse and are mostly restricted to soil microbiota.
The lack of being able to detect a long-lasting effect on both bacterial composition and leaf traits in the following growing season demonstrates the resilience of bacterial communities associated with plants as well as of plants in response to bacterial invasions. Coinciding with theoretical and experimental predictions (van Elsas et al. 2012; Vila et al. 2019) more diverse bacterial communities were more resilient than bacterial communities with low OTU numbers. The microbiomes’ and the plants’ resilience were likely supported by the fluctuating and unstable ephemeral environment of the phyllosphere in which annual plants complete their life cycle within a single growth season and perennial deciduous plants form and shed leaves every year. These factors clearly contributed to the lack of a signal of experimental bacterial additions in the second year of our investigation.
Our study demonstrates that inoculation experiments in the field with naturally occurring foliar microbial communities of wild plants can be used to investigate the resilience of bacterial communities as well as microbial effects on plant functional traits. In addition to laboratory-based studies in controlled environments, manipulative field experiments with complex natural communities are needed to disentangle the microbial effects in covariation with the environment. Bacterial communities associated with plants can exhibit distinct responses to host genotype and spatio-temporal variation and our study illustrates the importance of replicating microbiome experiments across space and time to reveal these relationships. A better understanding of the complex interactions between microbial communities, environment and plant traits is necessary for future investigations in microbial ecology and agricultural science.
Availability of data and materials
Raw data and figures supporting the results of this study can be found in the Online Resources (Supplementary Information 1–3). Bacterial sequences can be accessed in Fastaq files deposited at the NCBI Sequence Read Archive (SRA) under the BioProject accession PRJNA698437.
Code availability
Not applicable.
References
Abhilash P, Dubey RK, Tripathi V, Gupta VK, Singh HB (2016) Plant growth-promoting microorganisms for environmental sustainability. Trends Biotechnol 34:847–850. https://doi.org/10.1016/j.tibtech.2016.05.005
Angly FE, Dennis PG, Skarshewski A, Vanwonterghem I, Hugenholtz P, Tyson GW (2014) CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome 2:1–13. https://doi.org/10.1186/2049-2618-2-11
Archer E (2020) rfPermute: Estimate Permutation p-Values for Random Forest Importance Metrics. R package version 2.1.81. https://www.CRANR-projectorg/package=rfPermute
Beilsmith K, Thoen MP, Brachi B, Gloss AD, Khan MH, Bergelson J (2019) Genome-wide association studies on the phyllosphere microbiome: Embracing complexity in host–microbe interactions. Plant J 97:164–181. https://doi.org/10.1111/tpj.14170
Berg G, Köberl M, Rybakova D, Müller H, Grosch R, Smalla K (2017) Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol Ecol 93:fix050. https://doi.org/10.1093/femsec/fix050
Boachon B, Lynch JH, Ray S, Yuan J, Caldo KMP, Junker RR, Kessler SA, Morgan JA, Dudareva N (2019) Natural fumigation as a mechanism for volatile transport between flower organs. Nat Chem Biol 15:583–588. https://doi.org/10.1038/s41589-019-0287-5
Bonanomi G, Capodilupo M, Incerti G, Gaglione SA, Scala F (2014) Fungal diversity increases soil fungistasis and resistance to microbial invasion by a non resident species. Biol Control 72:38–45. https://doi.org/10.1016/j.biocontrol.2014.02.005
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chaudhry V, Runge P, Sengupta P, Doehlemann G, Parker JE, Kemen E (2021) Shaping the leaf microbiota: plant–microbe–microbe interactions. J Exp Bot 72:36–56. https://doi.org/10.1093/jxb/eraa417
Chen W, Simpson J, Levesque C (2018) RAM: R for Amplicon-Sequencing-Based Microbial-Ecology. R package version 1.2.1.7., https://CRAN.R-project.org/package=RAM
Compant S, Samad A, Faist H, Sessitsch A (2019) A review on the plant microbiome: ecology, functions and emerging trends in microbial application. J Adv Res 19:29–37. https://doi.org/10.1016/j.jare.2019.03.004
Copeland JK, Yuan L, Layeghifard M, Wang PW, Guttman DS (2015) Seasonal community succession of the phyllosphere microbiome. Mol Plant Microbe Interact 28:274–285. https://doi.org/10.1094/MPMI-10-14-0331-FI
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML (2015) Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J 9:968–979. https://doi.org/10.1038/ismej.2014.19
Escobar Rodríguez C, Antonielli L, Mitter B, Trognitz F, Sessitsch A (2020) Heritability and functional importance of the Setaria viridis bacterial seed microbiome. Phytobiomes J 4:40–52. https://doi.org/10.1094/PBIOMES-04-19-0023-R
Finkel OM, Castrillo G, Paredes SH, González IS, Dangl JL (2017) Understanding and exploiting plant beneficial microbes. Curr Opin Plant Biol 38:155–163. https://doi.org/10.1016/j.pbi.2017.04.018
Fischer A (2018) Die Bedeutung historischer und aktueller Gletscherforschungen des Alpenvereins für die Geowissenschaften. In: Gletscher im Wandel. Springer Spektrum, Berlin, Heidelberg, pp 97–116. https://doi.org/10.1007/978-3-662-55540-8_6
Friesen ML, Porter SS, Stark SC, Von Wettberg EJ, Sachs JL, Martinez-Romero E (2011) Microbially mediated plant functional traits. Annu Rev Ecol Evol Syst 42:23–46. https://doi.org/10.1146/annurev-ecolsys-102710-145039
Gaube P, Junker RR, Keller A (2020) Changes amid constancy: flower and leaf microbiomes along land use gradients and between bioregions. Basic Appl Ecol 50:1–15. https://doi.org/10.1016/j.baae.2020.10.003
Hassani MA, Durán P, Hacquard S (2018) Microbial interactions within the plant holobiont. Microbiome 6:58. https://doi.org/10.1186/s40168-018-0445-0
Hassani MA, Özkurt E, Seybold H, Dagan T, Stukenbrock EH (2019) Interactions and coadaptation in plant metaorganisms. Annu Rev Phytopathol 57:483–503. https://doi.org/10.1146/annurev-phyto-082718-100008
Hilger L, Dusik J-M, Heckmann T, Haas F, Glira P, Pfeifer N, Vehling L, Rohn J, Morche D, Baewert H (2019) A Sediment Budget of the Upper Kaunertal. In: Geomorphology of Proglacial Systems. Springer, Cham, pp 289–312. https://doi.org/10.1007/978-3-319-94184-4_17
Hubbard CJ, Li B, McMinn R, Brock MT, Maignien L, Ewers BE, Kliebenstein D, Weinig C (2019) The effect of rhizosphere microbes outweighs host plant genetics in reducing insect herbivory. Mol Ecol 28:1801–1811. https://doi.org/10.1111/mec.14989
Junker RR, Keller A (2015) Microhabitat heterogeneity across leaves and flower organs promotes bacterial diversity. FEMS Microbiol Ecol 91:fiv097. https://doi.org/10.1093/femsec/fiv097
Junker RR, Hanusch M, He X, Ruiz-Hernández V, Otto J-C, Kraushaar S, Bauch K, Griessenberger F, Ohler L-M, Trutschnig W (2020) Ödenwinkel: an Alpine platform for observational and experimental research on the emergence of multidiversity and ecosystem complexity. Web Ecol 20:95–106. https://doi.org/10.5194/we-20-95-2020
Kassambara A (2020) ggpubr: ‘ggplot2’ Based Publication Ready Plots. R package version 0.4.0. https://CRAN.R-project.org/package=ggpubr
Kazemi S, Hatam I, Lanoil B (2016) Bacterial community succession in a high-altitude subarctic glacier foreland is a three-stage process. Mol Ecol 25:5557–5567. https://doi.org/10.1111/mec.13835
Knelman JE, Legg TM, O’Neill SP, Washenberger CL, González A, Cleveland CC, Nemergut DR (2012) Bacterial community structure and function change in association with colonizer plants during early primary succession in a glacier forefield. Soil Biol Biochem 46:172–180. https://doi.org/10.1016/j.soilbio.2011.12.001
Knelman JE, Graham EB, Prevéy JS, Robeson MS, Kelly P, Hood E, Schmidt SK (2018) Interspecific plant interactions reflected in soil bacterial community structure and nitrogen cycling in primary succession. Front Microbiol 9:128. https://doi.org/10.3389/fmicb.2018.00128
Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA (2010) Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J 4:719–728. https://doi.org/10.1038/ismej.2010.9
Koskella B (2020) The phyllosphere. Curr Biol 30:R1143–R1146. https://doi.org/10.1016/j.cub.2020.07.037
Kuhn M (2020) caret: Classification and Regression Training. R package version 6.0-86. https://www.CRANR-projectorg/package=caret
Kumar M, Brader G, Sessitsch A, Mäki A, van Elsas JD, Nissinen R (2017) Plants assemble species specific bacterial communities from common core taxa in three arcto-alpine climate zones. Front Microbiol 8:12. https://doi.org/10.3389/fmicb.2017.00012
Laforest-Lapointe I, Messier C, Kembel SW (2016) Host species identity, site and time drive temperate tree phyllosphere bacterial community structure. Microbiome 4:27. https://doi.org/10.1186/s40168-016-0174-1
Lau JA, Lennon JT (2011) Evolutionary ecology of plant–microbe interactions: soil microbial structure alters selection on plant traits. New Phytol 192:215–224. https://doi.org/10.1111/j.1469-8137.2011.03790.x
Lieb G, Kellerer-Pirklbauer A (2020) Gletscherbericht 2019/20 Sammelbericht über die Gletschermessungen des Österreichischen Alpenvereins im Jahr 2020. Letzter Bericht: Bergauf 2/2020, Jg. 75 (145): 6–15
Liu H, Brettell LE, Singh B (2020) Linking the phyllosphere microbiome to plant health. Trends Plant Sci 25:841–844. https://doi.org/10.1016/j.tplants.2020.06.003
Massoni J, Bortfeld-Miller M, Jardillier L, Salazar G, Sunagawa S, Vorholt JA (2020) Consistent host and organ occupancy of phyllosphere bacteria in a community of wild herbaceous plant species. ISME J 14:245–258. https://doi.org/10.1038/s41396-019-0531-8
Massoni J, Bortfeld-Miller M, Widmer A, Vorholt JA (2021) Capacity of soil bacteria to reach the phyllosphere and convergence of floral communities despite soil microbiota variation. PNAS 118:e2100150118. https://doi.org/10.1073/pnas.2100150118
Matthews JA (1992) The ecology of recently-deglaciated terrain: a geoecological approach to glacier forelands. Cambridge Univ Press. https://doi.org/10.1002/jqs.3390080213
Mitter B, Pfaffenbichler N, Flavell R, Compant S, Antonielli L, Petric A, Berninger T, Naveed M, Sheibani-Tezerji R, von Maltzahn G (2017) A new approach to modify plant microbiomes and traits by introducing beneficial bacteria at flowering into progeny seeds. Front Microbiol 8:11. https://doi.org/10.3389/fmicb.2017.00011
Navrátilová D, Tláskalová P, Kohout P, Dřevojan P, Fajmon K, Chytrý M, Baldrian P (2019) Diversity of fungi and bacteria in species-rich grasslands increases with plant diversity in shoots but not in roots and soil. FEMS Microbiol Ecol 95:fiy208. https://doi.org/10.1093/femsec/fiy208
Nemergut DR, Anderson SP, Cleveland CC, Martin AP, Miller AE, Seimon A, Schmidt SK (2007) Microbial community succession in an unvegetated, recently deglaciated soil. Microb Ecol 53:110–122. https://doi.org/10.1007/s00248-006-9144-7
Oksanen J, Guillaume Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin P, O’Hara R, Simpson G, Solymos P, Henry M, Stevens H et al. (2019) vegan: Community Ecology Package. R package version 2.5–6. https://CRAN.R-project.org/package=vegan
Paulson JN, Stine OC, Bravo HC, Pop M (2013) Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10:1200–1202. https://doi.org/10.1038/nmeth.2658
Porazinska DL, Farrer EC, Spasojevic MJ, Bueno de Mesquita CP, Sartwell SA, Smith JG, White CT, King AJ, Suding KN, Schmidt SK (2018) Plant diversity and density predict belowground diversity and function in an early successional alpine ecosystem. Ecology 99:1942–1952. https://doi.org/10.1002/ecy.2420
Redford AJ, Fierer N (2009) Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb Ecol 58:189–198. https://doi.org/10.1007/s00248-009-9495-y
Remus-Emsermann MN, Kowalchuk GA, Leveau JH (2013) Single-cell versus population-level reproductive success of bacterial immigrants to pre-colonized leaf surfaces. Environ Microbiol Rep 5:387–392. https://doi.org/10.1111/1758-2229.12040
Rognes T, Flouri T, Nichols B, Quince C, Mahé F (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. https://doi.org/10.7717/peerj.2584
Saary P, Forslund K, Bork P, Hildebrand F (2017) RTK: efficient rarefaction analysis of large datasets. R package version 0.2.6.1, https://github.com/hildebra/Rarefaction
Schulz-Bohm K, Gerards S, Hundscheid M, Melenhorst J, de Boer W, Garbeva P (2018) Calling from distance: attraction of soil bacteria by plant root volatiles. ISME J 12:1252–1262. https://doi.org/10.1038/s41396-017-0035-3
Stone BW, Weingarten EA, Jackson CR (2018) The role of the phyllosphere microbiome in plant health and function. Annu Plant Rev. https://doi.org/10.1002/9781119312994.apr0614
Stubbs WJ, Wilson JB (2004) Evidence for limiting similarity in a sand dune community. J Ecol 92:557–567. https://doi.org/10.1111/j.0022-0477.2004.00898.x
Vacher C, Hampe A, Porté AJ, Sauer U, Compant S, Morris CE (2016) The phyllosphere: microbial jungle at the plant–climate interface. Annu Rev Ecol Evol Syst 47:1–24. https://doi.org/10.1146/annurev-ecolsys-121415-032238
van Elsas JD, Chiurazzi M, Mallon CA, Elhottovā D, Krištůfek V, Salles JF (2012) Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc Natl Acad Sci 109:1159–1164. https://doi.org/10.1073/pnas.1109326109
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A (2015) The importance of the microbiome of the plant holobiont. New Phytol 206:1196–1206. https://doi.org/10.1111/nph.13312
Vila JC, Jones ML, Patel M, Bell T, Rosindell J (2019) Uncovering the rules of microbial community invasions. Nat Ecol Evol 3:1162–1171. https://doi.org/10.1038/s41559-019-0952-9
Vorholt JA (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840. https://doi.org/10.1038/nrmicro2910
Wagner MR, Lundberg DS, Tijana G, Tringe SG, Dangl JL, Mitchell-Olds T (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun 7:1–15. https://doi.org/10.1038/ncomms12151
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer, New York
Yuan X, Knelman JE, Wang D, Goebl A, Gasarch E, Seastedt TR (2017) Patterns of soil bacterial richness and composition tied to plant richness, soil nitrogen, and soil acidity in alpine tundra. Arct Antarct Alp Res 49:441–453. https://doi.org/10.1657/AAAR0016-050
Zahn G, Amend AS (2017) Foliar microbiome transplants confer disease resistance in a critically-endangered plant. PeerJ 5:e4020. https://doi.org/10.7717/peerj.4020
Zumsteg A, Luster J, Göransson H, Smittenberg RH, Brunner I, Bernasconi SM, Zeyer J, Frey B (2012) Bacterial, archaeal and fungal succession in the forefield of a receding glacier. Microb Ecol 63:552–564. https://doi.org/10.1007/s00248-011-9991-8
Acknowledgements
We thank local stakeholders, namely the Agrargemeinschaft Birgalpe, Naturpark Kaunergrat, the municipality of Feichten, the operator of the Kaunertal glacier road (Kaunertaler Gletscherbahnen GmbH), the governing authority Bezirkshauptmannschaft Landeck (Land Tirol, Austria) for the permit to conduct our research (LA-NSCH/B-145/3-2018), and the Tyrolian Water Company (TIWAG) for allowing and supporting the field work necessary for data collection.
Funding
Open Access funding enabled and organized by Projekt DEAL. The present study is part of the PHUSICOS project, funded by the European Union’s Horizon 2020 research and innovation program under Grant agreement No. 776681.
Author information
Authors and Affiliations
Contributions
All authors contributed to study design and method development. LMO and SS performed material preparation and data collection. LMO and RRJ performed statistical analyses. LMO drafted the manuscript and all authors contributed to the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohler, LM., Seeleitner, S., Haselberger, S. et al. Manipulation of phyllosphere bacterial communities reversibly alters the plant microbiome and leaf traits in the field. Alp Botany 132, 301–314 (2022). https://doi.org/10.1007/s00035-021-00273-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-021-00273-8