
Abstract
Reliable biodiversity data are crucial for environmental research and management. Unfortunately, data paucity prevails for many regions and organismal groups such as aquatic invertebrates. High-throughput DNA-based identification, in particular DNA metabarcoding, has accelerated biodiversity data generation. However, in the process of metabarcoding, specimens are usually destroyed, precluding later specimen-based analyses. Metabarcoding of DNA released into the preservative ethanol has been proposed as a non-destructive alternative, but proof-of-principle studies have yielded ambiguous results, reporting variance in detection probability for various taxa and methodological biases. In this study, we tested the performance of preservative-based metabarcoding of aquatic invertebrates in comparison to a standard morpho-taxonomic assessment based on samples from one of Europe’s last free-flowing rivers, the Vjosa. Multi-habitat samples were collected at 43 sites in two seasons and stored in ethanol, after fixation in formaldehyde for morpho-taxonomic analyses. Preservative-based DNA metabarcoding detected three times more taxa but failed to detect other taxa found using the standard method. In addition to incomplete reference data and primer bias that likely precluded the detection of specific taxa, preservative-based DNA metabarcoding cannot provide accurate abundance estimates. However, the metabarcoding data revealed distinct small-scale and large-scale community patterns in the Vjosa river network, which were also recovered by quantitative data of the standard approach. Overall, our results indicate that preservative-based metabarcoding provides important biodiversity data, which could be further improved by quantitative validation. The method is robust and reliable, even though samples were taken under harsh field-conditions and stored without cooling. Further, our results emphasise the need for reliable DNA barcoding reference libraries. Building those may be supported by preservative-based metabarcoding that maintains intact vouchers for subsequent specimen-based analyses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biodiversity data are key for ecological research as well as ecosystem management. Stringent sampling and sample processing are necessary to obtain high-quality biodiversity data (Tessarolo et al. 2017). Which method is used to assess biodiversity depends on the habitat and taxa under investigation, and each method comes with its own limitations. Biodiversity of small-bodied freshwater benthic invertebrate fauna is best assessed by actively collecting specimens in bulk through the multi-habitat sampling approach. Bulk samples are fixed or preserved, and material is then manually sorted and separated from debris. Finally, the target specimens are identified and counted. Throughout this procedure, taxonomic expertise is pivotal for sampling, sorting and identification (Wägele et al. 2011).
Taxonomic expertise can be complemented or partly replaced by high-throughput sequencing, including DNA metabarcoding (Ji et al. 2013; Marquina et al. 2019; Bush et al. 2019). For most DNA metabarcoding analyses, specimens are homogenized to facilitate DNA extraction—preventing later validation of results (Elbrecht and Leese 2015; Hajibabaei et al. 2019). A potential solution to this problem is DNA extraction by incubating bulk samples in a lysis buffer and the subsequent DNA isolation from this buffer (Nielsen et al. 2019). Yet, this approach is costly and makes specimens fragile. An alternative is to extract DNA directly from the preservative ethanol. This strategy preserves voucher specimens for later use (e.g. to confirm the detection of a taxon) and takes advantage of time and cost-efficient high-throughput sequencing (Hajibabaei et al. 2012; Marquina et al. 2019; Martins et al. 2019; Zizka et al. 2019). However, the suitability of this method for routine biodiversity assessment remains to be evaluated.
First proof-of-principle studies on freshwater biodiversity compared the performance of preservative-based DNA metabarcoding with standard approaches (Martins et al. 2019; Erdozain et al. 2019) but also to metabarcoding of environmental DNA (eDNA) (Marquina et al. 2019; Persaud et al. 2021) or homogenized samples (Marquina et al. 2019; Zizka et al. 2019). These comparisons yielded encouraging yet incongruent results. Specifically, detection may be precluded by low biomass of rare or small taxa or by a particularly tough and dense anatomy (Marquina et al. 2019; Martins et al. 2021). Together with primer bias, PCR stochasticity, and environmental PCR inhibitors, these technical aspects potentially degrade the efficiency of preservative-based DNA metabarcoding. Additionally, these first studies were set in a context relating to ecological status assessment and did not explicitly aim to recover biodiversity turnover patterns along spatial gradients of dendritic fluvial networks. Thus, it is still unclear to what degree this method is suitable for routine stream ecology applications.
To illuminate this issue, we benchmark preservative-based DNA metabarcoding against a standardized assessment of benthic invertebrate fauna biodiversity in a large near-natural river network. Thereby we also assess the power of this method in the context of conservation biology: few near-natural, free-flowing river networks still exist in Europe and the majority lie in the Balkans (Schiemer et al. 2020; Schwarz 2022). The Vjosa is a particularly good example of a hydromorphologically minimally impaired system, where the natural flow and sediment dynamics of the main stem as well as of most of its tributaries are still intact. The Vjosa riverscape supports endemic flora as well as the critically endangered European eel and a highly diverse benthic invertebrate fauna (Schiemer et al. 2020). Ongoing hydropower development threatens the unique fluvial networks in the Balkans and conservation actions are urgently required (Schiemer et al. 2020). Consequently, intense research on biodiversity and ecological functioning of the Vjosa aims to collect benchmark data—as evidence for conservation needs, but also as a guide for river restoration elsewhere (Schiemer et al. 2020).
Assessing biodiversity in the Vjosa river network is particularly challenging. Sampling efforts are limited so far and taxonomic literature is lacking, yet the existing evidence from biodiversity surveys suggests the presence of many endemic and rare taxa (e.g. Martini et al. 2022). Consequently, DNA barcode reference data for the Vjosa fauna is less complete than for other European river networks, preventing accurate community characterisation based on DNA metabarcoding. But, preservative-based DNA metabarcoding may offer an enhanced resolution of both taxonomic richness and spatially structured community turnover in these circumstances. However, sampling the Vjosa catchment imposes logistic constraints and forces extended transportation and prolonged storage at high ambient temperatures. So far, only a few studies with optimally preserved samples (limited transport, cold storage) achieved an assessment of benthic invertebrate fauna communities through preservative-based DNA metabarcoding (Hajibabaei et al. 2012; Zizka et al. 2019; Erdozain et al. 2019). Suboptimal sample treatment may—in addition to technical challenges—also diminish the capacities of this method for freshwater biodiversity assessment in a monitoring context or in remote regions such as the Vjosa catchment.
In the present study, we assess the performance of preservative-based DNA metabarcoding under realistic conditions typical for large-scale biodiversity assessments in stream networks. We compare taxonomic inventories of benthic invertebrate fauna obtained by standard and preservative-based DNA methods. To this end, we use multi-habitat samples taken in parallel at 43 sites—one set fixed in formaldehyde for standard assessments, the other preserved in ethanol—across the Vjosa river network over two sampling seasons. Moreover, we contrast river network-scale community patterns to assess data sufficiency for ecological analyses. We achieve this by relating biodiversity turnover in the Vjosa network as measured by standard and preservative-based DNA methods to asymmetric eigenvector maps that formalize autocorrelated, hierarchical spatial gradients of stream networks, and thereby assess how well biodiversity data obtained through the two approaches depict directional fluvial processes.
Materials and methods
Study river network, sampling procedure and fixation/preservation strategy
The Vjosa river network is located in the southern Balkan [40.680°N, 19.320°E; 40.680°N, 21.180°E; 39.740°N, 19.320°E; 39.740°N, 21.180°E] and covers a catchment of approx. 6700 km2 ranging in elevation from 0 to 1600 m a.s.l. from the Adriatic Sea to the Pindos Mountains. Upstream, the river network comprises the Aoos and the Sarantaporos rivers that create the Vjosa at their confluence (at [40.071°N, 20.589°E]); a sizeable tributary of the Aoos is the Voidomatis, while the Sarantaporos receives only minor tributaries. Larger tributaries of the Vjosa are the Langarica, the Dishnica, the Drinos, the Bence, the Luftinja and the Shushica. Land use intensifies as the river grows, with downstream sections dominated by permanent crops and heterogeneous agricultural areas. Geological diversity is high: (Upper) Aoos and Sarantaporos catchments are dominated by ultramafic rock and sandstones, respectively, while the Vjosa main stem (as well as the Voidomatis catchment) is located mostly in clastic formations, flysch and limestone (Martini et al. 2022).
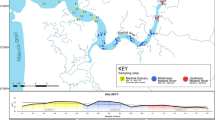
In total, 86 multi-habitat samples (MHS) were collected with a 500 µm mesh size quadratic net (Hering et al. 2003) across the entire catchment during a spring and an autumn campaign, i.e. 43 MHS in May and September/October 2018, respectively (Fig. 1). Sampling sites were selected to reflect the hierarchical structure of the river network and efficiently capture biodiversity. According to the MHS procedure, 20 single samples, each corresponding to an area of 0.0625 m2, were distributed proportionally across existing substrate types at each sampling site. In large-river meandering sections with exclusively mesolithal (6–20 cm) substrate, single samples were distributed to reflect a flow velocity gradient from the shoreline (no flow) to the stream centre (maximum flow). At each site, two MHS were collected in parallel by two investigators sampling the same substrate types while walking against the flow; in concert with a mostly homogeneous substrate type distribution this guaranteed maximum similarity between the two samples collected in parallel. To cater to different prerequisites of standard macroinvertebrate assessment and preservative-based DNA metabarcoding, one MHS was fixed in 3% unbuffered formaldehyde, and the second MHS spun dry and preserved in 1 L 96% denatured ethanol in factory-new 1 L sample containers. We opted for parallel sampling with two fixation/preservation methods because standard method experience shows that formalin-fixed bulk specimens retain some diagnostic characters better than ethanol-preserved ones. For the purpose of this study, we needed to evaluate species composition in the metabarcoding data based on the best possible taxonomic identification. Thus, we obtained morphologically intact specimens as well as preservative ethanol from our extensive MHS sampling effort. Samples were transported by car (2–3 weeks) and stored at ambient temperatures.

Location of the 43 sampling sites in the Vjosa river network
Standard MHS processing
Formaldehyde-fixed samples were rinsed (50 µm mesh size sieve) and analysed following the AQEM protocol (Hering et al. 2003). In brief, one-sixth of each sample was subsampled using a Caton Pan (500 µm mesh). Of this subsample, all specimens were then picked, identified to the lowest feasible taxonomic level (which varied between taxonomic groups) and enumerated. In cases where a sample did not comprise at least 700 specimens, the entire sample was processed. Raw count data were standardized to reflect abundance per m2.
Preservative-based DNA metabarcoding procedure
Ethanol-preserved MHS samples were stored in the dark at room temperature for 3 months before further treatment. Decantation and subsequent filtration of preservative ethanol followed Zizka et al. (2019): a 12% sodium hypochlorite-treated 50 µm sieve was used to retain specimens and particles, and the entire volume was filtered through mounted sterile 0.45 µm nitrocellulose filters (Nalgene Analytical Test Filter Funnel CN, ThermoScientific) using a vacuum pump. For the spring campaign, two filters per MHS were used and pooled for extraction. For the autumn campaign fine particle load was lower and one filter per MHS was sufficient and used for extraction. Filters were folded using sterilized forceps and stored in fresh 96% denatured ethanol at −20 °C until further analyses.
DNA was extracted following Zizka et al. (2019). In brief, we cut filters and dried them in 2 mL PCR-grade reaction vials (Eppendorf) at 56 °C or overnight in petri dishes at room temperature. Then, filters were incubated in 600 µL TNES buffer + 10 µL Proteinase K (10 mg mL−1) per filter at 55° C for at least 3 h. Subsequently, DNA was extracted using a modified salt precipitation protocol to pellet unwanted digestion products (Sunnucks and Hales 1996; Weiss and Leese 2016), followed by ethanol precipitation and centrifugation to pellet extracted DNA. After washing and drying, the pellet was resuspended in 50 µL PCR-grade water. Raw DNA extracts were cleaned up following RNase digestion using the MinElute Reaction CleanUp Kit (Qiagen) and final DNA concentration and quality was quantified on a Qubit 2.0 fluorometer and using agarose gel electrophoresis.
A 421 bp fragment of the mitochondrial cytochrome oxidase I unit (COI, barcode region) was targeted in amplification using 1 µL of the final DNA extracts as described in Zizka et al. (2019). A two-step PCR was conducted for amplification and tagging of the marker gene using the Multiplex PCR Plus Kit (Qiagen). First-step PCR was done using the primer pair BF2/BR2 (0.2 µM) (Elbrecht and Leese 2017) in 50 µl reactions at 95° C 5', 30 x (95° C 30", 50° C 90", 72° C, 120"), 68° C 10'. Second-step PCR to label amplicons was performed with tailed fusion primers BF2/BR2 (Elbrecht and Steinke 2018) at the same temperature conditions as in the first PCR step, but with 15 cycles. For each PCR reaction, a negative (1 µL water instead of DNA) and a positive control (DNA from bulk sample tissue homogenate) were included. Two technical PCR replicates of each sample were produced and uniquely labelled. The library was created by equimolar pooling of labelled and size selected (SPRIselect beads, Beckmann Coulter, ratio 0.76) PCR products and included the amplified samples, negative controls (15 µL of each negative control spiked in) and positive controls. Samples were sequenced at CeGat GmbH using a 250 bp paired-end kit with a sequencing depth of 12 million clusters on an Illumina MiSeq platform (5% PhiX).
Bioinformatic analysis, reference libraries and plausibility check
We excluded 6 autumn samples from our analyses because of sequencing failures (< 150 reads in at least one PCR replicate) and retained 43 spring and 37 autumn samples for downstream analyses (Online resource Tab. SI 1). Raw metabarcoding data were quality checked with FastQC v0.11.8 (Andrews 2010) and subsequently treated with the JAMP v0.67 (https://github.com/VascoElbrecht/JAMP) workflow in R (R Core Team 2018) (Online resource 2.4): first, raw data were demultiplexed and forward/reverse reads were merged with USEARCH v11.0.667 (Edgar 2010). Primer sequences were trimmed with Cutadapt v2.3 (Martin 2011). Next, data were size-selected allowing for ± 12 bp variation from the expected amplicon size (421 bp), and quality-filtered after singleton-removal with a maximum expected error of 1. After omitting sequences with unused label combinations, sequences were clustered to preliminary Molecular Operational Taxonomic Units (MOTUs) at 97% similarity through UPARSE-OTU. Preliminary MOTUs with relative read abundances < 0.01% in the respective sample (PCR replicates) were excluded. Then, technical replicates were merged by averaging their read abundances.
For the taxonomic assignment of preliminary MOTUs, we used BOLDigger v1.2.2 (Buchner and Leese 2020), implementing searches across the Barcode of Life Data system (BOLD; version 18.11.2019) (Ratnasingham and Hebert 2007). We enforced an assignment threshold of at least 85% sequence similarity to maximize taxonomic coverage and to account for potential limitations of reference data in the poorly known study river network. Thus, MOTUs assigned to a taxon based on sequence similarity of at least 85% were retained and subjected to a taxonomic plausibility test as follows: first, the taxonomically assigned MOTUs were filtered to obtain Metazoa-MOTUs, from which we selected those representing benthic invertebrate fauna (i.e., aquatic and semiaquatic invertebrate taxa with a body size ≥ 500 µm) using a catalogue of aquatic and semiaquatic taxa (Moog and Hartmann 2017). Then, the plausibility of benthic invertebrate fauna MOTUs was checked using a custom R script to flag taxonomic assignments if the 20 most similar sequences belonged to more than one taxonomic entity at a similarity > 90%. Flagged taxonomic assignments were then manually controlled, and, if necessary, corrected by using higher taxonomic levels—e.g. changing a species-level assignment to genus-level. In the final data set, we retained species-level assignments only if the 20 most similar reference sequences belonged to the same species at > 90% similarity, or if the best match to a reference sequence or a set of reference sequences had a similarity ≥ 98%.
Spatial biodiversity patterns
Descriptive statistics of taxa numbers recovered by the standard approach in comparison to DNA metabarcoding were produced and visualized in R (R Core Team 2018). To compare how well the methods recover spatial patterns, we first calculated Sørensen (presence/absence) and Bray–Curtis (abundances/read abundances) pairwise dissimilarity metrics to test for local differences in communities for both taxonomic data sets. Pairwise dissimilarities were computed based on taxonomic entities (i.e. assigned MOTUs for molecular data and lowest taxonomic level for morphological data) with the R package betapart (Baselga et al. 2021) and visualized with functions of the package ComplexHeatmap (Gu et al. 2016). Systematic differences in dissimilarity estimates due to the methodological approaches were tested with a two-sided paired t-test. Second, Principal Coordinate Analysis (PCoA) was based on the same indices and computed to assess catchment scale community turnover for each method and season. PCoA were calculated with the wcmdscale function of the R package vegan (Oksanen et al. 2019). Third, we computed Asymmetric Eigenvector Maps (AEM) according to the coordinates of sampling sites and weighted them by reach length (Blanchet et al. 2008, 2011), and obtained 12 spatial vectors reflecting large- and small-scale spatial patterns. These variables account for the dendritic structure of river networks that imply asymmetric directional fluvial processes (Brown and Swan 2010; Altermatt 2013). We finally computed a forward-selected distance-based redundancy analysis (dbRDA) to constrain and compare the generated spatial patterns of both approaches, seasons and dissimilarity indices, respectively.
Results
Benthic invertebrate detections and abundances
We detected 4030 taxonomically assigned MOTUs in total in the 43 spring and 37 autumn preservative-based DNA metabarcoding samples, corresponding to 1,775,177 reads. The spring samples contributed 2,608 assigned MOTUs and 1,197,650.5 reads. The autumn samples contributed 2,479 assigned MOTUs and 577,526 reads. For further information about sequencing success, refer to Online resource 3.1.
We recovered 638 and 491 target MOTUs corresponding to benthic invertebrate fauna in spring and autumn, respectively, with 789 shared target MOTUs. Taxonomic assignments ranged from class to species level with most assignments to species (236), genus (229) and family level (279) (Online resource Tab. SI 1). Benthic invertebrate fauna MOTUs dominated total read numbers (Online resource Fig. SI 1, Tab. SI 2), with 73% of all reads stemming from benthic invertebrate fauna in spring, and 64.3% in autumn. Highest per-site richness (as MOTU numbers) was observed at site 22 (135 MOTUs) in spring and site 70 (104 MOTUs) in autumn (Online resource Fig. SI 2, Tab. SI 3). Most target benthic invertebrate fauna MOTUs came from Diptera, Ephemeroptera, Plecoptera and Trichoptera in both seasons (Online resource Fig. SI 2). Diptera contributed the most target MOTUs in spring (350 MOTUs), followed by Ephemeroptera (96 MOTUs) and Plecoptera (64 MOTUs). Ephemeroptera contributed the most reads in spring (153,380 reads, 77% of all benthic invertebrate fauna reads) (Online resource Fig. SI 2). Diptera also contributed most target MOTUs in autumn (235 MOTUs), followed by Ephemeroptera (69 MOTUs), Plecoptera (52 MOTUs) and Haplotaxida (25 MOTUs) (Online resource Fig. SI 2). Again, read abundances were dominated by Ephemeroptera (195,668 reads, 54% of all autumn benthic invertebrate fauna reads). Additionally, preservative-based DNA metabarcoding detected 18 benthic invertebrate fauna orders with very low read numbers (< 467 reads) at individual sites (Online resource Fig. SI 2).
Standard benthic invertebrate fauna assessment detected 183 taxa in spring and 148 taxa in autumn, where in both seasons 43 samples were taken. The highest per-site taxon numbers were 70 in spring (site 31) and 55 in autumn (site 71); on average we found 35.39 ± 13.3 taxa in spring and 24.33 ± 11.58 taxa in autumn (Online resource Tab. SI 3). Taxonomic identification ranged from family to species levels (Online resource Tab. SI 1), with most taxa identified to family and genus levels (Chironomids were mostly identified at subfamily or tribe level). The majority of identified taxa belonged to the orders Diptera, Ephemeroptera, Plecoptera and Trichoptera. Moreover, we counted in total of 58,138 specimens in spring. The majority of these were Diptera (42.4%) and Ephemeroptera (32.7%). In autumn we counted more than twice the number of specimens (123,373), of which the majority were Trichoptera (30.7%), Ephemeroptera (30.7%) and Diptera (20.7%). We found a median of 705 (121—398.5—1,456.5—10,393; 0—25—75—100% quantiles) ind. m−2 in spring and 2,313 (92—687—4,591—13,857) ind. m−2 in autumn (Tab. 1).
In white (upper) standard and in blue preservative-based DNA metabarcoding approach. In autumn sites 2, 3, 11, 27, 26, and 79 were excluded.
Biodiversity across the Vjosa catchment estimated through different methods
In total, the standard approach yielded 32 species: 21 and 17 species were found in spring and autumn, respectively, with 6 species found in both seasons. A total of 236 MOTUs assigned to species level were detected through preservative-based DNA metabarcoding, representing 181 individual species: 132 and 134 species-level MOTUs were found in the spring and the autumn data set, respectively; 69 were found to occur in both spring and autumn with 63 species detected only in spring, and 65 exclusively in autumn. Compared to the standard approach, preservative-based DNA metabarcoding captured roughly 5.6 times more species (Online resource Tab. SI 1).
The set of taxa identified by the standard approach was not a subset of the taxa recovered from preservative-based DNA metabarcoding, and metabarcoding failed to detect taxa that were found in standard morphology-based assessments: Seven orders were discovered by the standard approach only (Anomopoda, Architaeniglossa, Gordioidea, Megaloptera, Physidae, Pulmonata, Unionida). Sixteen species were detected exclusively by the standard approach (Online resource Tab. SI 4), including Prosopistoma pennigerum (Ephemeroptera) and Ithytrichia lamellaris (Trichoptera) for which the Vjosa represents one of the last European refugia. For two of those 16 species (Wiedemannia fallaciosa, P. pennigerum), no reference sequence was available in BOLD (Online resource Tab. SI 4). Consequently, these species showed no match and were included at higher taxonomic levels only (Diptera, Insecta). Vice versa, 9 orders were detected by preservative metabarcoding only (Anthoatecata, Arhynchobdellida, Catenulida, Cycloneritida, Haplosclerida, Harpacticoida, Limnomedusae, Monostilifera, Plumatellida, Triplonchida) (Online resource Tab. SI 1).
River network scale diversity patterns
The overall higher taxa numbers lead to higher beta diversity estimates inferred with preservative-based DNA metabarcoding data. On average, pairwise Sørensen dissimilarities based on metabarcoding data were 0.7 ± 0.12 and 0.73 ± 0.12 for spring and autumn, respectively. The standard approach yielded average dissimilarities of 0.56 ± 0.13 in spring and 0.55 ± 0.13 in autumn (Fig. 2; see Online resource Fig. SI 3 for an analysis based on Bray–Curtis dissimilarities).

Compared to the standard approach, systematically higher Sørensen dissimilarities between sites were assessed with the preservative-based DNA metabarcoding method in spring and autumn, respectively. *** = p-values < 0.001
Low dissimilarities between sites in the Vjosa main stem and high dissimilarities between sites in tributaries were inferred for both approaches and seasons. Through the metabarcoding approach we estimated high dissimilarities among sites of the upper Aoos (44, 45, 46) and all other sites, but we also distinguished other tributaries like the Langarica, Bence or Shushica. This pattern could be partly observed with the standard approach, where single sites (i.e. 10, 63, 3) with relatively low taxa numbers show higher dissimilarities to all other sampling points (Fig. 3; see Online resource Fig. SI 4 for an analysis based on Bray–Curtis dissimilarities).

Pairwise Sorensen dissimilarities in spring (left) and autumn (right). The upper and lower diagonal represents beta diversity estimates based on molecular and morphological data, respectively
To visualise spatial community patterns and further compare both approaches PCoA dimensions were projected in the Vjosa river network. Relative eigenvalues drop from about 0.25 to > 0.05 within the first six to seven dimensions (Online resource Fig. SI 5 and SI 6). However, the first PCoA dimension indicates more heterogeneous communities in the headwater and more homogeneous communities in the Vjosa main stem. This general upstream-to-downstream gradient can be observed in both seasons and approaches. The second PCoA dimension differentiates Sarantaporos and the main stem from other tributaries for spring. But both the 1st and 2nd dimensions also capture smaller scale community turnover among tributaries (Fig. 4). Smaller scaled patterns, such as communities of single tributaries, triplet reaches or sites are shown in PCoA 3–6 (Online resources Fig. SI 7, SI 8). Similar patterns can be observed by the Bray–Curtis dissimilarity index based PCoAs 1–6 (Online resource Fig. SI 9—SI 11).

Sørensen-based PCoA dimension 1 (left) and 2 (right) values for spring (top, green) and autumn (bottom, red) samples, respectively. On each sampling side the community turnover comparison of standard morphological (left side half circles) and preservative-based DNA metabarcoding (right side half circles) approaches
In addition, both dissimilarity indices and approaches were compared by forward-selected dbRDAs constrained with AEMs as spatial predictors (Table 2). We observe (i) higher adjusted r2 values for preservative-based DNA metabarcoding dbRDAs, based on Sørensen, compared to the standard approach. However, the opposite is observed if both approaches are analysed based on Bray–Curtis indices. In addition, we observe (ii) a clear upstream-to-downstream gradient, with both approaches, indices and seasons. Generally higher adj. r2 are observed for the spring season where at least two dbRDA (in Sørensen, spring metabarcoding three) dimensions show significant spatial patterns. These patterns are explained by a set of AEMs (2, 3, 4, 6, 7, 11, 12) representing smaller-scale spatial patterns (e.g., AEM2: Drinos vs Vjosa; AEM4: Aoos vs Sarantaporos; Online resource Fig. SI 12).
We compare the output of the, respectively, forward selected dbRDA, to assess spatial community patterns (i.e. for Sørensen spring AEM1 represents an upstream to downstream spatial gradient, while remaining AEMs contribute to explain smaller scaled species turnover). *** = p < 0.001; ** = p < 0.01; * = p < 0.05.
Discussion
Our results demonstrate that even in a relatively remote area and under suboptimal sampling conditions, spatially explicit biodiversity data can be generated using a preservative-based DNA metabarcoding approach. Additionally, the estimated richness of benthic macroinvertebrates is higher compared to assessments through standard methods. This difference stems from the greater proportion of species- to genus-level identification achieved through preservative-based DNA metabarcoding. Abundances of benthic invertebrate fauna as estimated through standard means were reflected in metabarcoding results: highly abundant orders (Diptera, Ephemeroptera, Plecoptera, Trichoptera) also contributed most sequencing reads. However, preservative-based DNA metabarcoding failed to detect some taxa that were assessed through the standard approach, and vice versa.
When comparing between-site Sørensen dissimilarities, we found that preservative-based DNA metabarcoding recovered stronger turn-over between sites based on macroinvertebrate MOTUs than the standard approach. This is an effect of the higher taxa numbers inferred through metabarcoding, but the abundance of information from the standard approach partly compensates for the lack of taxonomic resolution.
Both standard and preservative-based DNA metabarcoding approaches accurately depicted spatial gradients in the Vjosa catchment as formalized through AEMs: strong longitudinal turn-over but also small-scale patterns of single tributaries are evident. Similar to turn-over across sites, we found that data obtained through preservative-based DNA metabarcoding recovered greater spatial details when omitting read abundance information. In contrast, information on benthic invertebrate fauna abundances of the standard approach improved spatial gradients. Thus, the main patterns were similar for both approaches. Therefore, preservative-based DNA metabarcoding provides sufficiently informative data for ecological analyses, and application for large-scale stream ecology studies should be encouraged. In this context, preservative-based DNA metabarcoding offers several advantages over other methods.
First, voucher specimens can be obtained from multi-habitat samples used for preservative-based DNA metabarcoding. Specifically in habitats with high degrees of endemism and uncharted biodiversity such as the Vjosa riverscape this is an important feature: voucher specimens can be used to validate taxa lists derived from metabarcoding (Hajibabaei et al. 2012; Zizka et al. 2019) and if necessary, ambiguous taxonomic assignments can be tracked and resolved (Erdozain et al. 2019). Additionally, estimates of abundances or body sizes of selected taxa can be generated from leftover voucher specimens and bridge this gap of PCR-based biodiversity assessment methods. While DNA metabarcoding approaches excel at appraising biodiversity and evidently detect more species, they fail at estimating abundances or biomass reliably (Elbrecht and Leese 2015; Elbrecht et al. 2017). Our results indicate that spatial community patterns of stream networks can be accurately depicted based on the presence/absence metabarcoding data. However, abundance data, if available from standard methods, can even further validate spatial patterns of a large river network.
Second, targeted taxonomic work in an integrative framework (Dayrat 2005; Schlick-Steiner et al. 2010; Hubert and Hanner 2015) can be accomplished based on voucher specimens retained during preservative-based DNA metabarcoding. Thus, dedicated barcoding efforts or preparation of taxonomic keys to a certain region may be feasible simply by using the retained specimens for further analyses.
Third, molecular biodiversity assessment in combination with multi-habitat sampling allows for representative characterisation of local diversity including different microhabitats as opposed to typical water-sample-based eDNA metabarcoding. Most benthic invertebrates do not venture far from their preferred substrate, although drift occurs (Waringer 1992). Moreover, benthic invertebrate fauna is not randomly distributed (Cummins and Klug 1979; Martini and Waringer 2021) and it is crucial to integrate habitat diversity into the sampling scheme. Such adequate representation of habitats is critical for the general detection of species and is decisive for the presence/absence of data. This information cannot be delivered reliably by eDNA metabarcoding of freshwater. Environmental DNA metabarcoding was shown to systematically miss several species found by multi-habitat sampling, introducing a taxonomic bias (Keck et al. 2022).
Fourth, preservative-based DNA metabarcoding can yield more comprehensive taxa lists compared to standard morphological identification as demonstrated by our results. This is due to the fact that the majority of benthic invertebrate fauna can only be identified with low error probability in a particular life stage. If mature larvae of certain groups are not available, identification may only be possible at the genus-level. Moreover, there are groups lacking diagnostic characters in the larval stages, such as Empididae. Here, preservative-based DNA metabarcoding can resolve biodiversity that cannot be described by standard identification methods. Further, standard identification crucially relies on the availability of taxonomic keys or material for comparison specifically for the region of interest. When these resources are lacking or limited—such as in the present study—preservative-based DNA metabarcoding may be a better method for exploring biodiversity patterns in the framework of ecological or monitoring studies with time constraints.
We consider the potential of preservative-based DNA metabarcoding in a conservation context as high: we were able to confirm the presence of dynamic-river-species whose ranges decline dramatically in the past century, such as Marthamea vitripennis or Xanthoperla apicalis, but also Balkan (micro-)endemics like Isoperla vjosae, Thremma anomalum, Perlodes floridus, or Hydropsyche mostarensis in the Vjosa catchment. Therefore, the metabarcoding data generated here could well be used to document the significance of the Vjosa catchment for regional and sub-continental conservation efforts—even if Prosopistoma pennigerum, a species that is today restricted to only 3 river systems in Europe, could not be detected due to reference library shortcomings.
Comparison of detection efficiency—limitations and future challenges of preservative-based DNA metabarcoding
While biodiversity patterns recovered by both approaches are largely congruent, there are some inconsistencies regarding the detection efficiency of several taxa. In our data, neither approach detected the whole community we would obtain by combining results of the standard and the preservative-based DNA metabarcoding methods.
Failure to detect some orders with the standard method may be due to life-stage or size-related detection bias, or lack of diagnostic characters. For instance, no adult leeches (Arhynchobdellida) were found in our samples but egg cocoons of leeches that were not identified further; can be detected and assigned through preservative metabarcoding. The same applies to Cycloneritida (a Theodoxus sp.). Other specimens may have been washed out or overlooked when processing the samples because of their size and this may be the reason why taxa like Catenulida, Harpacticoida or Triplonchida were not detected. A lack of diagnostic characters after fixation may have been the reason why certain soft-bodied taxa were not detected: without anaesthesia specimens may retract and shrivel up to unidentifiable lumps of tissue and we assume that this is why we missed polypoid stages of Anthoatecata or Limnomedusae, or Monostilifera or Plumatellida. Also, Plumatellida and Haplosclerida will have been sorted out in the field if encrusting large sticks or rocks but may have left tissue residues on smaller organic and inorganic particles that allowed for DNA amplification and detection. While there is evidence that regurgitated gut content can affect diversity estimates in ethanol-based DNA metabarcoding (e.g., Zizka et al. 2019), we consider the above-outlined factors as more important for detection bias in our study.
In contrast, the standard method fared better than preservative-based DNA metabarcoding in detecting molluscs. Architaeniglossa, Physidae, Pulmonata and Unioinida were detected using the standard method only. We assume that this is due to primer properties: our primers were specifically designed to capture the diversity of aquatic insects and we expected poor recovery of molluscs. This may also apply to Anomopoda that were detected by the standard method only. The detection of Gordioidea may be attributed to chance, as this order was observed in a single specimen only. We have no explanation why Megaloptera was not detected using DNA metabarcoding.
At the level of species, preservative-based DNA metabarcoding outperforms standard methods: we detected > 130 species-level MOTUs in each season, compared to around 20 species recovered by the standard approach. However, a small set of species was detected only through the standard approach in our study. We argue that this is due to the general limitations of DNA metabarcoding methods that still need to be overcome, despite the fact that our results show that preservative-based DNA metabarcoding is a useful tool for the assessment of benthic invertebrate fauna communities. Specifically, these are primer bias, detection bias, MOTU (over) splitting, taxonomic assignments of MOTUs, and the general lack of abundance estimates from PCR-based approaches.
Primer bias can be reduced by using multiple primers for the same marker or by using different molecular markers (Martins et al. 2019; Hajibabaei et al. 2019; Meyer et al. 2021), or switching to PCR-free metabarcoding approaches (Zhou et al. 2013; Kennedy et al. 2020; Cordier et al. 2021).
Detection bias can be introduced by strongly sclerotized specimens that may release less DNA into the preservative liquids. At the same time, subsampling of preservative ethanol may preclude detection because of too low DNA concentration (Marquina et al. 2019; Martins et al. 2019). For many soft-bodied benthic invertebrates this may be a negligible factor (Hajibabaei et al. 2012; Zizka et al. 2019) but benthic invertebrate communities comprise strongly sclerotized and very small forms as well (e.g. Elmidae, Hydraenidae, Nemouridae) that possibly are poorly recovered through preservative-based DNA metabarcoding. Another form of detection bias may be introduced by PCR stochasticity, but increasing the number of extraction and amplification replicates can forearm against this issue. PCR-free methods may also help in addressing detection bias from PCR stochasticity.
Molecular diversity estimates can be inflated because of over splitting that occurs when multiple MOTUs are defined that all actually belong to one species. Threshold-based approaches that cluster ASVs to MOTUs at 97% sequence similarity as used here may not be appropriate for all taxa. High intraspecific diversity may additionally inflate MOTU diversity estimates (see Beermann et al. (2018) for a discussion). A conservative approach to control for over splitting may be to aggregate MOTUs assigned to the same species (Elbrecht et al. 2017) or use adaptive clustering thresholds (Mahé et al. 2014). Alternatively, MOTUs defined based on a fixed sequence similarity threshold can be retained and used for downstream analyses. We opted for the latter approach because we thereby expected higher diversity estimates linked to stronger turnover among sites. In addition, MOTUs assigned to the same species are often indicators for unknown or cryptic biodiversity (Bálint et al. 2011; Weiss et al. 2014).
Taxonomic assignment of MOTUs is not trivial. Unguided automatized taxonomic assignment recovered rogue taxa in our dataset, including an antipodean genus of stoneflies, endemic gastropods of the Iberian Peninsula, or endemic sponges of Lake Baikal. To account for this error source, we checked unguided taxonomic assignments manually after script-based flagging of potential errors and ‘corrected’ implausible assignments by reducing taxonomic resolution—but this process is time-consuming and heavily depends on expert knowledge. Additionally, incompleteness of reference databases complicates taxonomic assignment. Preservative-based DNA metabarcoding failed to detect several taxa, including the rare mayfly P. pennigerum – despite the documented presence of a vital relict population in the Vjosa (Martini et al. 2022) and sizable abundances in our MHS samples. Only after the completion of our analyses a reference sequence for this species was made publicly available on NCBI in 2021 (Schletterer et al. 2021).
The lack of reliable abundance estimates in metabarcoding data is a disadvantage. Our results show that standard multi-habitat samples depicted spatial biodiversity turn-over as efficiently as the preservative-based DNA metabarcoding if abundance data were included—despite the fact that standard samples estimated lower taxonomic richness which complicated assessment of spatial turn-over patterns. However, it should be possible to combine preservative-based DNA metabarcoding and standard analyses of voucher specimens to obtain or correct abundance estimates. While this is a possibility to improve molecular assessment of biodiversity, it also would deprive metabarcoding approaches of two of their greatest assets: speed and cost efficiency.
To make full use of (preservative-based) DNA metabarcoding techniques, these challenges must be overcome. Methodological developments already address primer bias or PCR stochasticity, and target capture metabarcoding approaches apparently improve estimates of biomass through read numbers. Mostly MOTU definition and assignment issues remain to be resolved. We defined MOTUs based on 97% sequence similarity and obtained community data that was highly informative and could be used to assess spatial turn-over patterns in the Vjosa catchment. MOTU definition usually relies on distance-based measures, but alternative strategies have been proposed to improve MOTU definition. Amplicon sequence variant trees in combination with clade-specific molecular species delimitation are an especially promising approach but are computationally intensive and not easily implemented. At present, multiple bioinformatic pipelines for MOTU definition are available, and end-users must select a set of methods for their purpose. However, the results of the different bioinformatic pipelines are not congruent, and the decision for or against a specific approach to MOTU definition has consequences for downstream analyses of metabarcoding datasets (Yang et al. 2013; Pauvert et al. 2018; Bailet et al. 2020; Bonin et al. 2021; Creedy et al. 2022). MOTU assignment is additionally onerous. Here, it is the quality and comprehensiveness of reference libraries that remains an important problem. For instance, sequences of nine Prosopistoma species are deposited in BOLD, yet these are all from other continents and cannot be used to identify P. pennigerum, the European species. Additionally, if a single specimen is identified wrongly to species-level and its barcode is available and used in automatized assignment of MOTUs in further studies, taxonomic errors will propagate. Therefore, we argue that it is necessary to include certain criteria in addition to the best match of a query to a reference sequence, particularly for automatized biodiversity assessment of unknown faunas. Specifically, a centralized taxonomic curation of reference libraries should be achieved: quality scores for reference sequences aside the standard parameters should be developed, based on (i) the reliability (origin and taxonomic expertise) of a submitted barcode as well as (ii) how accurate taxa can be identified and resolved based on this particular barcode. In automatized taxonomic assignment of MOTUs, including distribution data from ZOBODAT, GBIF (Global Biodiversity Information Facility), iNaturalist, Map of Life (Jetz et al. 2012) or Freshwater Ecology Info can improve the plausibility of assignment (Macher et al. 2020). This conservative approach may increase the risk of neglecting poorly documented species as well as neobiota or neonatives — but less so in a preservative-based DNA metabarcoding context as such records may be confirmed with voucher specimens.
Conclusions and outlook
Given the ongoing biodiversity decline, management and conservation efforts need to be geared towards the protection and conservation of our near-natural aquatic habitats. To this end, temporally, spatially and taxonomically highly resolved biodiversity data are necessary to monitor the effects of global change and protection measures. Our results demonstrate that non-destructive preservative-based DNA metabarcoding can produce such data efficiently at large spatial scales in regions with taxonomically underexplored fauna. Although preservative metabarcoding cannot fully replace other methods of biodiversity assessment, we interpret our data as indicative of general suitability of preservative-based DNA metabarcoding in the context of ecosystem assessment and macroecology that require resolution of spatially distinct biodiversity signals, but also in a nature conservation context. While the reference libraries are far from complete, they seemingly can be used in their current states to obtain high-resolution taxonomical data at the European scale. Exploiting the full potential of (preservative-based) DNA metabarcoding data in fundamental and applied ecological research will require the completion and curation of reference libraries beyond the current state. While the Central European aquatic and semiaquatic freshwater invertebrate fauna is covered quite well, other regions, including the Southern Balkans, are clearly data-deficient. This highlights the need to intensify the current efforts to fill these knowledge gaps and curate reference data for an improved biodiversity assessment.
Data availability
The raw metabarcoding data is available at the EMBL Nucleotide Sequence Database (ENA) repository under project PRJEB51480.
References
Altermatt F (2013) Diversity in riverine metacommunities: a network perspective. Aquat Ecol 47:365–377. https://doi.org/10.1007/s10452-013-9450-3
Andrews S (2010) FastQC A Quality Control tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 19 Apr 2022
Bailet B, Apothéloz-Perret-Gentil L, Baričević A et al (2020) Diatom DNA metabarcoding for ecological assessment: comparison among bioinformatics pipelines used in six European countries reveals the need for standardization. Sci Total Environ 745:140948. https://doi.org/10.1016/j.scitotenv.2020.140948
Bálint M, Ujvárosi L, Theissinger K et al (2011) The Carpathians as a major diversity hotspot in Europe. In: Zachos FE, Habel JC (eds) Biodiversity hotspots. Springer, Berlin Heidelberg, Berlin
Baselga A, Orme D, Villeger S, et al (2022) betapart: Partitioning Beta Diversity into Turnover and Nestedness Components. R package version 1.5.6. https://CRAN.R-project.org/package=betapart
Beermann AJ, Zizka VMA, Elbrecht V et al (2018) DNA metabarcoding reveals the complex and hidden responses of chironomids to multiple stressors. Environ Sci Eur 30:26. https://doi.org/10.1186/s12302-018-0157-x
Blanchet FG, Legendre P, Borcard D (2008) Modelling directional spatial processes in ecological data. Ecol Model 215:325–336. https://doi.org/10.1016/j.ecolmodel.2008.04.001
Blanchet FG, Legendre P, Maranger R et al (2011) Modelling the effect of directional spatial ecological processes at different scales. Oecologia 166:357–368. https://doi.org/10.1007/s00442-010-1867-y
Bonin A, Guerrieri A, Ficetola F (2021) Optimal sequence similarity thresholds for clustering of molecular operational taxonomic units in DNA metabarcoding studies. Authorea Preprints
Brown BL, Swan CM (2010) Dendritic network structure constrains metacommunity properties in riverine ecosystems. J Anim Ecol 79:571–580. https://doi.org/10.1111/j.1365-2656.2010.01668.x
Buchner D, Leese F (2020) BOLDigger – a Python package to identify and organise sequences with the Barcode of Life Data systems. Metabarcoding Metagenomics 4:e53535. https://doi.org/10.3897/mbmg.4.53535
Bush A, Compson ZG, Monk WA et al (2019) Studying ecosystems with DNA metabarcoding: lessons from biomonitoring of aquatic macroinvertebrates. Front Ecol Evol 7:434. https://doi.org/10.3389/fevo.2019.00434
Cordier T, Alonso-Sáez L, Apothéloz-Perret-Gentil L et al (2021) Ecosystems monitoring powered by environmental genomics: a review of current strategies with an implementation roadmap. Mol Ecol 30:2937–2958. https://doi.org/10.1111/mec.15472
Creedy TJ, Andújar C, Meramveliotakis E et al (2022) Coming of age for COI metabarcoding of whole organism community DNA: towards bioinformatic harmonisation. Mol Ecol Resour 22:847–861. https://doi.org/10.1111/1755-0998.13502
Cummins KW, Klug MJ (1979) Feeding ecology of stream invertebrates. Annu Rev Ecol Syst 10:147–172. https://doi.org/10.1146/annurev.es.10.110179.001051
Dayrat B (2005) Towards integrative taxonomy. Biol J Lin Soc 85:407–415. https://doi.org/10.1111/j.1095-8312.2005.00503.x
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Elbrecht V, Vamos EE, Meissner K et al (2017) Assessing strengths and weaknesses of DNA metabarcoding-based macroinvertebrate identification for routine stream monitoring. Methods Ecol Evol 8:1265–1275. https://doi.org/10.1111/2041-210X.12789
Elbrecht V, Leese F (2015) Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10:e0130324. https://doi.org/10.1371/journal.pone.0130324
Elbrecht V, Leese F (2017) Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Front Environ Sci. https://doi.org/10.3389/fenvs.2017.00011
Elbrecht V, Steinke D (2018) Scaling up DNA metabarcoding for freshwater macrozoobenthos monitoring. Freshw Biol. https://doi.org/10.1111/fwb.13220
Erdozain M, Thompson DG, Porter TM et al (2019) Metabarcoding of storage ethanol vs. conventional morphometric identification in relation to the use of stream macroinvertebrates as ecological indicators in forest management. Ecol Ind 101:173–184. https://doi.org/10.1016/j.ecolind.2019.01.014
Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32(18):2847–2849. https://doi.org/10.1093/bioinformatics/btw313
Hajibabaei M, Spall JL, Shokralla S, van Konynenburg S (2012) Assessing biodiversity of a freshwater benthic macroinvertebrate community through non-destructive environmental barcoding of DNA from preservative ethanol. BMC Ecol 12:28. https://doi.org/10.1186/1472-6785-12-28
Hajibabaei M, Porter TM, Wright M, Rudar J (2019) COI metabarcoding primer choice affects richness and recovery of indicator taxa in freshwater systems. PLoS ONE 14:e0220953. https://doi.org/10.1371/journal.pone.0220953
Hering D, Buffagni A, Moog O et al (2003) The development of a system to assess the ecological quality of streams based on macroinvertebrates – design of the sampling programme within the AQEM project. Int Rev Hydrobiol 88:345–361. https://doi.org/10.1002/iroh.200390030
Hubert N, Hanner R (2015) DNA Barcoding, species delineation and taxonomy: a historical perspective. DNA Barcodes. https://doi.org/10.1515/dna-2015-0006
Jetz W, McPherson JM, Guralnick RP (2012) Integrating biodiversity distribution knowledge: toward a global map of life. Trends Ecol Evol 27:151–159. https://doi.org/10.1016/j.tree.2011.09.007
Ji Y, Ashton L, Pedley SM et al (2013) Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol Lett 16:1245–1257. https://doi.org/10.1111/ele.12162
Keck F, Hürlemann S, Locher N et al (2022) A triad of kicknet sampling, eDNA metabarcoding, and predictive modeling to assess aquatic macroinvertebrate biodiversity. Metabarcoding Metagenomics 6:117–131. https://doi.org/10.3897/mbmg.6.79351
Kennedy SR, Prost S, Overcast I et al (2020) High-throughput sequencing for community analysis: the promise of DNA barcoding to uncover diversity, relatedness, abundances and interactions in spider communities. Dev Genes Evol 230:185–201. https://doi.org/10.1007/s00427-020-00652-x
Macher JN, van der Hoorn BB, Peijnenburg KTCA et al (2020) Metabarcoding reveals different zooplankton communities in northern and southern areas of the North Sea. bioRxiv. https://doi.org/10.1101/2020.07.23.218479
Mahé F, Rognes T, Quince C et al (2014) Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2:e593. https://doi.org/10.7717/peerj.593
Marquina D, Esparza-Salas R, Roslin T, Ronquist F (2019) Establishing arthropod community composition using metabarcoding: Surprising inconsistencies between soil samples and preservative ethanol and homogenate from Malaise trap catches. Mol Ecol Resour 19:1516–1530. https://doi.org/10.1111/1755-0998.13071
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12. https://doi.org/10.14806/ej.17.1.200
Martini J, Waringer J (2021) Dynamic microhabitat shifts in space and time of caddisfly larvae (Insecta: Trichoptera) in a first-order calcareous mountain stream. Biologia 76:2527–2541. https://doi.org/10.1007/s11756-021-00741-w
Martini J, Walther F, Schenekar T, Birnstiel E, Wüthrich R, Oester R et al (2022) The last hideout: Abundance patterns of the not-quite-yet extinct mayfly Prosopistoma pennigerum in the Albanian Vjosa River network. Insect Conserv Divers. https://doi.org/10.1111/icad.12620
Martins FMS, Galhardo M, Filipe AF et al (2019) Have the cake and eat it: optimizing nondestructive DNA metabarcoding of macroinvertebrate samples for freshwater biomonitoring. Mol Ecol Resour 19:863–876. https://doi.org/10.1111/1755-0998.13012
Martins FMS, Porto M, Feio MJ et al (2021) Modelling technical and biological biases in macroinvertebrate community assessment from bulk preservative using multiple metabarcoding markers. Mol Ecol 30:3221–3238. https://doi.org/10.1111/mec.15620
Meyer A, Boyer F, Valentini A et al (2021) Morphological vs. DNA metabarcoding approaches for the evaluation of stream ecological status with benthic invertebrates: testing different combinations of markers and strategies of data filtering. Mol Ecol 30:3203–3220. https://doi.org/10.1111/mec.15723
Moog O, Hartmann A (2017) Fauna Aquatica Austriaca, 3rd edition 2017. A Comprehensive Species Inventory of Austrian Aquatic Organisms with Ecological Notes. Abt. IV/3, Stubenring 1, A-1010; Bundesministerium für Land-und Forstwirtschaft. Umwelt und Wasserwirtschaft: Vienna, Austria.
Nielsen M, Gilbert MTP, Pape T, Bohmann K (2019) A simplified DNA extraction protocol for unsorted bulk arthropod samples that maintains exoskeletal integrity. Environ DNA 1:144–154. https://doi.org/10.1002/edn3.16
Oksanen J, Blanchet FG, Friendly M, et al (2020) vegan: Community Ecology Package. R package version 2.5–7. https://CRAN.R-project.org/package=vegan
Pauvert C, Buee M, Laval V, et al (2018) Its all fun guys: a comparison of bioinformatic pipelines for metabarcoding plant and soil fungal communities. In: International Phytobiomes Conference 2018. Montpellier, France, p np. https://hal.archives-ouvertes.fr/hal-01938707
Persaud SF, Cottenie K, Gleason JE (2021) Ethanol eDNA reveals unique community composition of aquatic macroinvertebrates compared to bulk tissue metabarcoding in a biomonitoring sampling scheme. Diversity 13:34. https://doi.org/10.3390/d13010034
R Core Team (2018) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria
Ratnasingham S, Hebert PDN (2007) Bold: the barcode of life data system. Mol Ecol Notes 7:355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Schiemer F, Beqiraj S, Drescher A et al (2020) The Vjosa River corridor: a model of natural hydro-morphodynamics and a hotspot of highly threatened ecosystems of European significance. Landscape Ecol 35:953–968. https://doi.org/10.1007/s10980-020-00993-y
Schletterer M, Weiss SJ, Kuzovlev VV et al (2021) The rare and enigmatic mayfly Prosopistoma pennigerum (Müller, 1785): Habitat characteristics, recent records from the Volga (Russia) and Vjosa (Albania) rivers, and a proposal for flagship species status. Aquat Conserv Mar Freshwat Ecosyst 31:3636–3643. https://doi.org/10.1002/aqc.3748
Schlick-Steiner BC, Steiner FM, Seifert B et al (2010) Integrative taxonomy: a multisource approach to exploring biodiversity. Annu Rev Entomol 55:421–438. https://doi.org/10.1146/annurev-ento-112408-085432
Schwarz U (2022) Hydropower Projects on Balkan Rivers – 2022 Update. RiverWatch & EuroNatur, Vienna/Radolfzell, 37 pp. https://balkanrivers.net/uploads/files/3/Balkan_HPP_Update_2022.pdf
Sunnucks P, Hales DF (1996) Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in Aphids of the Genus Sitobion (Hemiptera: Aphididae). Mol Biol Evol 13(3):510–524. https://doi.org/10.1093/oxfordjournals.molbev.a025612
Tessarolo G, Ladle R, Rangel T, Hortal J (2017) Temporal degradation of data limits biodiversity research. Ecol Evol 7:6863–6870. https://doi.org/10.1002/ece3.3259
Wägele H, Klussmann-Kolb A, Kuhlmann M et al (2011) The taxonomist—an endangered race. A practical proposal for its survival. Front Zool 8:25. https://doi.org/10.1186/1742-9994-8-25
Waringer JA (1992) The drifting of invertebrates and particulate organic matter in an Austrian mountain brook. Freshw Biol 27:367–378. https://doi.org/10.1111/j.1365-2427.1992.tb00546.x
Weiss M, Leese F (2016) Widely distributed and regionally isolated! Drivers of genetic structure in Gammarus fossarum in a human-impacted landscape. BMC Evol Biol 16:153. https://doi.org/10.1186/s12862-016-0723-z
Weiss M, Macher JN, Seefeldt MA, Leese F (2014) Molecular evidence for further overlooked species within the Gammarus fossarum complex (Crustacea: Amphipoda). Hydrobiologia 721:165–184. https://doi.org/10.1007/s10750-013-1658-7
Yang C, Ji Y, Wang X et al (2013) Testing three pipelines for 18S rDNA-based metabarcoding of soil faunal diversity. Sci China Life Sci 56:73–81. https://doi.org/10.1007/s11427-012-4423-7
Zhou X, Li Y, Liu S et al (2013) Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience 2:4. https://doi.org/10.1186/2047-217X-2-4
Zizka VMA, Leese F, Peinert B, Geiger MF (2019) DNA metabarcoding from sample fixative as a quick and voucher-preserving biodiversity assessment method. Genome 62(3):122–136. https://doi.org/10.1139/gen-2018-0048
Acknowledgements
We thank Sajmir Beqiraj, Franziska Walther and the FLEE Lab for their considerable contribution during fieldwork. This work is part of the project ERC-2016-STG 716169, FLUFLUX—Fluvial meta-ecosystem functioning: Unravelling regional ecological controls behind fluvial carbon fluxes (PI: Gabriel Singer). Lab work at the University of Duisburg-Essen was supported by a grant from the Rüdiger Kurt Bode Foundations (Deutsches Stiftungszentrum). Netzwerk ImFluss acknowledges support from Georg und Bertha Schwyzer-Winiker-Stiftung, Hydrobiologie-Limnologie-Stiftung and Parrotia-Stiftung. Open access funding is provided by the University of Natural Resources and Life Sciences Vienna (BOKU).
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU).
Author information
Authors and Affiliations
Contributions
GS, SV planned and organized fieldwork; SV, JM, OW, RW, EB, GS took and processed samples; JM, OW, RW, EB, RO picked, sorted and identified benthic macroinvertebrates; SV supervised identification of benthic macroinvertebrates; SV, OW preprocessed MHS for molecular lab work; FL, MB, VZ planned and organized molecular lab work; SV, MB conducted molecular lab work; MB conducted bioinformatic analyses; SV conducted plausibility checks with support from JM; SV, MB, JM prepared summary statistics on conventional and preservative metabarcoding data; SV, MB, FL, GS developed the conceptual framework of the manuscript; SV, MB, JM produced a draft version with support from FL, GS; all authors contributed to draft versions of the manuscript and approved of the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brasseur, M.V., Martini, J., Wilfling, O. et al. Exploring macroinvertebrate biodiversity in the dynamic southern Balkan stream network of the Vjosa using preservative-based DNA metabarcoding. Aquat Sci 85, 51 (2023). https://doi.org/10.1007/s00027-023-00948-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00027-023-00948-w