
Abstract
Chronic obstructive pulmonary disease (COPD) is a complex syndrome with poorly understood mechanisms driving its early progression (GOLD stages 1–2). Elucidating the genetic factors that influence early-stage COPD, particularly those related to airway inflammation and remodeling, is crucial. This study analyzed lung tissue sequencing data from patients with early-stage COPD (GSE47460) and smoke-exposed mice. We employed Weighted Gene Co-Expression Network Analysis (WGCNA) and machine learning to identify potentially pathogenic genes. Further analyses included single-cell sequencing from both mice and COPD patients to pinpoint gene expression in specific cell types. Cell–cell communication and pseudotemporal analyses were conducted, with findings validated in smoke-exposed mice. Additionally, Mendelian randomization (MR) was used to confirm the association between candidate genes and lung function/COPD. Finally, functional validation was performed in vitro using cell cultures. Machine learning analysis of 30 differentially expressed genes identified 8 key genes, with CLEC5A emerging as a potential pathogenic factor in early-stage COPD. Bioinformatics analyses suggested a role for CLEC5A in macrophage-mediated inflammation during COPD. Two-sample Mendelian randomization linked CLEC5A single nucleotide polymorphisms (SNPs) with Forced Expiratory Volume in One Second (FEV1), FEV1/Forced Vital Capacity (FVC) and early/later on COPD. In vitro, the knockdown of CLEC5A led to a reduction in inflammatory markers within macrophages. Our study identifies CLEC5A as a critical gene in early-stage COPD, contributing to its pathogenesis through pro-inflammatory mechanisms. This discovery offers valuable insights for developing early diagnosis and treatment strategies for COPD and highlights CLEC5A as a promising target for further investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a persistent inflammatory lung condition characterized by respiratory distress and restricted airflow, primarily triggered by smoking or prolonged exposure to harmful gases and particulates [1]. Globally, COPD has emerged as a significant public health concern, profoundly impacting patient quality of life and life expectancy [1, 2]. The pathophysiological mechanisms of COPD are multifaceted, involving various cell types and signaling pathways, among which chronic airway inflammation is one of the core pathological alterations [3]. Throughout the course of COPD, inhaled harmful substances, such as tobacco smoke, can provoke damage to the airway epithelial cells and incite inflammatory responses, persistently altering the structure and function of the airways, leading to the thickening of airway walls, increased mucus secretion, and smooth muscle contraction, culminating in airway obstruction and difficulty breathing [4, 5].
Moreover, COPD progression typically follows a protracted course, and its etiological mechanisms are complex, encompassing multiple cell types and signaling pathways [3]. Currently, our comprehension of the early pathological mechanisms of COPD is limited, impeding the design of early diagnostic and therapeutic strategies [6]. The clinical classification of COPD primarily aligns with the Global Initiative for Chronic Obstructive Lung Disease (GOLD), where stages 1–2 indicate the early-stage COPD. During this period, despite the absence of overt symptoms in patients, pathological lung alterations have been documented, involving processes such as airway inflammation, small airway disease, and emphysema [7].
During the pathological progression of COPD, macrophages assume a crucial role as a primary cell type within the immune system. These cells initiate inflammatory responses to eliminate pathogens while also modulating immune reactions to preserve tissue homeostasis [8]. During the early stages of COPD, dysfunction in macrophages, characterized by reduced clearance and an exaggerated inflammatory response, serve as significant contributing factors to disease progression [9]. Clec5a, identified as a C-type lectin receptor, is predominantly expressed on myeloid cells such as macrophages and neutrophils [10]. Its activation has been demonstrated to induce inflammatory responses in various diseases, exacerbating the underlying conditions. For instance, in Dengue Fever and Japanese Encephalitis, Clec5a can recognize and bind to the virus, activating macrophages and neutrophils, thereby initiating inflammation [11]. Concurrently, Clec5a has been observed to identify dead cells, promoting macrophage phagocytosis and mitigating excessive inflammatory responses [12, 13].
In this investigation, we employed lung tissue sequencing from COPD patients with GOLD stages 1 and 2, coupled with single-cell sequencing from mice exposed to smoke at an early stage, to reveal potential pathogenic genes in the initial phases of COPD. Our screening efforts identified upregulated expression of CLEC5A in the lung tissue of patients with GOLD stages 1–2 and early smoke-exposed mice, predominantly within macrophages. We further confirmed that CLEC5A-positive macrophages in both mouse and human lungs exhibited high expression of pro-inflammatory factors. These findings suggest that CLEC5A may play a role in the early pathological processes of COPD by influencing macrophage function, particularly in their interactions with other immune cells. This offers novel insights into understanding the early pathological mechanisms of COPD and holds the potential to contribute to the identification of innovative early diagnostic markers and therapeutic targets.
Materials and methods
Animal experiment and COPD modeling
C57BL/6 male mice (n = 20), 5–6 weeks old, weighing 20–24 g, were acquired from Gempharmatech Co., Ltd. (Jiangsu, China). A chronic CS-induced COPD model was established following previously described protocols [14]. Briefly, 8-week-old mice were exposed to cigarette smoke for 2 h per session, twice daily, six days a week. Mice were exposed for 2 or 6 months before collection for analysis. Ethical approval for the animal experiment was obtained from The First Affiliated Hospital of Guangzhou Medical University (Reference number: SYXK 2020-0227).
Cell culture
THP-1 cells, sourced from Procell (Wuhan, China), were cultured in RPMI-1640 medium enriched with 0.05 mM β-mercaptoethanol, 10% fetal bovine serum (FBS, Gibco, USA), and 1% penicillin/streptomycin (P/S, Invitrogen, USA). Differentiation into M0 macrophages was induced with 100 ng/ml phorbol 12-myristate 13-acetate (PMA), following established protocols. Cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere.
RNA extraction, cDNA synthesis, and qRT-qPCR
Total RNA was isolated from lung tissues and cells using the SteadyPure RNA kit (AG21022, AG, China) following an established protocol [15]. To synthesize cDNA, the Evo M-MLV RT Kit with gDNA Clean for qPCR (AG, China) was employed, utilizing 1000 ng of total RNA in accordance with the manufacturer's instructions. Subsequently, qRT-PCR was performed using the SYBR® Green Premix Pro Taq HS qPCR Kit (AG, China) and analyzed on a CFX Connect real-time PCR detection system (Bio-Rad, USA). The relative expression levels of the target genes were determined using the 2−∆∆CT method, with GAPDH serving as the internal control. Supplementary Table 19 provides a comprehensive list of the specific primers utilized in this study.
Double immunofluorescence (IF) assay
Lung tissue sections underwent IF staining as per modified existing protocols. Sections were treated with primary antibodies—CD68 Polyclonal antibody (1:300, 66231-2-IG, Proteintech) and CLEC5A (1:300, ab313337, Abcam)—for 2 h at 37 °C, followed by incubation with Alexa Fluor 488 goat anti-rabbit IgG (H + L) (1:500, Invitrogen) and Alexa Fluor 594 rabbit anti-mouse IgG (H + L) (1:500, Invitrogen) secondary antibodies for 40 min. Imaging was performed on a Leica DM6 M microscope.
Western blot (WB)
Western blot analysis was performed following previously established methods [16]. In brief, protein samples from cell lines and lung tissue were prepared using RIPA lysis buffer (89901, Thermo, USA) with added protease inhibitors (78430, Thermo, USA), and incubated at 4 °C for 20 min. Proteins were then resolved by 10% SDS-PAGE and transferred to PVDF membranes (BioRad, USA). Membranes were blocked and probed overnight at 4 °C with primary antibodies against CLEC5A (ab313337, Abcam, USA) and β-ACTIN (66009-1-Ig, Proteintech, China). Subsequent to washing, membranes were treated with HRP-linked secondary antibody (Proteintech) and detected using enhanced chemiluminescence on an Amersham Imager 680 (Thermo Fisher Scientific, USA).
Hematoxylin–Eosin (HE) and Masson staining
Paraffin-embedded tissue sections were deparaffinized, rehydrated, and stained using HE and Masson's trichrome methods [17]. Digital images of the stained tissues were captured with an Aperio CS2 Digital Pathology Scanner (Leica). The mean linear intercept (MLI), reflecting alveolar diameter, was calculated using the equation: MLI = total length of tissue line/number of alveolar septa intersected. Airway wall thickness was quantified by segmental airway wall area percentage, using the formula: Segmental WA% = ([total outer area of the bronchus—luminal area] / total outer area of the bronchus) × 100. Quantitative analysis of airway morphometry and collagen deposition was performed using ImageJ software. Specifically, measurements of the airway and Masson's trichrome-positive areas were obtained, enabling the calculation of the proportion of collagen deposition relative to the total airway wall area.
Enzyme-linked immunosorbent assay (ELISA) assay
Concentrations of TNF-α, CXCL1, IL-1B, IL-8 and CCL2 in THP-1 cell culture supernatants were quantified using ELISA kits for CXCL1 (BMS2122), IL-1B (88-7261-22), IL-8 (88-8086-88), CCL2 (88-7399-22) and TNF-α (88-7346-88) from Invitrogen (USA). Supernatants were harvested via centrifugation at 2000 × g for 10–15 min at room temperature.
mRNA microarray chip datasets and bioinformatics
Microarray datasets GSE47460 [18], GSE103174 [19] and GSE125521 [20] were retrieved from the Gene Expression Omnibus (GEO) database. For GSE47460, analyses were limited to GOLD stage 1–2 patients and healthy individuals utilizing the GPL14550 platform. For GSE125521, the GPL6885 platform was applied. Volcano plot construction was carried out with the "ggplot2" R package. Principal component analysis (PCA) and visualization were executed using the "ggbiplot" R package, while gene correlation assessments were performed with the "corrplot" R package. The CIBERSORTx database was employed for immune infiltration analysis of sequencing data. Peripheral blood microarray datasets from two independent COPD cohorts, GSE71120 [21] and GSE76705 [22], were analyzed, focusing on controls and GOLD 1–2 stage patients (GSE71120: control = 45, GOLD1/2 = 188; GSE76705: control = 136, GOLD1/2 = 116). ROC analysis was conducted via GraphPad Prism. Additionally, microarray datasets from macrophages in bronchoalveolar lavage fluid (BALF) of two other independent COPD cohorts, GSE13896 [23] (nonsmokers = 91, smokers = 91, COPD = 82) and GSE130928 [24] (nonsmokers = 24, smokers = 42, COPD = 22), were utilized for comparative analysis. Based on CLEC5A expression levels, the GSE47460 dataset was stratified into three groups: the lowest 0–33.3% as Low CLEC5A, 33.3–66.6% as Moderate CLEC5A, and 66.6–100% as High CLEC5A.
Differentially expressed genes (DEGs) were identified via the limma package from R/Bioconductor. Significant DEGs were identified based on p-values < 0.05 and fold changes > 0.25. The genes were subjected to gene ontology enrichment analysis utilizing the online Gene Ontology resource (http://geneontology.org) in order to categorize gene function. Additionally, Kyoto Encyclopedia of Genes and Genomes pathway analysis (http://kegg.jp) was performed on the genes to elucidate associated biological pathways and processes. Gene Set Enrichment Analysis (GSEA) was conducted using GSEA software.
Overlapped genes were used to construct PPI networks via the Search Tool for the Retrieval of Interacting Genes/Proteins database [25] (STRING, http://string-db.org). The networks were visualized with Cytoscape software version 3.8.1. The CytoHubba plugin within Cytoscape was employed to determine the central core genes within these networks. The 'WGCNA' R package facilitated weighted gene co-expression network analysis (WGCNA [26]) on GSE47460. Networks were then correlated with COPD clinical status and forced expiratory volume in 1 s (FEV1) % predicted (FEV1%pre) values.
Machine learning
Five machine learning algorithms were employed to identify gene signatures associated with COPD: Support Vector Machine Recursive Feature Elimination (SVM-RFE), LASSO Model, Ridge Regression, Elastic Net, and Random Forest Model. The optimal lambda value, defined as the value within one standard error of the minimum error, was used to select genes associated with COPD. Following the identification of hub genes, all machine learning and statistical analyses were performed using R (version 4.0.3) with the "e107", "glmnet", and "ggplot2" packages.
Single-cell RNA-seq analysis
Single-cell RNA-sequencing (scRNA-seq) data for mouse lung tissue were obtained from the GEO database (dataset GSE185006). This dataset encompasses 16 samples across four groups (FA2M = 3, SMK2M = 5, FA4M = 3, SMK4M = 5) and includes a total of 46,122 cells and 28,213 detected genes. Cell type-specific marker genes for this dataset have been established in previous studies [27]. Similarly, scRNA-seq data for human lung tissue were retrieved from the GEO database (dataset GSE173869 [28]). This dataset consists of 12 samples across two groups (non-smoker samples = 3, COPD samples = 9). Cell type-specific marker genes have also been characterized in prior studies. Single-cell RNA-seq data analysis and visualization were performed with R and Seurat Package (https://satijalab.org/seurat/). Briefly, the single-cell analysis procedure and the markers, including data normalization, dimensionality reduction, and clustering, were as carried out as previously described. Macrophages were categorized into Clec5a-positive (Clec5a > 0.5) and Clec5a-negative (Clec5a = 0) groups. Additionally, macrophages were stratified into Clec5a low, median, and high groups based on Clec5a expression percentiles: 0–33.3% as Low CLEC5A, 33.3–66.6% as Median CLEC5A, and 66.6–100% as High CLEC5A.
The DEGs from different cell subsets were subjected to Reactome [29] enrichment analysis using https://reactome.org/. The "CellChat" R package (1.1.3 version) was performed to analyze intercellular communication. The pseudotime analysis was validated using the "Monocle 2" R package. Visualization of single-cell analysis was performed using R packages "ggplot2".
RNA-seq and bioinformatics
RNA samples were sequenced at Wekomo (China) using the Illumina system (San Diego, USA). The resulting RNA-seq data were aligned to the Ensembl v105 transcript annotations. To identify DEGs, the "Limma" package in R software was employed. The differentially expressed (DE) mRNAs were further subjected to GSEA.
Transfection
The cells were transfected as described in a previous study. THP-1 cells were differentiated into M0 macrophages by PMA and were transfected with siRNA using Lipofectamine RNAiMax (Invitrogen) following the manufacturer's instructions. siRNAs of CLEC5A were designed and constructed from RiboBio (Guangzhou, China). Cells were harvested for experiments 72 h after transfection.
Cigarette smoke extract (CSE) extraction and preparation
In our methodology, we established a system for generating and capturing CSE using a setup comprising two active core gas sampling bottles connected by a latex tube (ZJ4383). This setup allowed the transfer of gas from the left to the right bottle under negative pressure conditions. We added 5 ml of DMEM culture medium (C11995500BT) to each bottle. A “Hongmei” brand filter cigarette was then connected to the inlet of the left bottle and ignited. Using a 20 ml syringe, gas from within the apparatus was drawn out from the inlet of the right bottle, facilitating the passage and dissolution of tobacco smoke into the medium. This process was repeated until 10 ml of medium had absorbed the smoke from two cigarettes. The medium was then thoroughly mixed and filtered through a 0.22 µm filter (SLGP033RB) to remove any bacteria and particulate matter. The filtered solution was considered to be 100% CSE, which was subsequently diluted with DMEM medium for use, stored at − 80 °C.
GWAS Data sources
SNPs associated with FEV1/FVC, FEV1, emphysema/chronic bronchitis, and COPD diagnosis were downloaded from the IEU OpenGWAS project [30] as outputs from a GWAS pipeline analyzing variables from the UK Biobank. All SNP data were adjusted for age, sex, and genotype measurement batch. Data for the FEV1/FVC ratio (n = 321,047) came from one GWAS [31], including 321,047 individuals, sourced from the UK Biobank [32], while separate GWAS from the UK Biobank provided data for FEV1 (n = 321,047) and FVC (n = 321,047). Finally, GWAS data for COPD diagnosis (cases: 26,710, controls: 334,484; total n = 361,194) were also obtained from the UK Biobank.
An eQTL analysis was performed for ENSG00000258227 using a dataset of 31,684 individuals obtained from the IEU GWAS database. eQTLs associated with CLEC5A served as proxies for increased CLEC5A expression. Notably, all GWAS datasets used in this study were downloaded from the IEU GWAS database (https://gwas.mrcieu.ac.uk/) [33].
Summary-data-based Mendelian (SMR) randomization analyses
We employed SMR and HEIDI (Heterogeneity in Dependent Instruments) tests within cis-regulatory regions using the SMR software [34]. This approach, based on the original SMR publication, leverages a single-nucleotide variant (SNV) at a primary cis-eQTL as an instrumental variable (IV). This IV, combined with summary-level eQTL and GWAS data, allowed us to investigate potential causal or pleiotropic relationships between gene expression and traits of interest. However, SMR cannot definitively distinguish between causal and pleiotropic effects due to its reliance on a single IV. The HEIDI test helps address this limitation by differentiating causality from linkage, a less biologically relevant association. We additionally performed SMR and HEIDI analyses using summary-level sQTL data as the exposure. We maintained the standard settings in the SMR software, employing a p-value threshold of 5.0 × 10−8 for selecting the top eQTLs and a 1 Mb window around the probe center for cis-eQTL selection. Importantly, all SMR analyses were restricted to cis-regulatory regions. For these tests, a p-value ≤ 0.05, considering the use of a single probe, indicated statistical significance. In the case of HEIDI, a p-value < 0.05 suggested significant linkage, implying that the association was not purely causal.
Two sample MR
Our Mendelian randomization (MR) analysis primarily employed the inverse-variance weighted (IVW) method. We complemented this approach with MR Egger, weighted median, simple mode, and weighted mode methods [33, 35]. Heterogeneity across individual causal effects was assessed using the Cochran's Q statistic, implemented in both MR Egger and IVW methods. A p-value < 0.05 for the Cochran's Q test indicated significant heterogeneity. Horizontal pleiotropy was evaluated using two methods: MR Egger regression and MR-PRESSO (Pleiotropy Residual Sum and Outlier). MR Egger regression assessed for directional horizontal pleiotropy, with an intercept near zero and p-value > 0.05 suggesting its absence. MR-PRESSO evaluated overall genetic pleiotropy within the MR model, performing outlier removal and re-running the analysis to minimize bias. A p-value > 0.05 for the MR-PRESSO Global test indicated no evidence of horizontal pleiotropic outliers. Leave-one-out sensitivity analyses were conducted to ensure robustness. Positive estimates for remaining variants after excluding a single variant supported the overall findings. Additionally, Steiger filtering was employed to verify the causal direction between the two attributes of interest. All statistical analyses were performed in R using the 'TwoSampleMR' package [30] for IVW, weighted median, simple mode, weighted mode, and MR Egger regression methods. A significance level of p-value < 0.05 was applied. In the presence of significant heterogeneity based on Cochran's Q test, a random effects model would be utilized to calculate the IVW estimates.
Colocalization analysis
We employed the 'coloc' R package [36] with default settings to perform Bayesian colocalization analysis. This method estimates the probability of two traits sharing a common causal variant. As mentioned earlier, Bayesian colocalization calculates four posterior probabilities (PPs) to assess the likelihood of a single variant influencing both traits. Our study focused on testing hypotheses 3 (PPH3) and 4 (PPH4), which investigate the relationship between a protein and asthma risk through distinct or shared genetic variants within a specific region. A gene-based PPH4 score exceeding 80% served as the primary criterion for colocalization in our analysis.
Statistical analyses
All statistical analyses were performed using GraphPad Prism v8.0.1. The two-tailed paired Student's t-test, two-tailed Mann–Whitney test, and two-tailed Pearson Correlation were used to determine the significance between means. The P-values were represented as follows: ns (not significant), *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Analysis of GOLD stage 1–2 patients and early-stage smoke-exposed mice
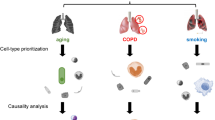
We conducted a comprehensive analysis utilizing data from GOLD stages 1–2 COPD patients and early-stage smoke-exposed mice (Fig. 1A). Initially, sequencing data from human lung tissue samples in the GEO dataset GSE47460, encompassing both healthy control groups and patients with GOLD stage 1–2 COPD (nonCOPD = 91; COPD = 82), were employed. Subsequent differential expression and PCA analyses did not reveal significant differences between healthy controls and COPD GOLD stage 1–2 cohorts (Fig. 1B). A total of 514 upregulated and 557 downregulated genes were identified using a fold change cutoff of 0.25 (Fig. 1B).

Combined Analysis of GOLD 1/2 Patients in GSE47460 and 4–6 Month Smoke-Exposed Mice in GSE125521. A The diagrammatic representation illustrates the analysis process of GSE47460 at GOLD stages 1 and 2 (n = 91 controls and n = 82 GOLD1/2 patients) and GSE125521 (n = 3 Air and n = 3 smoke). B The PCA analysis and volcano plot of GOLD stages 1 and 2 in GSE47460. C The PCA analysis and volcano plot of smoked mice at 4 months and 6 months in GSE125521 in GSE125521. D WGCNA analysis of the correlation of 17 gene modules with both the occurrence of COPD and the association with FEV1%pre and FVC% in GSE47460. E–F KEGG/GO enrichment analysis of DEGs in Negatively (E) and Positively (F) correlated DEGs using Metascape database.
To isolate genes crucial in COPD pathogenesis, sequencing data from smoke-exposed mice in the GEO dataset GSE125501 were also utilized, including samples at 4, 6, and 15 months of smoke exposure. Focusing on the 4- and 6-month datasets, differential expression and PCA analyses demonstrated distinct clustering between smoke-exposed and air-exposed mice (Fig. 1C). Specifically, 569 and 494 genes were upregulated and downregulated in the 4-month smoke-exposed mice, while 713 and 780 genes were correspondingly upregulated and downregulated in the 6-month cohort. Utilizing CIBERSORTx, we investigated immune cell infiltration in patients at GOLD stage 1–2, revealing significant reductions in resting CD4 memory T-cells and elevations in M0 macrophages (Figure S1A). Similar trends in immune cell changes were observed in the 4- and 6-month smoke-exposed mice (Figure S1B-C).
Further analysis using WGCNA identified 22 gene modules (Figure S1D). Correlation analysis with COPD status and FEV1% predicted value revealed two positively correlated modules (yellow and red) and three negatively correlated modules (light yellow, green, and light cyan) (Fig. 1D). Different gene sets, including upregulated DEGs from GSE47460, genes from the yellow and red modules in WGCNA, and upregulated DEGs from 4- to 6-month smoke-exposed mice were selected for negative gene sets. For positive gene sets, downregulated DEGs from GSE47460, genes from the light yellow, green, and light cyan modules in WGCNA, and downregulated DEGs from 4- to 6-month smoke-exposed mice were curated. Unified enrichment analysis using Metascape indicated that both negative and positive gene sets were enriched in similar biological pathways and phenotypes. Negative gene sets were significantly enriched in pathways such as positive regulation of MAPK cascade, positive regulation of immune response, neutrophil degranulation, inflammatory response, and regulation of leukocytes (Fig. 1E). Conversely, positive gene sets were mainly enriched in pathways like cell–cell adhesion, tissue morphogenesis, negative regulation of cell differentiation, and extracellular matrix organization (Fig. 1F). These findings suggest that in the early stages of COPD, genes associated with immunological functions play a pivotal role in disease onset, while genes critical for maintaining pulmonary tissue homeostasis are downregulated.
Next, both negative- and positive-correlated gene sets were intersected. Remarkably, 23 genes were intersected across all four subcategories of the negative-correlated gene set, while 7 genes were consistently present in the positive-correlated sets (Fig. 2A and Tables S1 and S2). Leveraging this 30-gene intersection, we performed analyses on these genes for GO enrichment in Biological Processes (BP) and Molecular Functions (MF). Additionally, Reactome and KEGG enrichment analyses were conducted, suggesting significant enrichment in immune-inflammatory pathways such as CCR Chemokine Receptor Binding and Myeloid Leukocyte Activation (Fig. 2B). Simultaneously, we constructed a Protein–Protein Interaction (PPI) network using the STRING [25] database and utilized CytoHubba for core gene identification (Fig. 2C). This analysis identified FCER1G, ITGB2, TREM2, FCGR2B, CD53, CLEC5A, and CTSS as central nodes in the PPI network (Fig. 2C).

Eight differential genes were selected as potential pathogenic genes for COPD. A 23 overlapping genes were identified at the intersection of the negatively correlated gene set, and seven overlapping genes were found in the positively correlated gene set. B Display of the enrichment results for Molecular Function (MF), Biological Process (BP), Reactome, and KEGG pathways of 30 overlapping genes, with p-values represented as log10 values. C Schematic representation of the PPI network of RRA genes according to the 30 overlapping genes. Top 15 hub genes screened by Cytobubba plugin with Cytoscape according to the PPI network. D Gene selection by the LASSO regression according to the occurrence of COPD in GSE47460. E Eight overlapping genes were identified after intersecting the results from five machine learning methods (SVM-RFE, LASSO Model, Ridge Regression, Elastic Net, and Random Forest Model). F The coefficient values of the 8 genes from the LASSO Model, Ridge Regression, Elastic Net, and Random Forest Model results. G The expression of 8 overlapping genes in GSE47460 (left) and GSE125521 (right). Data are expressed as mean ± SD. P values shown in charts determined by a two-tailed Mann–Whitney test (G)
To identify key genes associated with COPD pathology and lung function decline, this study employed five machine learning methods: SVM-RFE, LASSO Model, Ridge Regression, Elastic Net, and Random Forest Model (Tables S3-S7). These methods were applied to the GSE47460 dataset, analyzing variables related to COPD (Fig. 2D, Figs. S2A–D, and E). By cross-validating the results from these five methods, we identified eight genes (CD53, CLEC5A, CXCL13, FCER1G, HK3, MRC1, CDH16, and IYD) that consistently appeared across all models (Fig. 2E, F and Table S8). This finding suggests that these eight genes could potentially be pathogenic factors in COPD, and they were therefore selected for further analysis. Our data indicated upregulation of CD53, CLEC5A, CXCL13, FCER1G, and MRC1 in COPD patients, while CDH16, IYD, and PGC were downregulated. These findings were corroborated by sequencing results from mice exposed to smoke for 4 and 6 months (Fig. 2G).
Significant changes in Cd53, Clec5a, and Fcer1g expression in early smoke-exposed mice
To further explore the expression profiles of the identified 8 key genes across distinct pulmonary cell types and their alterations during early-stage COPD, we utilized single-cell sequencing data from GSE185006 [27], encompassing 2- (SMK2M) and 4-month smoke-exposed mice (SMK4M) along with their controls (Flash Air group, FA2M, and FA4M, n = 4; Fig. 3A and Figure S3A). The cells were re-clustered into 17 distinct groups, including alveolar type I (AT1), AT2, gCAP, aCAP, T cells, B cells, Alveolar Macrophages (AMs), Interstitial Macrophages (IMs), Dendritic Cells (DCs), NK cells, Fibroblasts, Neutrophils, Ciliated cells, Monocytes, Smooth Muscle Cells (SMCs), Vascular Endothelial Cells (VECs), and Proliferating Cells (Fig. 3B). The top two unique markers for each cell type were identified (Figure S3B).

Identification of Clec5a, Cd53, and Fcer1g as Potential COPD Pathogenic Genes Associated with Macrophage Inflammation. A Schematic representation of the mouse samples of GSE186005. B Umap plot of single-cell transcriptomes for 4 groups mice in GSE186005 (n = 4 FA2M, n = 4 SMK2M, n = 4 FA4M, n = 4 SKM4M). C-D The enrichment was demonstrated by the bubble chart, which shows the distribution and expression of hub genes (Cd53, Clec5a, Cxcl13, Fcer1g, Mrc1, Cdh16, Lyd, Pgc) in lung tissues from air group mice and smoke group mice at 2 months (C) and 4 months (D). E Schematic representation of the DEGs in Cleac5, CD53 and Fcer1g positive cells in air group and smoke group mice at 2 months (up) and 4 months (down). F The heatmap displays the significant DEGs in CLEC5A-positive cells of AMs and IMs in mice exposed to smoke for 2 and 4 months using Metascape database, with p-values represented as log10 values.
Proportional analysis of different sub-populations revealed a significant increase in T cells, B cells, AMs, IMs, and Neutrophils in 2-month smoke-exposed mice, accompanied by a noticeable decrease in AT2 cells. Similar trends were observed in the 4-month smoke-exposed cohort, suggesting pivotal roles for immune cells such as T cells, B cells, and macrophages during early-stage COPD, characterized by an initial attrition in alveolar epithelial cells (Figure S3C).
We then assessed the expression changes and primary cellular locations of the 8 selected genes within the single-cell sequencing data (Fig. 3C, D). CD53 exhibited ubiquitous expression across multiple immune cell types, including T cells, NK cells, Neutrophils, Monocytes, AMs, IMs, DCs, and B cells, with upregulation in the smoke-exposed group. Clec5a was predominantly expressed in AMs and IMs, showing upregulation in the smoke-exposed group. Cxcl13 was expressed in IMs, with a non-significant reduction in the smoke-exposed group. Fcer1g was widely expressed in NK cells, Neutrophils, Monocytes, IMs, and AMs, with upregulation in the smoke-exposed group. Mrc1 was mainly found in Active and Alveolar Macrophages, with no significant differential expression between FA and SMK groups. Cdh16 and Pgc were predominantly expressed in AT2 cells, with no noticeable changes between the two groups (Figure S4-6). Iyd was expressed in ciliated cells and, although slightly downregulated in the SMK group, the change was not statistically significant. Given the crucial role of immune cell activation and quantitative shifts in the early stages of COPD and considering the absence of significant changes in Mrc1 and Cxcl3, we focused on Cd53, Clec5a, and Fcer1g for further research, as they were predominantly expressed in immune cells (Fig. 3C, D).
Considering that all upregulated genes, including Cd53, Clec5a, Cxcl13, Fcer1g, and Mrc1, were expressed in macrophages, and given the existing literature indicating that lung macrophages account for over half of pulmonary immune cells, it is highly conceivable that lung macrophages play a crucial role in the early stages of COPD. Accordingly, we focused on active and alveolar macrophages. Initially, we explored alterations occurring in alveolar macrophages and interstitial macrophages in both 2-month and 4-month groups exposed to either filtered air (FA) or smoke (SMK). Enrichment analysis on the DEGs in AMs and IMs for both time points revealed that in 2-month mice, IMs and AMs primarily triggered inflammatory and oxidative stress responses (Figure S7A). In 4-month mice, hypoxic effects were additionally observed (Figure S7B).
Further classification of AMs and IMs based on the expression of Cd53, Clec5a, and Fcer1g into either positive or negative expression sub-groups was conducted, followed by differential and enrichment analysis. In mice exposed to smoke for 2 and 4 months, Clec5a-positive AMs and IMs both exhibited responses related to leukocyte and cell migration, notably with neutrophil degranulation (Fig. 3F). Similarly, Cd53-positive AMs and IMs were primarily associated with neutrophil degranulation and the adaptive immune system (Figure S7C). Furthermore, Fcer1g-positive AMs and IMs in both 2-month and 4-month smoke-exposed mice were chiefly associated with endocytosis, leukocyte activation, and PTEN regulation (Figure S7D).
Clec5a identified as a potential pathogenic gene in early smoke exposure mice
To validate the results from mouse single-cell sequencing concerning the expression of specific genes in macrophages, we used publicly available sequencing data of alveolar macrophages isolated from BALF in COPD patients (GSE13896 [23] and GSE130928 [24]). In the GSE13896 dataset, 91 non-COPD, 91 smokers, and 82 COPD patients were included. For GSE130928, the dataset comprised 24 non-COPD, 42 smokers, and 22 COPD patients (Fig. 4A). The results showed that CLEC5A expression was comparable between non-COPD and smoker groups but was significantly elevated in COPD patients. FCER1G showed decreased expression in COPD patients, whereas CD53 remained comparable (Fig. 4B, C).

Clec5A upregulation in early-stage COPD and its impact on lung function and macrophage inflammation. A Schematic representation of the aveolar macrophage samples of humans from GSE13896 (n = 91 nonCOPD, n = 91 smokers and n = 82 COPD) and GSE130928 (n = 24 nonCOPD, n = 42 smokers and n = 22 COPD). B–C The relative expression of CLEC5A, CD53, and FCER1G in alveolar macrophage samples from human subjects, comparing nonCOPDs, smokers, and COPD patients across three groups, in datasets GSE13896 (B) and GSE130928 (C). D The relative expression of CLEC5A, CD53, and FCER1G in peripheral blood samples of humans from GSE71220 (left) (n = 83 Controls, n = 146 GOLD1/2 patients) and GSE76705 (right) (n = 44 Controls, n = 188 GOLD1/2 patients) at GOLD stages 1 and 2. E ROC curve analysis depicting the diagnostic performance of CLEC5A expression levels in the peripheral blood of COPD patients at GOLD stages 1 and 2, as presented in datasets GSE71220 and GSE76705. F, G Correlation analysis was conducted between the expression levels of Clec5A in peripheral blood and FEV1%pre and the FEV1/FVC ratio in patients at GOLD stages 1 and 2 from datasets GSE71220 (F) and GSE76705 (G). H Expression of relative DEGs in CLEC5A-positive and negative IM and AM from mice exposed to smoke for 2 months and 4 months, as analyzed in the GSE186005 dataset. I, J Network graphs compare pulmonary cell interactions in Clec5c- (left) and Clec5c + (right) cohorts from 2 M (I) and 4 M (J) datasets. Each vertex represents a cellular subpopulation; edges signify ligand-receptor interactions. Edge thickness quantifies cumulative ligand-receptor gene expression, and vertex size reflects Kleinberg centrality, indicating a cell's role in signaling. Cells are differentiated by color and number. K, L Secretory-type intercellular interactions among various cell types within Clec5c + macrophage clusters (left) and within Clec5a- macrophage clusters (right) from the 2 M dataset (K) and the 4 M dataset (L). Data are expressed as mean ± SD. P values shown in charts determined by a two-tailed Mann–Whitney test (B–D), Wilson/Brown test (E), and Pearson Correlation two-tailed (F, G)
We also examined the expression of CD53, CLEC5A, and FCER1G in two peripheral blood sequencing datasets of COPD patients (GSE71200 [21] and GSE76705 [22]), with emphasis on control subjects and individuals classified under GOLD stages 1–2. Among these genes, only CLEC5A consistently exhibited statistically significant upregulation in both datasets (Fig. 4D). Additionally, we performed an analysis of Receiver Operating Characteristic (ROC) curves for CLEC5A across both datasets, revealing statistically significant Area Under the Curve (AUC) values of 0.6269 and 0.6098 (Fig. 4E). Concurrently, we conducted a correlation analysis between the expression levels of these three genes and pulmonary function parameters FEV1%pre and FEV1/FVC ratio in human populations. The results indicated that CLEC5A displayed a statistically significant negative correlation with both FEV1%pre and FEV1/FVC in both cohorts (Fig. 4F, G and Figure S8A-B). In contrast, CD53 and FCER1G showed no significant correlation with FEV1%pre and FEV1/FVC in the GSE71220 dataset, while in GSE76705, they exhibited a positive correlation with these parameters (Figure S8A-B). Furthermore, we validated the expression of CLEC5A, FCER1G, and CD53 in another COPD patient cohort (GSE103174) that includes patients with GOLD stage 1 and 2. The results indicated that only CLEC5A expression was significantly elevated in the lung tissues of GOLD stage 1 and 2 patients (Figure S8C). Additionally, we re-grouped the patients in the GSE47460 dataset according to their smoking status to investigate the distribution of CLEC5A expression. The results indicated that there was no significant difference in CLEC5A expression between non-smokers and non-COPD smokers. However, in COPD patients who had ever smoked, CLEC5A expression was significantly higher compared to non-COPD ever smokers, and it was further elevated in current smokers with COPD (Figure S8D). These findings suggest that CLEC5A is inherently upregulated in COPD and may play a pathogenic role in the development of the disease, prompting us to prioritize further investigation into CLEC5A.
Furthermore, in the mouse single-cell data, we observed that Clec5a-positive interstitial macrophages or alveolar macrophages exhibited elevated expression levels of inflammatory cytokines and metalloproteinases (Ccl2, Ccl3, Ccl9, Ccl12, Ccr1, Ccr5, Mmp12, Mmp14, Mmp19, and IL-1b), some of which have been previously implicated in COPD (Fig. 4H). Additionally, we observed that the M1 macrophage marker TNF was elevated in both Clec5a-positive IMs and AMs, whereas the primary M2 macrophage marker Mrc1 was decreased in both cell types (Figure S8E). Furthermore, we stratified macrophages into low, median, and high Clec5a expression groups and similarly found that Tnf expression was elevated in the median and high groups, while Mrc1 expression was decreased in these groups (Figure S8F). Using the GEPIA2 database, we assessed the correlation of CLEC5A with these related genes in lung tissue, revealing a significant positive correlation with all except CCR5 (Figure S9A).
Based on these comprehensive analyses, Clec5a was the most closely associated with COPD progression among the three candidates. It is highly likely that it plays a pathogenic role in the early stages of the disease, prompting us to designate Clec5a as the focus of our subsequent research.
Clec5a + IMs/AMs interaction with monocytes/neutrophils in smoke-exposed mice
We categorized AMs and IMs from 2 and 4 M smoke-exposed mice into Clec5a-negative and Clec5a-positive groups, followed by CellChat analyses to identify differences in intercellular communication between these subsets. In 2 M smoke-exposed mice, Clec5a-positive macrophages exhibited significantly enhanced interaction with neutrophils, DCs, and monocytes compared to their Clec5a-negative counterparts (Fig. 4I). In 4 M mice, Clec5a-positive cells predominantly interacted with neutrophils, DCs, and fibroblasts (Fig. 4J). To further characterize cellular interactions induced by smoke exposure, Clec5a-positive cells from 2 and 4 M smoke-exposed mice were subdivided into FA and SMK groups. Cell-chat analyses in 2 M mice indicated that Clec5a-positive AMs displayed no interactions with other cells under normal conditions, while Clec5a-positive IMs minimally interacted with AMs. Upon external stimulation, these cells predominantly interacted with neutrophils, monocytes, and DCs (Figure S9B, C). In 4 M control mice, Clec5a-positive AMs and IMs already interacted with neutrophils, and this interaction was amplified upon stimulation, introducing interactions with DCs and T cells. Similar findings were observed in Clec5a-positive IMs (Figure S9D, E).
Comparing the signaling patterns between Clec5a-positive and -negative cells, we observed an upregulation of CCL signaling in Clec5a + AMs compared to Clec5a- AMs in 2-month mice. Notably, in IMs, not only was the CCL signaling enhanced, but also the IL-16 and CXCL signaling pathways were upregulated (Fig. 4K). In 4-month mice, an increase was detected only in the TNF and IGF signaling pathways within certain cell subpopulations (Fig. 4L). Ligand-receptor analyses were conducted to elucidate the mechanisms through which Clec5a-positive cells, serving as ligand-producing entities, modulate these cellular interactions. In 2 M smoke-exposed mice, Clec5a-positive cells elevated pathways mediated by ligands such as Lgalas9, Ccl9, Cxcl4, and Tgfb1 and specifically enhanced communication with monocytes and T cells via Ccl6 and Cxcl16 (Figure S10A and Figure S11A, B). In 4 M mice, enhanced Cxcl16 signaling pathways were observed, along with increased ligand-mediated signaling via Ccl3 and Cxcl2, among others (Figure S10B).
Further stratification of Clec5a-positive macrophages into FA and SMK groups revealed that smoke exposure led to enhanced intercellular communication (Fig. 5A, B). Integrating these findings with previous ligand-receptor analyses, we concluded that in 2 M smoke-exposed mice, Clec5a-positive AMs enhanced communication with monocytes through the Ccl6-Ccr2 axis, whereas Clec5a-positive IMs elevated communication with monocytes and T cells via Ccl6 and Ccl9, introducing additional interactions mediated by ligands such as Lgalas9 and Cxcl4 (Figure S11C). In 4 M smoke-exposed mice, Clec5a-positive IMs exhibited increased communication with T cells and neutrophils via the Cxcl16-Cxcr2 and Ccl9-Ccr2 axes, respectively, and introduced new ligand-mediated interactions through Igf1, Gas6, and Cxcl2 (Fig. 5C, D).

Clec5a + IMs and AMs Interaction with Monocytes, T Cells, and Neutrophils May Enhance Pulmonary Inflammation. A, B Network graphs depict interactions between pulmonary Clec5c + macrophages and other cell types in FA (left) and SMK (right) cohorts, derived from 2 M (A) to 4 M (B) datasets. C, D Bubble chart visualizes cell-to-cell interactions involving Clec5c + AMs or Clec5c + IMs clusters with immune cells in FA (red) and SMK (blue) cohorts, sourced from 2 M (C) to 4 M (D) datasets. Clec5c + AMs or Clec5c + IMs function as ligand-expressing cells in these interactions, with significant differences denoted by red boxes. E UMAP plot illustrates the distribution of AMs and IMs in FA and SMK mouse cohorts, derived from 2 to 4 M datasets. AMs are categorized into 5 distinct subgroups (AM_0, AM_2, AM_4, AM_5, and AM_6), and IMs are segregated into 4 distinct subgroups (IM_0, IM_3, IM_7, and IM_8). F UMAP plot illustrates the distribution of Clec5c + and Clec5c- macrophages. G, H Pseudotime trajectory analysis illustrates the developmental progression of IMs and AMs (G), color-coded by five distinct States (H). The analysis identifies three branching pathways leading to two developmental fates. I Pseudotime trajectory analysis delineates the developmental progression of Clec5c + and Clec5c- macrophages (left) and the relative proportions of Clec5c + and Clec5c- macrophages within five distinct States of Pseudotime trajectory (right); red boxes highlight State5 clusters with significantly different cell proportions. J Reactome enrichment of the marker genes in five distinct States of Pseudotime trajectory; red boxes indicate functional annotations of the State5 cluster cell populations. K Expression variations of the genes Ccl2, Marco, Ccl6, and Tnf across different pseudotime states (State_1–5) of macrophages. L Reactome enrichment of DEGs in macrophage subpopulations of the AM_4, AM_6, and State_5 clusters
Furthermore, we conducted additional analyses to assess which cells regulate Clec5a-positive cells under smoking conditions. Using IMs and AMs as target cells for intercellular communication, we found that in 2 M mice, monocytes and neutrophils regulate Clec5a-positive IMs and AMs through ligands such as Csf1, Ccl3, Ccl6, and C3 after smoke exposure (Figure S12A). In 4 M mice, only neutrophils maintained regulatory control over Clec5a-positive macrophages (Figure S12B). These findings complement the role of Clec5a-positive IMs and AMs as ligand cells in intercellular communication, further substantiating the role of Clec5a during early-stage COPD.
Clec5a induced pro-inflammatory macrophage differentiation
Additionally, we performed unsupervised clustering on IMs and AMs from the single-cell sequencing data, categorizing them into five (AM_0, AM_2, AM_4, AM_5, and AM_6) and four (IM_0, IM_3, IM_7, and IM_8) distinct subgroups, respectively (Fig. 5E). We analyzed the proportion of Clec5a-positive cells within each subgroup (Fig. 5F) and found that subgroups AM_4 and AM_6 had higher proportions of Clec5a-positive cells, while IM_7 and IM_8 showed a reduced proportion (Figure S13A-B). We performed differential expression analysis between Clec5a + and Clec5a- macrophages within the AM_4 and AM_6 clusters. The analysis revealed an upregulation of Ccl2, Cxcl16, Tlr4, Mmp12, and Mmp14 in Clec5a + cells of the AM_4 cluster (Figure S12C). In the Clec5a + cells of the AM_6 cluster, there was an increased expression of Ccl2, Tlr2, and S100A8 (Figure S13C).
Pseudotemporal analyses were conducted on macrophages (Fig. 5G), categorizing them into five distinct states (State_1 to State_5; Fig. 5H). An increased proportion of Clec5a-positive cells was observed in the more differentiated State_5 (Fig. 5I). Functional annotation of these cellular states revealed that State_5 is predominantly associated with the innate immune system, neutrophil degranulation, and the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in phagocytes (Fig. 5J). Temporal gene expression changes in macrophages showed that genes such as Ccl2, Ccl6, Tnf, and Marco were most highly expressed in the State_5 condition, indicating a pro-inflammatory phenotype of macrophages during this temporal stage (Fig. 5K). Additionally, in the pseudotime analysis of macrophages at different stages, we similarly found that the M1 marker gene Tnf is expressed at higher levels in CLEC5A-positive macrophages, while the M2 marker gene Mrc1 is expressed at lower levels (Figure S13D).
Further differential expression analysis and enrichment analyses on Clec5a-positive and -negative cells in State_5, AM_4, and AM_6 indicated that State_5 was primarily enriched in pathways such as Interleukin_12, Toll-like receptor cascades, and the innate immune system; AM_6 was primarily enriched in Cell cycle, the innate immune system, and neutrophil degranulation; and AM_4 was enriched in Toll-like receptor cascades, Chronic Myeloid Leukemia, and Oxidative Phosphorylation (Fig. 5L).
To explore the translational potential of our findings on the Clec5a gene from mice to humans, we analyzed single-cell RNA sequencing data from human lung tissues (GSE173869). This dataset comprised samples from 3 non-smokers and 9 COPD patients. The cells were reclassified into 20 distinct groups, including AT1 cells, AT2 cells, mast cells, alveolar fibroblasts, CD8 + T cells, CD4 + T cells, B cells, Mono_Macrophages, dendritic cells (DCs), natural killer (NK) cells, adventitial fibroblasts, B plasma cells, ciliated cells, basal/club cells, neutrophils, vascular endothelial cells (capillaries), pericytes/smooth muscle actin (SMA) cells, arterial and venous endothelial cells, and proliferating immune cells (Fig. 6A). Cell type-specific markers were identified (Figure S13E), and CLEC5A expression was found to be primarily localized to Mono_macrophages and macrophages, with minimal expression also observed in neutrophils (Fig. 6A and B).

CLEC5A + macrophages enhanced pro-inflammatory expression and amplified TNF pathway interactions with other cells. A UMAP plot of single-cell transcriptomes for human lung tissues in the GSE173869 dataset (non-smokers samples = 3, COPD samples = 9). B Bubble chart demonstrating the enrichment of CLEC5A in human lung tissues from the GSE173869 dataset, showcasing its distribution and expression. C Pathway enrichment results for CLEC5A-positive and negative Mono_Macrophages and Macrophages, with P-Values in log10 Scale. D Expression of relative DEGs in CLEC5A-positive and negative Mono_Macrophages and Macrophages, as analyzed in the GSE173869 dataset. E Secretory-type intercellular interactions among various cell types within CLEC5A + macrophage clusters and CLEC5A- macrophage clusters in control and COPD groups, as analyzed in the GSE173869 dataset. F Network graphs compare pulmonary cell interactions in CLEC5A- (left) and CLEC5A + (right) cohorts from GSE173869 datasets. Each vertex represents a cellular subpopulation; edges signify ligand-receptor interactions. Edge thickness quantifies cumulative ligand-receptor gene expression, and vertex size reflects Kleinberg centrality, indicating a cell's role in signaling. Cells are differentiated by color and number. G Comparison of TNF-TNFRSF1A signaling interactions between CLEC5A- and CLEC5A + macrophages and other cell subpopulations, as analyzed in the GSE173869 dataset
To investigate the functional role of CLEC5A in human macrophages, we categorized the Mono_Macrophages and Macrophages from the single-cell RNA-sequencing data (GSE173869) into CLEC5A-positive and -negative populations (Figure S14A). Differential gene expression analysis followed by enrichment analysis revealed that CLEC5A-positive cells upregulated genes associated with inflammatory pathways, while downregulating genes related to metabolism, lysosomes, and ferroptosis (Fig. 6C). Notably, genes like IL1B, TNF, and CXCL8 exhibited higher expression in CLEC5A-positive Mono_Macrophages, whereas CCL2, CCL3, and CCL4 were more highly expressed in CLEC5A-positive macrophages (Fig. 6D). Further cell-type interaction analysis demonstrated increased enrichment of MIF and TNF signaling pathways in CLEC5A-positive cells compared to negative cells (Fig. 6E). Additionally, CLEC5A-positive Mono_Macrophages and Macrophages interacted more frequently with other cell types compared to negative cells (Fig. 6F). This effect was particularly pronounced in the TNF-TNFRSF1A pathway, where CLEC5A-negative Mono_Macrophages displayed minimal interaction (Fig. 6G). These findings from human lung single-cell data corroborate our observations in mouse models, supporting the potential of CLEC5A in amplifying inflammatory responses within macrophages.
Elevated CLEC5A expression facilitates COPD progression through MR analysis
To clarify whether CLEC5A expression is associated with the progression of COPD, we examined its expression in lung tissue and peripheral blood among healthy control subjects, patients with GOLD stages 1/2, and patients with GOLD stages 3/4. While a gradual increase in CLEC5A expression was noted with more advanced GOLD stages, statistical significance was not observed in the GSE71220 dataset (Fig. 7A, B). Correlation analyses with pulmonary function parameters revealed a significant negative association of CLEC5A with FEV1%, FEVC%pre, Dlco%pre, and a positive correlation with %emphysema (f-950) (Fig. 7C–E). Subgroup analyses showed the positive correlation of CLEC5A with %emphysema (f-950) in GOLD 1/2 and GOLD 3/4 patients, with statistical significance observed in the GOLD 1/2 cohort (Fig. 7E). Although gene expression levels exhibited a negative trend with FEV1% predicted, the correlation was not statistically significant (Figure S14B). In peripheral blood, CLEC5A expression exhibited a significant negative correlation with FEV1% predicted and FEV1/FVC ratio (Figure S14C-D). These results suggest a continuous and significant role for CLEC5A in COPD progression, particularly in the development of emphysema, notably in GOLD stages 1/2.

Elevated CLEC5A Expression Facilitates COPD Progression through MR Analysis. A, B The expression of CLEC5A in lung tissue (GSE47460) (n = 91 Controls, n = 82 GOLD1/2 patients and n = 62 GOLD3/4 patients) (A) and peripheral blood (GSE71220 and GSE76705) (B) (GSE71220: n = 83 Controls, n = 146 GOLD1/2 patients and n = 213 GOLD3/4 patients) (GSE76705: n = 44 Controls, n = 188 GOLD1/2 patients and n = 115 GOLD3/4 patients) among control groups, patients with GOLD stages 1/2, and patients with GOLD stages 3/4. C, D Correlation analysis of CLEC5A in GSE47460 with FEV1%pre (C), FVC%pre, and Dlco% pre (D). E Correlation analysis of CLEC5A in GSE47460 with the percentage of emphysema (f-950), encompassing the entire population (left), and specifically with healthy controls, patients with GOLD stages 1/2, and patients with GOLD stages 3/4 (right). F MR analyses of CLEC5A expression and COPD outcomes, after conditioning on the effects of SNPs on other cell types. Effect sizes (beta, 95% CI) are represented as SD change in COPD outcome per SD CLEC5A expression. Points on the forest plot represent the effect size estimate, and whiskers denote 95% CIs. G This forest plot depicts the eQTLs of CLEC5A as exposure and COPD diagnosis, as well as FEV1, FVC and FEV1/FVC as outcomes. An odds ratio (OR) greater than 1 suggests that the respective mediator increases the risk of COPD outcome, while an OR less than 1 indicates a protective effect. H This forest plot depicts the eQTLs of CLEC5A as exposure and COPD-related respiratory insufficiency, as well as later on COPD and early on COPD as outcomes. I This forest plot depicts the eQTLs of CLEC5A as exposure and the protein level of CCL2, as well as CXCL1, CXCL8, IL-1B, IL-6 and TNF as outcomes. An odds ratio (OR) greater than 1 suggests that the respective mediator increases the risk of protein level outcome, while an OR less than 1 indicates a negative effect
To further explore the potential causal relationship between the CLEC5A gene and COPD, as well as its impact on lung function, we employed Mendelian randomization analyses (Fig. 7F). We focused on four COPD-related phenotypes from IEU GWAS database (COPD diagnosis, FEV1/FVC ratio, FEV1, and FVC) and three COPD-related phenotypes from FinnGen database (COPD-related respiratory insufficiency, early on COPD and later on COPD, Table S9). Summary-level data were analyzed using SMR and HEIDI tests. Notably, CLEC5A exhibited a significant causal association with COPD diagnosis and FEV1 according to the SMR test and did not show evidence of horizontal pleiotropy based on the HEIDI test (P > 0.05, Table. S10).
We further complemented our analysis with two-sample Mendelian randomization [30, 33] (TSMR). This method offers a cost-effective approach to estimate causal effects between gene expression and outcomes by leveraging SNP summary statistics from two independent datasets. In our study, we identified and employed approximately 229 CLEC5A-associated SNPs from the eQTLGen database as IVs (Instrumental variables) in the MR analysis. These IVs exhibited no evidence of directional pleiotropy after rigorous evaluation, and all instruments possessed F-statistics exceeding 10, indicative of strong instrumental validity, a key assumption for MR.
Our investigation into CLEC5A's role in COPD risk, as defined by diagnosis, revealed horizontal pleiotropy and outlier IVs based on MR-PRESSO analysis (Table S11). However, the MR-PRESSO outlier test identified no evidence of horizontal pleiotropy after removing outlier variants. Subsequently, the IVW method indicated a significant positive association between CLEC5A and COPD risk. Notably, no heterogeneity was observed between the IVs, and the MR Egger intercept suggested minimal evidence of pleiotropy. Additionally, consistency in OR values across most methods (Fig. 7G, Figure S15A) strengthens the robustness of our findings (Tables S11–S13).
We further investigated the impact of CLEC5A on lung function parameters, including FEV1/FVC, FEV1, and FVC. MR-PRESSO analysis identified outlier IVs for some analyses. After removing these outliers, the IVW method revealed a significant positive association of CLEC5A with both FEV1 and FVC (without heterogeneity, P-value = 1). However, the IVW analysis did not detect a significant effect on FEV1/FVC, with evidence of heterogeneity. The alignment of OR values from supplementary methods with the IVW results for FEV1/FVC, FEV1, and FVC (Fig. 7G, Figure S15B–D, Tables S11–S13) further strengthened these findings. Additionally, we investigated the influence of CLEC5A on COPD-related phenotypes in the FinnGEN database, including COPD-associated respiratory insufficiency, early on COPD, and later on COPD. After eliminating any outliers, the results of the IVW indicated a positive correlation between CLEC5A expression and COPD-related respiratory insufficiency and early on COPD, while a negative correlation was observed with later on COPD. The consistency of odds ratio values obtained from supplementary methods with the IVW results for COPD-related respiratory insufficiency, early on COPD, and later on COPD (Fig. 7H, Figure S15E–G, Tables S11–S13) further bolstered these findings. Collectively, all MR analyses suggested that CLEC5A acts upstream of the investigated COPD outcomes (Table S14). Although Bayesian colocalization analysis did not identify a shared genetic variant between CLEC5A and COPD outcomes (Table S15), it’s worth noting that the candidate colocalizing SNPs, rs13235900 and rs62479376, have passed the colocalization analysis (SNP.PP.H4 > 0.8, Table S16). This discovery suggests that rs13235900 and rs62479376 may be potential pathogenic SNP loci that jointly contribute to both changes in CLEC5A expression and influence on the progression of COPD. We also conducted reverse MR analysis, which indicated that COPD, lung function, and early/later on COPD do not influence the expression of CLEC5A (Table S17).
Further investigation was conducted to explore the causal relationship between CLEC5A expression and the expression of COPD-related inflammatory factors, we analyzed the impact of CLEC5A on the elevation of prominent inflammatory factors in peripheral blood from deCODE genetic datacase, including CCL2, IL-6, IL-1B, CXCL1, CXCL8, and TNF (Table S9). The IVW method revealed a positive correlation between CLEC5A and the inflammatory factors, although IL-6 and IL-1B did not reach statistical significance (Fig. 7I and Figure S15H). The alignment of odds ratio values obtained from supplementary methods with the IVW results for the aforementioned protein levels further corroborated these findings (Table S18).
These results highlight the effectiveness of Mendelian randomization in establishing a causal relationship between CLEC5A expression and key factors like COPD risk and lung function, as well as a significant positive correlation with COPD-related inflammatory factors. From a genetic perspective, this evidence supports the potential role of CLEC5A as a pathogenic factor in COPD development.
CLEC5A knockout in THP-1-derived macrophages reduces inflammatory mediators
We next established mouse models exposed to smoke for 2 and 6 months to validate the earlier analytical findings (Fig. 8A). Histological examination (HE staining) revealed significant alveolar septal destruction in mice exposed to smoke for 2 months, with a more pronounced effect observed in those exposed for 6 months (Fig. 8B). Although collagen deposition around small airways was not discernible in the 2-month smoke-exposed mice, an increase was noted in the 6-month group, albeit without statistical significance (Fig. 8B). Elevated Clec5a expression was observed in mice exposed to smoke for 2 and 6 months, with a more significant increase in the 2-month cohort (Fig. 8C). Similarly, post-smoke exposure, the protein levels of Clec5a were significantly increased (Fig. 8D). We also measured the expression of Fcer1g in 2- and 6-month smoke-exposed mice and found it to be elevated in both, with a more pronounced increase at 2 months, consistent with Clec5a, which contrasts with the earlier findings (Figure S16A). Fluorescent dual-staining demonstrated an increase in the abundance of Clec5a-positive macrophages in 2-month smoke-exposed mice, including both interstitial and alveolar macrophages. Similar observations were made in mice exposed to smoke for 6 months, with these Clec5a-positive macrophages primarily localized in the alveolar septa (Fig. 8E).

CLEC5A Knockout in THP-1-Derived Macrophages Reduces Inflammatory mediators. A Schematic representation of the experimental design: Mice were exposed to cigarette smoke and sampled at 2 months and 6 months. B HE and Masson's trichrome staining of lung tissue from mice exposed to smoke for a duration of 2 months (B) and 6 months (C). (n = 5). C Clec5a RNA expression levels were assessed in murine pulmonary tissues after 2 and 6 months of cigarette smoke exposure. (n = 5). D The protein expression of Clec5a was analyzed in the lung tissues of mice subjected to cigarette smoke exposure. (n = 5). E IF double staining was performed by using anti-Clec5a/anti-Cd68 antibodies from mice exposed to smoke for a duration of 2 months and 6 months. Quantification of the relative abundance of Clec5a and Cd68 double-positive cells to Cd68 single-positive cells within the observed field. (n = 5). F Efficiency of three CLEC5A siRNAs assessed by Western blot analysis 48 h post-transfection in PMA-differentiated THP-1 cells. (n = 3). G Diagrammatic representation of RNA-seq following CLEC5A knockdown and CSE stimulation in PMA-differentiated THP-1 cells. H GSEA of Hallmark performed on upregulated and downregulated DEGs. I The expression of TNF, CXCL1, CXCL2, CCL2, IL-1B, and IL-8 was measured after knockdown of CLEC5A by qRT-PCR assy. (n = 4). J The secretion of TNF, CCL2, CXCL1, IL-1B and IL-8 in cell supernatant was measured after the knockdown of CLEC5A by ELISA assay. (n = 3). Data are expressed as mean ± SD. P values shown in charts determined by two-tailed Student's t-test (B, D, E, F, J) and Multiple paired t test (I)
To further validate the in vitro expression changes of CLEC5A, PMA-differentiated THP-1 cells were stimulated with CSE for 72 h, resulting in increased CLEC5A expression (Figure S16B). To investigate the potential functions of CLEC5A, we treated THP-1 cells with PMA to induce their differentiation into macrophages and designed three CLEC5A-specific siRNAs to evaluate silencing efficiency. Results indicated that si-CLEC5A-2 and si-CLEC5A-3 significantly knocked down CLEC5A expression (Fig. 8F and Figure S16C), and si-CLEC5A-3 was selected for subsequent experiments. Knockdown of CLEC5A in differentiated THP-1 macrophages, followed by stimulation with 0.5% CSE for 72 h and subsequent RNA-seq (Fig. 8G), resulted in a distinct separation between the siNC + CSE and si-CLEC5A + CSE groups in PCA analysis (Figure S16D). Differential gene analysis revealed 382 upregulated genes and 337 downregulated genes (Figure S16E).
Our analysis of the sequencing data further explored expression changes within gene families associated with inflammation and extracellular matrix remodeling, including CCL, CXCL, IL, and MMPs. Notably, the majority of these inflammatory and MMP genes exhibited reduced expression in the siCLEC5A group (Figure S16F). GSEA showed that downregulated genes were mainly enriched in TNF Signaling via NFKB, Inflammatory Response, and KRAS signaling pathways. Conversely, upregulated genes were primarily enriched in Interferon Alpha Response, Interferon Gamma Response, Wnt/beta-catenin signaling, TGF-beta signaling, and Apoptosis pathways (Fig. 8H and Figure S16G).
Knockdown of CLEC5A in PMA-induced differentiated THP-1 cells was analyzed for the expression of COPD-related factors such as TNF, CXCL1, CXCL2, CCL2, IL1B, and IL-8. These results suggested that CLEC5A knockdown in PMA-induced differentiated THP-1 cells could suppress the expression of TNF, CXCL1, CXCL2, and IL-8, though the change in IL1B was not statistically significant (Fig. 8I). Furthermore, we evaluated the secretion levels of TNF, CCL2, CXCL1, IL-8, and IL-1B. Upon CLEC5A knockdown, there was a significant reduction in TNF, IL-1B, and IL-8 secretion, while no significant alteration was observed in CCL2 and CXCL1 secretion (Fig. 8J). Analysis of sequencing results using the CIBERSORTx database showed reduced M0 and M1 macrophage scores and increased M2 scores upon CLEC5A knockdown, suggesting a potential pro-inflammatory role of CLEC5A in macrophages (Figure S16H). Furthermore, we conducted a joint enrichment analysis of genes significantly downregulated after CLEC5A knockdown with genes upregulated in macrophages from both COPD patients and smoke-exposed mice using single-cell sequencing data. The results indicate that CLEC5A-positive macrophages and COPD macrophages share similarities in pathways such as Signaling by Interleukins and Regulation of Inflammatory Response in humans. Additionally, they share similarities with smoke-exposed mouse genes in pathways related to Oxidative Phosphorylation and Cellular Response to Cytokine Stimulus (Figure S17A-B).
These results indicated that the knockdown of CLEC5A expression could suppress the expression of related inflammatory factors in macrophages, suggesting a pro-inflammatory role for CLEC5A in macrophages.
Statistically significant factors identified from CLEC5A-knockdown sequencing (CXCL5, CXCL8, CXCL6, CXCL1, CXCL2, CXCL3, CCL20, CCL4, IL5, IL1B, MMP7, MMP8, MMP10) were selected and analyzed for expression changes in COPD patients from GSE47460. Most genes showed increased expression with COPD progression, with CXCL8, CXCL1, CXCL2, CXCL3, CCL20, CCL4, and IL-1B being notably significant between GOLD1/2 patients and GOLD3/4 patients (Figure S17C). Populations were categorized based on CLEC5A expression into Low-CLEC5A, Moderate-CLEC5A, and High-CLEC5A groups, with most factors showing higher expression in the High-CLEC5A group (Figure S17D). Correlation analysis between CLEC5A and selected factors in dataset GSE47460 corroborated these findings, further substantiating the role of CLEC5A in the observed phenomena (Figure S17E).
Discussion
The 2023 GOLD guidelines have redefined COPD as a heterogeneous pulmonary condition characterized by chronic respiratory symptoms, including dyspnea, cough, sputum production, and exacerbations [37]. It is now understood that this condition is associated with airway abnormalities such as bronchitis and bronchiolitis and/or alveolar abnormalities like emphysema, resulting in persistent, often progressive airflow obstruction. Therefore, the identification of early-stage COPD and timely intervention at this phase is essential. Importantly, early-stage COPD may initiate before overt symptoms such as significant respiratory distress appear [38]. Characteristics of this phase may include minor airway diseases, mild airflow limitations, and early inflammatory responses [39].
However, there is currently a lack of clarity regarding the definition of early-stage COPD. Over the years, different studies have used divergent diagnostic criteria and methodologies, leading to issues of consistency and comparability. Additionally, there is a gap in sensitive and specific diagnostic methods to accurately identify early-stage COPD, complicating its diagnosis [6]. Moreover, the etiology of early-stage COPD remains elusive; while interactions among inflammation, genetic predispositions, and environmental factors are likely involved, the specific processes remain largely unexplored.
Investigating the evolutionary patterns of early-stage COPD can provide profound insights into its pathophysiological processes and reveal potential early intervention targets. Our study integrated sequencing data from lung tissues of COPD patients at GOLD stages 1 and 2 with early-stage smoke-exposure mouse models to identify CLEC5A as a putative pathogenic gene in the early evolution of COPD. Early studies have identified CLEC5A in the context of dengue fever. During dengue virus infection, CLEC5A interacts directly with dengue viral particles, leading to the phosphorylation of DAP12 [11]. While the interaction between CLEC5A and the dengue virus does not facilitate viral entry, it does stimulate the release of pro-inflammatory cytokines [40, 41]. Inhibiting CLEC5A-mediated signaling can reduce the production of these cytokines in macrophages infected with the dengue virus, including the NLRP3 inflammasome. Additional studies have demonstrated that blocking CLEC5A can attenuate pseudomonas aeruginosa-induced neutrophil extracellular traps (NETs), lung injury, and collagen deposition [42].
Subsequent research has confirmed the pivotal role of CLEC5A in the generation of NETs and the production of pro-inflammatory cytokines in response to microbial stimuli. CLEC5A exhibits maximal expression on pro-inflammatory M1 macrophages, while alveolar macrophages from healthy donors demonstrate minimal levels of CLEC5A expression [43]. In the present study, CLEC5A-positive macrophages were nearly absent in the air control group of the smoke-exposure animal model [43]. In summary, CLEC5A can be regarded as an innate immune checkpoint capable of enhancing pro-inflammatory signals, thereby contributing to either infectious or sterile inflammation.
In peripheral blood samples from two COPD patient cohorts, we observed no significant differences in Clec5A expression between non-smokers and smokers. However, in BALF samples from another two COPD patient cohorts, Clec5A levels were significantly elevated in GOLD stage 1–2 patients compared to healthy controls and were further elevated in GOLD stage 3–4 patients. Additionally, the expression of CLEC5A in both peripheral blood and lung tissue was correlated with pulmonary function indicators such as FEV1%pre; it also showed a significant correlation with the emphysema index, especially in early-stage COPD patients. These observations suggest that the aberrant expression of CLEC5A in response to external stimuli could be one of the determinants for the progression to COPD, and it may primarily be involved in the development and progression of emphysema. Despite increased FCER1G expression in lung tissues of COPD patients and smoke-exposed mice, its expression in human BALF and peripheral blood decreases with disease severity. The relationship between FCER1G and COPD has not been reported at present, and FCER1G may have a complex regulatory role in the progression of COPD.
MR leverages the random allocation of genes during meiosis, thereby reducing the impact of confounding factors on genetic effects. This approach allows for a more accurate elucidation of the causal relationships between genetic variations and diseases or other health conditions [33]. Our study employed MR techniques, including SMR, 2SMR, and MR colocalization, to strengthen the evidence for CLEC5A's involvement in COPD and its impact on lung function. These approaches were crucial in elucidating the genetic influence of CLEC5A on COPD susceptibility and its functional consequences. Our findings suggest that higher CLEC5A expression is associated with an increased risk of COPD and has detrimental effects on lung function. Additionally, in another GWAS dataset, FinnGEN, the results suggest that increased CLEC5A expression promotes the incidence of early-stage COPD and COPD-related respiratory insufficiency, while showing a negative correlation with later-stage COPD. The above MR Results highlights the potential role of CLEC5A in early-stage COPD detection and severity assessment, considering the importance of FEV1 as a key marker for COPD staging. While colocalization analysis did not detect a shared genetic variant, the eQTLs for CLEC5A, rs13235900 and rs62479376, emerged as potential causal variants associated with COPD diagnosis. Additionally, we found that CLEC5A positively regulates common inflammatory factors in COPD, such as CCL2, CXCL8, and TNF.
Despite identifying CLEC5A as a potential early-stage pathogenic gene in COPD evolution, our study did not investigate its specific functional mechanisms, especially in the early stages of COPD. Our hypothesis, based on the observed localization of CLEC5A-positive macrophages, suggests that these cells may contribute to COPD pathology through alveolar septal degeneration. This hypothesis is further supported by the correlation between CLEC5A-positive macrophage expression and markers of lung function (FEV1% predicted) and emphysema. Our mouse model further suggested that the onset of alveolar septal damage in smoking conditions considerably precedes airway remodeling. Furthermore, we knocked out the CLEC5A in THP-1-derived macrophages and conducted RNA sequencing. We observed a downregulation in the expression of most chemokines and pro-inflammatory cytokines, including TNF, CXCL1, CXCL2, CXCL8, and IL-1B, which have been previously reported in association with COPD [4, 44]. The combined enrichment analysis of DEGs from CLEC5A knockdown experiments and single-cell sequencing data of macrophages suggests that CLEC5A predominantly contributes to the inflammatory response in COPD-associated macrophages.
Furthermore, Wortham and colleagues demonstrated that CLEC5A mediates macrophage function and the pathologies associated with chronic obstructive pulmonary disease [43]; Zhang et al. found that expressions of CLEC5A were upregulated in macrophages derived from THP1 cells and lung tissue from COPD patients [45], combined with our findings, it suggests that the negative feedback regulatory mechanism controlling CLEC5A expression in response to external stimuli may be one of the distinguishing factors between smokers and COPD patients. This advancement substantiates the pro-inflammatory role of CLEC5A in macrophages. Nonetheless, these speculations require further empirical validation, representing a limitation of our current study. Future investigations should employ macrophage-specific Clec5A knockout mice to elucidate this aspect.
In summary, the concept of early-stage COPD is intricate and multifaceted. The identification and timely intervention in the early-stage COPD are imperative yet challenging due to indistinct definitions, unclear pathogenic mechanisms, and diagnostic complexities. Through the utilization of sequencing data derived from lung tissues of COPD patients at GOLD stages 1–2, integrated with an early-stage smoking mouse model, we have identified CLEC5A as a potential pathogenic gene involved in the initial progression of COPD. Additionally, Mendelian randomization analysis further confirms its association with COPD onset and declining lung function, particularly in its positive correlation with early on COPD and negative correlation with later on COPD. Subsequent research endeavors should focus on elucidating the specific role of CLEC5A in shaping the early progression of COPD and uncovering its underlying mechanisms. Moreover, concerted efforts should be directed towards evaluating the potential of CLEC5A as a prospective biomarker and an early therapeutic target.
Data availability
The datasets supporting the conclusions of this article are available in online repositories. Names of repositories/repositories and accession number(s) are provided in the article/Additional Material. All other relevant materials are available from the corresponding author upon reasonable request.
References
Christenson SA, Smith BM, Bafadhel M, Putcha N (2022) Chronic obstructive pulmonary disease. Lancet 399:2227–2242. https://doi.org/10.1016/S0140-6736(22)00470-6
Yang IA, Jenkins CR, Salvi SS (2022) Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med 10:497–511. https://doi.org/10.1016/S2213-2600(21)00506-3
Agusti A, Hogg JC (2019) Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med 381:1248–1256. https://doi.org/10.1056/NEJMra1900475
Brightling C, Greening N (2019) Airway inflammation in COPD: progress to precision medicine. Eur Respir J. https://doi.org/10.1183/13993003.00651-2019
Wang Y, Xu J, Meng Y, Adcock IM, Yao X (2018) Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis 13:3341–3348. https://doi.org/10.2147/COPD.S176122
Choi JY, Rhee CK (2020) Diagnosis and treatment of early chronic obstructive lung disease (COPD). J Clin Med. https://doi.org/10.3390/jcm9113426
Fazleen A, Wilkinson T (2020) Early COPD: current evidence for diagnosis and management. Ther Adv Respir Dis 14:1753466620942128. https://doi.org/10.1177/1753466620942128
Wu J et al (2022) The role of lung macrophages in chronic obstructive pulmonary disease. Respir Med 205:107035. https://doi.org/10.1016/j.rmed.2022.107035
Woo YD, Jeong D, Chung DH (2021) Development and functions of alveolar macrophages. Mol Cells 44:292–300. https://doi.org/10.14348/molcells.2021.0058
Bakker AB, Baker E, Sutherland GR, Phillips JH, Lanier LL (1999) Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Natl Acad Sci U S A 96:9792–9796. https://doi.org/10.1073/pnas.96.17.9792
Chen ST et al (2008) CLEC5A is critical for dengue-virus-induced lethal disease. Nature 453:672–676. https://doi.org/10.1038/nature07013
Wu MF et al (2013) CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood 121:95–106. https://doi.org/10.1182/blood-2012-05-430090
Tosiek MJ et al (2022) Activation of the innate immune checkpoint CLEC5A on myeloid cells in the absence of danger signals modulates macrophages’ function but does not trigger the adaptive t cell immune response. J Immunol Res 2022:9926305. https://doi.org/10.1155/2022/9926305
Yi E et al (2023) Genetic screening of MMP1 as a potential pathogenic gene in chronic obstructive pulmonary disease. Life Sci 313:121214. https://doi.org/10.1016/j.lfs.2022.121214
Yi E et al (2021) Long noncoding RNA IL6-AS1 is highly expressed in chronic obstructive pulmonary disease and is associated with interleukin 6 by targeting miR-149-5p and early B-cell factor 1. Clin Transl Med 11:e479. https://doi.org/10.1002/ctm2.479
Zheng M et al (2019) Long noncoding RNA COPDA1 promotes airway smooth muscle cell proliferation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 61:584–596. https://doi.org/10.1165/rcmb.2018-0269OC
Yi E et al (2023) Smad3-mediated lncRNA HSALR1 enhances the non-classic signalling pathway of TGF-beta1 in human bronchial fibroblasts by binding to HSP90AB1. Clin Transl Med 13:e1292. https://doi.org/10.1002/ctm2.1292
Kim S et al (2015) Integrative phenotyping framework (iPF): integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genomics 16:924. https://doi.org/10.1186/s12864-015-2170-4
Cruz T et al (2019) Smoking impairs the immunomodulatory capacity of lung-resident mesenchymal stem cells in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 61:575–583. https://doi.org/10.1165/rcmb.2018-0351OC
Conlon TM et al (2020) Inhibition of LTbetaR signalling activates WNT-induced regeneration in lung. Nature 588:151–156. https://doi.org/10.1038/s41586-020-2882-8
Lesluyes T et al (2016) RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur J Cancer 57:104–111. https://doi.org/10.1016/j.ejca.2015.12.027
Bedekovics T, Hussain S, Feldman AL, Galardy PJ (2016) UCH-L1 is induced in germinal center B cells and identifies patients with aggressive germinal center diffuse large B-cell lymphoma. Blood 127:1564–1574. https://doi.org/10.1182/blood-2015-07-656678
Shaykhiev R et al (2009) Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183:2867–2883. https://doi.org/10.4049/jimmunol.0900473
O’Beirne SL et al (2020) Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 201:735–739. https://doi.org/10.1164/rccm.201908-1683LE
Szklarczyk D et al (2023) The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 51:D638–D646. https://doi.org/10.1093/nar/gkac1000
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformat 9:559. https://doi.org/10.1186/1471-2105-9-559
Spix B et al (2022) Lung emphysema and impaired macrophage elastase clearance in mucolipin 3 deficient mice. Nat Commun 13:318. https://doi.org/10.1038/s41467-021-27860-x
Watanabe N et al (2022) Anomalous epithelial variations and ectopic inflammatory response in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 67:708–719. https://doi.org/10.1165/rcmb.2021-0555OC
Jassal B et al (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48:D498–D503. https://doi.org/10.1093/nar/gkz1031
Burgess S, Thompson SG (2015) Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 181:251–260. https://doi.org/10.1093/aje/kwu283
Shrine N et al (2019) New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 51:481–493. https://doi.org/10.1038/s41588-018-0321-7
Sudlow C et al (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12:e1001779. https://doi.org/10.1371/journal.pmed.1001779
Hemani G et al (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife. https://doi.org/10.7554/eLife.34408
Zhu Z et al (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48:481–487. https://doi.org/10.1038/ng.3538
Han Z et al (2018) Parkinson’s disease and Alzheimer’s disease: a Mendelian randomization study. BMC Med Genet 19:215. https://doi.org/10.1186/s12881-018-0721-7
Giambartolomei C et al (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10:e1004383. https://doi.org/10.1371/journal.pgen.1004383
Sin DD et al (2023) Air pollution and COPD: GOLD 2023 committee report. Eur Respir J. https://doi.org/10.1183/13993003.02469-2022
Dona E et al (2023) Exploring current concepts and challenges in the identification and management of early-stage COPD. J Clin Med. https://doi.org/10.3390/jcm12165293
Laucho-Contreras ME, Cohen-Todd M (2020) Early diagnosis of COPD: myth or a true perspective. Eur Respir Rev. https://doi.org/10.1183/16000617.0131-2020
Sung PS, Hsieh SL (2019) CLEC2 and CLEC5A: pathogenic host factors in acute viral infections. Front Immunol 10:2867. https://doi.org/10.3389/fimmu.2019.02867
Sung PS, Huang TF, Hsieh SL (2019) Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun 10:2402. https://doi.org/10.1038/s41467-019-10360-4
Sung PS, Peng YC, Yang SP, Chiu CH, Hsieh SL (2022) CLEC5A is critical in Pseudomonas aeruginosa-induced NET formation and acute lung injury. JCI Insight. https://doi.org/10.1172/jci.insight.156613
Wortham BW et al (2016) Cutting edge: CLEC5A mediates macrophage function and chronic obstructive pulmonary disease pathologies. J Immunol 196:3227–3231. https://doi.org/10.4049/jimmunol.1500978
Barnes PJ (2016) Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 138:16–27. https://doi.org/10.1016/j.jaci.2016.05.011
Zhang Z et al (2023) A macrophage-related gene signature for identifying COPD based on bioinformatics and ex vivo experiments. J Inflamm Res 16:5647–5665. https://doi.org/10.2147/JIR.S438308
Acknowledgements
We are grateful to Professors Pixin Ran, Yumin Zhou, and Bing Li for their guidance and support throughout this study.
Funding
The National Natural Science Foundation of China (82200045), China Postdoctoral Science Foundation (2022M710900), Youth Foundation of the National Key Laboratory of Respiratory Diseases (SKLRD-Z-202326), National Key R&D Program of China (2021YFC2301101), Postdoctoral Startup Foundation of Guangzhou City (No. E.K.Y., No. Q.Y.L.) supported this study.
Author information
Authors and Affiliations
Contributions
E.Y., and Q.L. conceived and designed the experiments. E.Y., Q.L., X.W., Y.L., W.C., X.M., J.D., X.L., and X.X. performed the experiments. E.Y., C.X., and Q.L. performed bioinformatic analyses. E.Y. and Q.L. wrote the manuscript and analyzed the data. E.Y., Q.L., and C.Z. supervised the research and acquired the funding.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethical approval
Ethical approval for the animal experiment was obtained from The First Affiliated Hospital of Guangzhou Medical University (Reference number: SYXK 2020–0227).
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Q., Liu, Y., Wang, X. et al. The influence of CLEC5A on early macrophage-mediated inflammation in COPD progression. Cell. Mol. Life Sci. 81, 330 (2024). https://doi.org/10.1007/s00018-024-05375-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05375-0