
Abstract
Drug modulation of the α7 acetylcholine receptor has emerged as a therapeutic strategy for neurological, neurodegenerative, and inflammatory disorders. α7 is a homo-pentamer containing topographically distinct sites for agonists, calcium, and drug modulators with each type of site present in five copies. However, functional relationships between agonist, calcium, and drug modulator sites remain poorly understood. To investigate these relationships, we manipulated the number of agonist binding sites, and monitored potentiation of ACh-elicited single-channel currents through α7 receptors by PNU-120596 (PNU) both in the presence and absence of calcium. When ACh is present alone, it elicits brief, sub-millisecond channel openings, however when ACh is present with PNU it elicits long clusters of potentiated openings. In receptors harboring five agonist binding sites, PNU potentiates regardless of the presence or absence of calcium, whereas in receptors harboring one agonist binding site, PNU potentiates in the presence but not the absence of calcium. By varying the numbers of agonist and calcium binding sites we show that PNU potentiation of α7 depends on a balance between agonist occupancy of the orthosteric sites and calcium occupancy of the allosteric sites. The findings suggest that in the local cellular environment, fluctuations in the concentrations of neurotransmitter and calcium may alter this balance and modulate the ability of PNU to potentiate α7.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are found throughout the central and peripheral nervous systems where they elicit post-synaptic excitation or enhance pre-synaptic release of neurotransmitters [1, 2]. They contribute to diverse physiological processes, including voluntary muscle contraction, autonomic control of blood pressure and heart rate, cortical excitability, cognition, attention, and reward [3]. Dysfunctions of nAChRs underpin neurological diseases such as congenital myasthenia and nocturnal epilepsy, and they have also been implicated in neurodegenerative, neuroinflammatory, and neuropsychiatric disorders [4,5,6]. Consequently, the development of nAChR-targeted drugs holds significant therapeutic potential [7, 8].
The α7 nAChR is among the most abundant nAChR subtypes within the central nervous system, and is widely expressed in non-neuronal cells where it has anti-inflammatory and neuroprotective roles [9,10,11]. Modulation of α7 has thus emerged as a therapeutic strategy for neurological, neurodegenerative and neuroinflammatory disorders [5, 12].
As a member of the Cys-loop receptor superfamily, α7 shares the pentameric architecture found in other family members in which five homologous subunits form a central ion-conducting channel [13,14,15,16]. Each subunit comprises extracellular (ECD), transmembrane (TMD), and intracellular domains (ICD), which, across different family members, harbor sequence differences conferring receptor-specific agonist binding, ion permeability, ion selectivity, and drug sensitivity. Toward development of therapeutics, structural investigations have revealed sites to which agonists, allosteric modulators, and calcium bind [14, 16,17,18]. These sites are topographically distinct from each other and occur in multiple copies depending on the types of subunits within the receptor’s pentameric structure; in the case of a homo-pentameric receptor like α7, there are five binding sites for each type of ligand. However, relationships between occupancy of each type of binding site and the receptor’s ionotropic response remain poorly understood.
The homo-pentameric architecture of α7 makes it an appealing model receptor to explore relationships between ligand occupancy and the resulting ionotropic response. While α7 is weakly activated by agonist alone, its activity is augmented by the divalent cations calcium or barium [19], and strongly potentiated by the positive allosteric modulator PNU-120596 (PNU) [20,21,22,23]. Here, we manipulate the number of agonist and intact calcium binding sites to probe relationships between ligand occupancy and PNU potentiation. By monitoring PNU potentiation in the presence or absence of calcium, our findings at the level of single receptor channels reveal a robust interplay between the number of agonist binding sites, the presence or absence of calcium, and PNU potentiation. Leveraging recent structural data, we exploit this interplay to dissect the contributions to PNU potentiation by residues that form a calcium binding site.
Materials and methods
Chemicals
Acetylcholine chloride (ACh) was purchased from Sigma-Aldrich (St. Louis, MO, USA), and PNU-120596 (N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea) from Tocris Biosciences (Bristol, UK).
Expression of human wild-type and mutant α7 nAChRs
Mutations were introduced using either the QuikChange™ Site-Directed Mutagenesis Kit (Agilent Technologies) or an oligo-cloning method. In the later method, unique restriction sites flanking the targeted mutation were identified, and a synthetic double stranded oligonucleotide harboring the mutation was ligated between those sites using standard molecular biological methods [24]. The low-conductance form of α7 (LC-α7) was constructed by substituting three residues in the intracellular domain with arginine residues (Q428R, E432R, S436R) [25]. Concatemeric (α7)5 composed of five monomers with each flanked by unique restriction sites was constructed as previously described [26]. Mutations in a chosen monomer within (α7)5 were introduced using the QuikChange method, and the cDNA monomer carrying the mutant was cloned between the corresponding unique restriction sites using standard molecular biological methods. Validation of all constructs was achieved by sequencing the entire coding region.
Bosc-23 cells, a variant of the HEK 293 fibroblast cell line [27], were maintained at 37 °C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum until they reached ∼30% confluence. Thereafter, cDNAs encoding wild-type or mutant α7, the chaperone NACHO, and GFP were co-transfected by calcium phosphate precipitation; the ratio of α7 to NACHO cDNAs was 4:1. GFP was included to allow fluorescent identification of transfected cells for patch-clamp recording [28]. For co-expression of the high-conductance (HC) and low-conductance (LC) forms of α7 a 1:1 ratio of the cDNAs was used. For the combinations HC-Y188V and either LC, LC-E45V, LC-D42N or LC-D44N the ratio was 2:1, and for the combination HC-Y188V and LC-E173Q the ratio was 1:4. Recordings were obtained 2 to 3 days after transfection, except for the combination HC-Y188V and LC-E173Q the recordings were obtained 4 to 5 days after transfection.
Single-channel recordings
Recordings were obtained in the cell-attached patch configuration at room temperature (21–23 °C) as described [19, 22, 25]. A membrane potential of −100 mV was applied to enable detection of the lowest amplitude classes of channel openings. The bath and pipette solution contained (in mM) 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, 10 HEPES, while the calcium-free bath and pipette solutions contained 80 KF, 20 KCl, 40 K-aspartate, 2 MgCl2, 1 EGTA, and 10 HEPES [29]; in this solution Mg2+ forms a complex with F− resulting in a low ‘free Mg2+ concentration of approximately 2.4 µM. Solutions were adjusted to pH 7.4 with KOH. For recordings with CaCl2, MgCl2 or SrCl2 the bath and pipette solutions contained 142 KCl, 5.4 NaCl, 10 HEPES and either 1.8 CaCl2 and 1 EDTA, 1.7 MgCl2 and 1 EGTA or 1.8 SrCl2 and 1 EDTA (pH 7.4). Pipette solutions also contained 100 μM ACh and 10 μM PNU-120596.
Single-channel currents were digitized at 20 μs intervals, low-pass filtered at a bandwidth of 10 kHz using an Axopatch 200B patch-clamp amplifier (Molecular Devices Corp., CA), and channel openings were detected using the program TAC (Bruxton Corporation, Seattle, WA, USA) with the Gaussian digital filter set at 5 kHz. A key step in our analysis of potentiation was identification of clusters, which were recognized as a series of closely spaced openings flanked by closures lasting longer than a critical duration, denoted tcrit. This duration was determined from the point of intersection between successive exponential components in the closed duration histogram. The optimal tcrit was determined by comparing histograms of cluster durations generated following application of successively greater intersection points, adopting the tcrit beyond which no further change in the cluster duration histogram occurred (Fig. S1). In recordings without distinct clusters, tcrit typically ranged from 1 to 4 ms, whereas in recordings with clusters tcrit ranged from 20 to 80 ms. To quantify cluster duration histograms, they were fitted by the sum of exponential functions by maximum likelihood using the program TACFit (Bruxton Corporation, Seattle, WA, USA).
Electrical fingerprinting
To determine subunit stoichiometry we used a method based on single channel amplitude, known as electrical fingerprinting, as detailed previously [25, 30, 31]. Briefly, channel openings recorded from cells co-expressing both HC and LC forms of α7, or mutations thereof, were detected using a combination of the fixed amplitude and track-events options within TAC. First, the mean amplitude of each amplitude class was determined by fitting multiple Gaussian functions to all points histograms generated from segments of the recording in which all amplitudes were represented. Then, using the mean amplitude of each Gaussian, the fixed-amplitude option was used to determine the duration of each opening in a given amplitude class; only openings long enough to form flat tops were accepted for analysis. Nevertheless, some fully resolved openings had amplitudes slightly higher or lower than the mean amplitude, but still within one SD of the mean of that amplitude class. For these openings we used the track-events option to reset the amplitude followed by detection using the fixed-amplitude option. For each amplitude class, which indicates the stoichiometry of HC and LC subunits, cluster duration histograms were constructed.
Data and statistical analysis
Data from 3 to 4 independent transfections and 4 to 7 separate recordings from different cells were subjected to analysis. To capture the full range of PNU potentiation, including both potentiated and un-potentiated clusters, the mean duration of all clusters was determined for each amplitude class and experimental condition. The data presented represent the mean and SD of the cluster durations obtained from different patches for each subunit composition and amplitude class. Each plot of the fraction of clusters with greater than N re-openings per cluster against N was constructed with data from different patches under the same conditions and fitted by single or double exponential decays using GraphPad Prism software. The time constants and 95% confidential intervals are reported in the Tables. Group sizes were determined based on prior experiments and literature within the field. Furthermore, no data were excluded from the analysis, no randomization or blinding procedures were performed during the experiments, and group sizes were not predetermined by statistical methods.
Results
We begin with the premise of topographically distinct binding sites for agonist, calcium and PNU, each present in five copies per α7 pentamer (Fig. 1). Our goal is to investigate the functional interplay between the agonist and calcium binding sites and PNU potentiation. The investigations toward this goal are divided into three sections, each addressing a different facet of PNU potentiation using single channel patch clamp recordings in the presence or absence of calcium.

Structure of α7 with bound agonist, calcium and PNU. Cryo-EM structure of α7 (PDB: 8V8A) showing the protein backbone (blue lines), the agonist epibatidine (green sticks), PNU (magenta sticks) and calcium (red spheres). Views from the top and side are shown. Horizontal lines indicate the approximate location of the cell membrane. The intracellular domain is omitted for clarity
The first section employs either concatemeric receptors or an electrical fingerprinting approach to manipulate the number of agonist binding sites while monitoring PNU potentiation. In the second section, we generate mutants of key residues at an inter-domain calcium binding site, manipulate the number of copies of each mutant per receptor, and monitor PNU potentiation. The third section manipulates both the number of agonist and intact calcium binding sites and monitors PNU potentiation.
PNU potentiation depends on the number of agonist binding sites and divalent cations
Previous studies demonstrated that in the presence of ACh alone, α7 activates as brief submillisecond channel openings flanked by long closings, or occasionally, several openings in quick succession [19, 25]. However, in the presence of ACh and PNU channel openings become prolonged and form clusters of many openings lasting from hundreds of milliseconds to several seconds [22, 30].
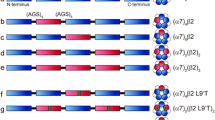
To investigate the interplay between agonist, PNU, and calcium binding sites, we employed a concatemeric α7 receptor composed of five α7 subunits connected by short polypeptide linkers. This construct, designated (α7)5, mimics the wild-type α7 receptor in its response to ACh and potentiation by PNU [26]. Using (α7)5 as a template, we introduced the mutation Y188V into one or more subunits to manipulate the number of agonist binding sites, with precise control over both the number of mutant subunits and their positions within the pentamer. Previous studies showed that substituting threonine for Y188, a residue essential for agonist binding, abolished agonist-elicited macroscopic or single channel currents and binding of α-bungarotoxin [31]. These observations showed that substituting threonine for Y188 disabled the agonist binding site. Here we substituted valine for Y188 to mirror the geometry of threonine and eliminate potential interactions with polar groups within ACh. Hereafter, a subunit that contains the native Y188 is assumed to form a functional agonist binding site, whereas a subunit that contains the Y188V mutation is assumed to form a disabled binding site.
We expressed (α7)5 in Bosc 23 cells, a clonal cell line derived from HEK cells, and recorded single channel currents elicited by ACh in the presence of PNU either without calcium or with physiological concentrations of calcium and magnesium; hereafter these two solutions are designated either without or with calcium, respectively. To maximize PNU occupancy a concentration of 10 µM PNU was used throughout. The recordings reveal long clusters of potentiated channel openings either without or with calcium (Fig. 2a). Additionally, a modest decrease in the unitary current amplitude is observed with calcium, as described previously [19].

PNU-potentiation of concatemeric (α7)5 depends on the number of agonist binding sites and divalent cations. Single-channel currents were recorded from Bosc-23 cells expressing concatemeric receptors with (a) five agonist binding sites, (α7)5, or (b) one binding site, (α7)1(α7Y188V)4, without or with Ca2+ and Mg2+ or with only Ca2+, Mg2+ or Sr2+ as the divalent cation. Schematic diagrams depict wild type α7 subunits in white and subunits with the mutation Y188V in red. For each receptor and condition, representative traces of single channel currents and the corresponding cluster duration histograms fitted by multiple exponentials are shown. Single channel currents were elicited by 100 µM ACh in the presence of 10 µM PNU at a membrane potential of − 100 mV, displayed with a Gaussian filter of 5 kHz. Channel openings are upward deflections from baseline
To quantify the mean durations of the clusters, we define a cluster as a series of channel openings separated by brief closings shorter than a defined critical time, tcrit, as outlined in Materials and Methods. We then constructed histograms of cluster durations and fitted multiple exponential components to the histograms; the exponential component with the longest mean duration corresponds to clusters of fully potentiatiated channel openings. The results show that the cluster duration histograms are similar without or with calcium (Fig. 2a), indicating that in receptors with five agonist binding sites, PNU potentiation does not depend on calcium.
However, in concatemeric receptors with four of the five agonist binding sites inactivated by the mutation Y188V, designated (α7)1(α7Y188V)4, potentiated channel openings are observed with but not without calcium (Fig. 2b). Without calcium, channel openings appear as single brief spikes flanked by long closings, leading to a marked shift of the cluster duration histogram by several orders of magnitude toward shorter durations (Fig. 2b). Thus, in α7 receptors with only one agonist binding site calcium is pivotal in conferring potentiation by PNU.
To assess divalent cation specificity for PNU potentiation, we individually tested calcium, magnesium, and strontium on the (α7)1(α7Y188V)4 concatemer. The recordings reveal long clusters of channel openings in the presence of each divalent cation, with each cluster duration histogram showing a major exponential component with prolonged mean duration corresponding to potentiated channel openings (Fig. 2b). Thus, for α7 receptors with a single agonist binding site divalent cations of differing sizes enable PNU potentiation.
Next we increased the number of agonist binding sites within the the concatemeric receptor and monitored PNU potentiation. In contrast to receptors with a single agonist binding site, receptors with two or three sites, designated (α7)2(α7Y188V)3 and (α7)3(α7Y188V)2, respectively, exhibit potentiated channel openings either without or with calcium, as shown by long clusters of channel openings and a major exponential component of clusters with prolonged mean duration (Fig. S2). Thus increasing the number of agonist binding sites beyond one overcomes the requirement of calcium for PNU potentiation. Given the novel interdependence between the number of agonist binding sites, PNU potentiation and calcium, we sought to determine the molecular and mechanistic underpinnings of this phenomenon.
Studies of receptors with unlinked subunits-
The preceding studies manipulated the number of agonist binding sites using concatemeric receptors in which the subunits were connected via short peptide linkers. To exclude possible influences from the peptide linkers, we employ an electrical fingerprinting approach. This approach involves co-expression of two different unlinked subunit types (Fig. 3a): one with arginine substitutions for anionic or polar residues flanking intracellular portals, designated LC for low conductance, and another retaining the original anionic or polar residues, designated HC for high conductance [25, 26, 30,31,32]. Located within the intracellular domain, these portals are far from the calcium binding sites near the extracellular membrane leaflet, and thus are not expected to impact binding of calcium.

Electrical fingerprinting strategy to distinguish single channel currents from receptors with different subunit stoichiometries. (a) Close up view of a region of α7 encompassing the intracellular domain, with the protein backbone in blue lines and residues mutated to generate the LC form of α7 in red sticks. For clarity only three subunits that bracket two intracellular portals are shown; one portal is visible as the open space flanked by red sticks to the left. For reference the location of the intracellular membrane leaflet is indicated by horizontal lines. (b) Segments from a representative recording from a cell co-expressing HC and LC subunits with Ca2+. ACh, 100 µM; PNU, 10 µM; membrane potential, − 100 mV; Gaussian filter, 5 kHz. (c) Plot of the mean current amplitude of each amplitude class against the inferred number of HC subunits per pentamer; HC subunits are depicted in grey with black outline and LC subunits in white with red outline. The data correspond to the mean ± SD of 5 independent recordings. (d) Plot of current amplitude of each cluster against its duration from a representative recording. The plot shows six stripes of points, each corresponding to a different amplitude class and stoichiometry of HC and LC subunits
Recordings from control receptors, generated from HC and LC subunit mixtures, reveal long clusters of potentiated channel openings. However, the current amplitudes of the openings exhibit discrete, evenly spaced values owing to variations in the stoichiometric ratio of HC to LC subunits within individual pentameric receptors (Fig. 3b, c). Accordingly, a plot of current amplitude of each cluster against its duration reveals six horizontal stripes of points, corresponding to receptors with zero to five HC subunits (Fig. 3d). Notably, each horizontal stripe spans a similar brief to long time range, indicating cluster duration is independent of the stoichiometric ratio of HC to LC subunits.
To manipulate the number of agonist binding sites per receptor, we co-expressed HC-Y188V and LC subunits and recorded single channel currents elicited by ACh in the presence of PNU, either without or with calcium. In this experiment, receptors with five HC-Y188V subunits remain electrically silent because all five ACh binding sites are disabled, while receptors with four HC-Y188V subunits and one LC subunit exhibit the maximum observed current amplitude, indicating these receptors have one ACh binding site. Exemplar traces of channel openings from receptors with four HC-Y188V and one LC subunit, distinguished by current amplitude, show that in the absence of calcium channel openings are brief and occur in isolation, whereas in their presence channel openings are prolonged and occur in clusters (Fig. 4a, b), mirroring the observations from concatemeric receptors. A plot of current amplitude of each cluster against its duration exhibits five distinct horizontal stripes of points corresponding to receptors with zero to four HC-Y188V subunits. Notably, a sixth stripe of points corresponding to receptors with five HC-Y188V subunits is absent, confirming this stoichiometry, if present, is electrically silent. Moreover, without calcium, receptors with four HC-Y188V and one LC subunit show a compressed stripe of points spanning only brief durations (Fig. 4a), whereas with calcium, these receptors show an extended stripe spanning durations from brief to long (Fig. 4b). Receptors with three or fewer HC-Y188V subunits show stripes spanning durations from brief to long regardless of the presence of calcium (Fig. 4a, b). Bar graphs of the mean duration of all clusters affirm that receptors with one agonist binding site show brief unpotentiated channel openings without calcium, while all other combinations of binding site number and the presence or absence of calcium show long clusters of potentiated channel openings.

PNU-potentiation of α7 nAChRs composed of unlinked subunits depends on the number of agonist binding sites and calcium. Single channel currents were recorded from cells co-expressing HC-Y188V and LC subunits without (a) or with (b) Ca2+. ACh: 100 µM, PNU: 10 µM, membrane potential: −100 mV, Gaussian filter, 5 kHz. For each condition, the panels from left to right show: Representative traces of single channel currents of the highest amplitude class corresponding to receptors with one agonist binding site without (a) and with (b) Ca2+. In the traces in panel (a), brief openings with reduced amplitude are not fully resolved and thus are not included in any amplitude class (see Materials and Methods). Plots of current amplitude of each cluster against its duration from a representative recording without (a) or with (b) Ca2+. Each plot shows five discrete amplitude classes of clusters corresponding to receptors with different subunit stoichiometries. The highest amplitude class corresponds to receptors with one agonist binding site comprised of four HC-Y188V subunits and one LC subunit; Mean cluster durations for receptors with the indicated numbers of LC subunits in the absence (white bars, a) or presence (grey bars, b) of Ca2+. Each symbol corresponds to a recording from a different membrane patch. The data correspond to the mean ± SD of 5–6 independent experiments recorded from different cells. -Plots of the fraction of clusters with greater than N re-openings against the number of re-openings per cluster in the absence (a) or presence (b) of Ca2+. Data correspond to receptors containing either one (black) or two (red) LC subunits, each forming an ACh binding site, for each experimental condition. Data are fitted by a single or double exponential decay, with the fitted parameters in Table 1. Each plot includes data from 5 recordings for each condition
To further quantify potentiation, we plot the fraction of clusters with more than N channel re-openings, where N is an integer, against the number of times the channel re-opened per cluster. Here, a cluster with zero re-openings corresponds to a single channel opening flanked by long closings, while a cluster with one re-opening corresponds to two channel openings separated by a brief closure. A plot of the fraction of clusters with greater than N re-openings against N is expected to decay from one to zero, with a rapid decay indicating few re-openings per cluster and a slow decay indicating numerous re-openings per cluster. The number of exponential components in the decay indicates the minimum number of kinetically distinct closed states to which a cluster may transition, while the relative weight of each component indicates the probability of transition to a specific closed state. For receptors with one agonist binding site, without calcium, we observe a rapid decay in the number of re-openings per cluster, well fitted by a single brief exponential (Fig. 4a). Conversely, with calcium, the decay markedly slows, requiring a fit by the sum of two prolonged exponentials (Fig. 4b). By contrast, receptors with two binding sites exhibit a slow biexponential decay both without and with calcium (Fig. 4a, b). The mean number of re-openings per cluster for each exponential component, subunit stoichiometry and experimental condition is presented in Table 1. To summarize, our studies using both concatemeric receptors and electrical fingerprinting reveal that unlike receptors with multiple agonist binding sites, receptors with just one agonist binding site require calcium for PNU potentiation.
Structural basis of the effect of calcium on occupancy dependent PNU potentiation
To evaluate calcium binding sites linked to PNU potentiation, we took advantage of recent cryo-electron microscopic structures of the α7 nAChR. These structures revealed a ball-shaped density consistent with a calcium ion positioned between the extracellular and transmembrane domains of each subunit [14] (Fig. 1). Notably, an analogous density was absent in a subsequent α7 structure obtained without added calcium [16]. Surrounding this calcium ion density are four anionic residues, E45, D44, D42, and E173 originating from the extracellular domain (Fig. 5a). To determine the impact of these residues on PNU potentiation, we engineered mutations of each residue individually within the HC subunit. We then co-expressed each mutant subunit with the LC subunit and recorded single channel currents elicited by ACh together with PNU, either without or with calcium. We then determined the stoichiometric ratio of subunits based on current amplitude and analyzed mean cluster duration and channel re-opening for each amplitude class.

Contributions of key anionic residues to PNU potentiation. (a) Close up view of the structural region of α7 that forms a calcium binding site, with the protein backbone shown as blue lines, calcium as a red ball, and flanking electron-rich residues as sticks (PDB: 7KOQ). Single channel currents were recorded either without or with Ca2+ from cells expressing a HC-E45V (b) or HC-D44N (c) subunit together with the LC subunit. ACh: 100 µM, PNU: 10 µM, membrane potential: 100 mV, Gaussian filter, 5 kHz. For both mutant receptors, the panels from left to right show: representative traces of currents of the highest amplitude class corresponding to receptors with five mutant HC subunits (black circle, zero intact calcium sites), currents from receptors with four mutant HC subunits and one LC subunit (red circle, one intact calcium site), in the absence or presence of Ca2+. Bar graphs showing the mean cluster duration for each amplitude class corresponding to receptors with the indicated numbers of unaltered calcium binding sites recorded with (grey bars) or without Ca2+ (white bars). The data correspond to the mean ± SD of 5–6 independent experiments recorded from different cells. Plots of the fraction of clusters with greater than N re-openings against the number of re-openings per cluster without (top) or with (bottom) Ca2+. Data correspond to receptors containing zero (black line) or one (red line) unaltered calcium binding site. Each plot includes data from 5 recordings for each condition. Data were fitted by either a single or double exponential decay, with the fitted parameters in Table 2
Among the four mutants at the calcium site, only E45V had a discernable impact on mean cluster duration and channel re-opening. Receptors harboring five HC-E45V subunits (zero intact calcium sites) show brief channel openings flanked by long closings, regardless of the presence of calcium, indicating block of PNU potentiation (Fig. 5b). However, receptors with four HC-E45V subunits and one LC subunit show brief unpotentiated channel openings without calcium, but with calcium they show long clusters of potentiated openings, indicating the single copy of E45 within the LC subunit restores calcium-dependent PNU potentiation. In contrast, receptors with three or fewer HC-E45V subunits and two or more LC subunits show long potentiated clusters regardless of the presence of calcium, showing that two or more copies of E45 per pentamer enable calcium-independent PNU potentiation. Analyses of channel re-opening reveals changes parallel to those in mean cluster duration: a rapid, single exponential decay of channel re-opening corresponding to brief cluster duration, and a slow, biexponential decay corresponding to prolonged cluster duration (Table 2). Thus, in receptors with five agonist binding sites, E45 is pivotal in determining PNU potentiation either without or with calcium.
In contrast to receptors harboring E45V, those containing D44N, regardless of the number of mutants per receptor, show long clusters of channel openings either without or with calcium (Fig. 5c; Table 2). Similarly, PNU potentiation is largely preserved in receptors with the mutants D42N and E173Q either without or with calcium (Fig. S3; Table 2). Thus, in receptors harboring five agonist binding sites, D42N, D44N and E173Q are dispensable for PNU potentiation, regardless of the presence of calcium.
Interdependence of agonist and calcium binding sites in PNU potentiation
To investigate the dynamic interplay among agonist binding sites, calcium binding sites, and PNU potentiation, we manipulated the number of agonist and calcium binding sites and recorded single channel currents from the resultant receptors. Inherent to this experiment, mutations of the agonist and calcium sites are located in different subunits so that any changes compared to the control subunit combination would manifest via inter-subunit interactions. We first recap the results for the control subunit combination HC-Y188V plus LC, which alters the number of agonist sites while preserving all five calcium sites (Fig. 4). Receptors with one agonist site and five intact calcium sites, denoted 1/5 in Fig. 6a, show brief mean cluster durations without calcium but long mean cluster durations with calcium. Analyses of channel re-opening confirms that brief clusters remain unpotentiated, exhibiting a single rapid exponential decay, while long clusters are potentiated, exhibiting a prolonged biexponential decay (Table 1).

Interdependence between agonist and calcium binding sites in PNU potentiation. Receptors were formed by co-expressing the HC-Y188V subunit with an LC subunit (a) or the HC-Y188V subunit with an LC subunit carrying a mutation of the indicated anionic residue (b–e). Single channel currents were recorded with 100 µM ACh and 10 µM PNU without (left) or with (right) Ca2+. The bar graphs show the mean cluster durations ± SD for receptors with the indicated ratios of agonist to intact calcium binding sites without (white bars) or with (grey bars) Ca2+ (n = 5–7 independent recordings for each condition from different cells). The re-opening plots show the fraction of clusters with greater than N re-openings against the number of re-openings per cluster in either the absence or presence of Ca2+. For each curve, the color represents the stoichiometry of the receptor indicated by the black, blue or red circle in the corresponding bar graph. Each plot includes data from 5 to 7 recordings for each condition. Data were fitted by a single or double exponential decay, with the fitted parameters in Table 3
For receptors with one agonist binding site, four intact calcium sites, and one calcium site containing any of the four mutants, denoted 1/4 in Fig. 6b–e, we observe profiles similar to those of the control receptor with one agonist site and five calcium sites: brief unpotentiated channel openings without calcium and long potentiated openings with calcium. Furthermore, analyses of channel re-opening reveals a rapid decay fitted by a predomintly single exponential without calcium, but a slow decay fitted by a prolonged single or double exponential with calcium (Fig. 6b–e; Table 3). Thus, for receptors with one agonist binding site, introducing a single copy of a mutation of any of the five residues at the calcium site has little effect either without or with calcium.
For the control subunit combination HC-Y188V plus LC, receptors with two agonist sites and five intact calcium sites, denoted 2/5, long clusters of potentiated openings are observed either without or with calcium (Fig. 6a). Analyses of channel re-opening confirms that the long clusters consist of many openings in quick succession and that the decay is prolonged and biexponential (Fig. 6a, red decay curves; Table 1). In contrast, receptors with two agonist sites, three intact calcium sites, and two calcium sites with either E45V, D42N, or D44N mutations, denoted 2/3, show brief unpotentiated clusters without calcium (Fig. 6b–e; Table 3). However, these receptors show long clusters of potentiated channel openings with calcium. Notably, the analogous 2/3 receptors with the mutation E173Q differ from the others in showing long clusters of potentiated channel openings regardless of the presence of calcium. Thus, in receptors with two agonist sites, increasing the number of mutant calcium sites unmasks calcium-independent contributions of E45, D42, and D44 to PNU potentiation, and distinguishes these contributions from those by E173.
For the control subunit combination HC-Y188V plus LC, receptors with three agonist sites and five intact calcium sites, denoted 3/5, long clusters of potentiated channel openings are observed either without or with calcium (Fig. 6a). Conversely, receptors with three agonist sites, two intact calcium sites, and three calcium sites containing the E45V mutation, denoted 3/2, show brief clusters without calcium and prolonged clusters with calcium (Fig. 6b). Analyses of channel re-openings confirm that the brief clusters remain unpotentiated, primarily occurring as solitary brief openings, while the long clusters are potentiated, occurring as many openings in quick succession (Table 3). In contrast, the analogous 3/2 receptors with either D42N, D44N, or E173Q show long clusters either without or with calcium (Fig. 6c–e; Table 3). Thus, in receptors with three agonist sites, increasing the number of mutant calcium sites further differentiates among the residues at the calcium binding site: E45V abolishes calcium-independent PNU potentiation, whereas D42N, D44N, and E173Q have no discernable effect.
Lastly, for the control subunit combination HC-Y188V plus LC, receptors harboring four or five agonist sites and five intact calcium sites, denoted 4/5 or 5/5, respectively, long potentiated clusters of channel openings are observed either without or with calcium (Fig. 6a). In contrast, receptors containing four or five agonist sites, one or zero intact calcium sites, and four or five calcium sites with E45V, denoted 4/1 or 5/0, show brief unpotentiated openings either without or with calcium (Fig. 6b). Thus, increasing the number of agonist sites while increasing the number of mutant calcium sites reveals that at least two copies of E45 are required for calcium-dependent PNU potentiation. However, the analogous 4/1 and 5/0 receptors with either D42N, D44N, or E173Q mutations show long potentiated clusters of openings regardless of the presence of calcium. Furthermore, analysis of channel re-opening with calcium distinguishes among the D42N, D44N, and E173Q mutations, revealing a distinct signature of time constants and relative weights of the components of the biexponential decay for each mutant (Fig. 6c–e; Table 3). Our overall observations of the interplay between agonist and calcium binding sites suggests PNU potentiation relies on a delicate balance between agonist occupancy of the orthosteric sites and calcium occupancy of the allosteric sites.
Extracellular calcium binding sites do not impact PNU potentiation
Previously, we showed that calcium enhances α7 activity in the presence of low concentrations of ACh alone [19]. Calcium increased the frequency and mean duration of ACh elicited channel openings, effects that required a pair of anionic residues from the principal and complementary faces of each agonist binding site. Consequently, we asked whether this extracellular calcium binding site impacts the ability of calcium to modulate agonist occupancy dependent PNU potentiation. To investigate the possible impact of this extracellular calcium binding site, we co-expressed the HC subunit containing the mutation E185Q at the principal face with the wild-type LC subunit and analysed PNU potentiaton across all stoichiometric combinations of subunits. In contrast to our observations for mutations of E45, long clusters of channel openings are observed for all stoichiometric forms containing E185Q regardless of the presence of calcium (Fig. S4a). Next we co-expressed the HC-Y188V subunit with the LC-E185Q subunit and recorded single channel currents in the presence of ACh and PNU (Fig. S4b). Receptors with one agonist binding site show brief unpotentiated channel openings without calcium and long clusters of potentiated openings with calcium. In contrast, receptors with two or more agonist binding sites show long clusters of channel openings regardless of the presence of calcium. These observations with receptors comprised of the HC-Y188V plus LC-E185Q subunits mirror those for control receptors comprised of HC-Y188V plus LC subunits, indicating that although E185 is essential for potentiation of α7 activated by ACh alone, it does not impact the relationships between agonist binding sites, calcium binding sites, and PNU potentiation.
Discussion
We describe a novel interplay between agonist and calcium binding sites in modulating PNU potentiation of the α7 nAChR. We find that in receptors with all five agonist binding sites, PNU potentiates regardless of the presence of calcium. However, in receptors with only one agonist binding site, PNU potentiates in the presence but not the absence of calcium. When present individually, calcium, magnesium, or strontium are sufficient to elicit this all-or-none modulatory effect. We demonstrate this interplay in concatemeric receptors where the subunits are covalently linked, and in receptors with unlinked subunits where the subunit stoichiometry is determined via electrical fingerprinting. Furthermore, we identify several electron-rich residues, previously shown to coordinate calcium in α7, that impact the interplay between agonist occupancy, calcium, and PNU potentiation. Overall, our results suggest PNU potentiation depends on a balance between agonist occupancy of the orthosteric sites and calcium occupancy of the allosteric sites. Alterations in the occupancy of each type of site alters this balance, and consequently the ability of PNU to potentiate α7.
A fundamental relationship in pharmacology is that between drug concentration and its biological response. A physico-chemical counterpart for this relationship began with the empirical Hill equation [33], which was subsequently formalized by the Langmuir equation describing the reversible association between ligand and receptor. The Langmuir equation then gave rise to the concept of occupation theory wherein fractional ligand occupancy was proportional to the fractional biological response [34]. Since then, pharmacologists have been intrigued by the numerous experimental and theoretical deviations from occupation theory. These include phenomena such as receptor reserve [35], partial agonism [36], inverse agonism [37], desensitization [38], and positive [39] or negative [40] cooperativity. In common with all of these departures is the recognition of additional processes distinct from the initial drug receptor interaction. A more recently appreciated departure from occupation theory is that by the α7 nAChR, which harbors five orthosteric binding sites, some of which are dispensible in eliciting a maximal biological response. Here, within the context of the positive allosteric modulator PNU, we find that α7 potentiation exhibits binding site reserve that is modulated by an additional process: calcium association with a site topographically distinct from the sites to which agonist or potentiator bind.
The present work builds upon previous studies from our laboratories relating the number of agonist binding sites in α7 to its biological response monitored at the level of single receptor channels. In our initial study, we employed a chimeric α7/5HT3 receptor to relate the number and relative locations of agonist binding sites required to achieve maximal mean channel open time. For this chimeric receptor, we found that three non-consecutive agonist binding sites afforded maximal channel open time, whereas two consecutive or just one binding site afforded brief channel open time [31]. In our subsequent study, we examined the relationship between the number of agonist binding sites and channel open time in wild type α7 receptors [25]. In contrast to the α7/5HT3 chimeric receptor, wild type α7 achieved maximal mean channel open time with only one agonist binding site available for occupancy. Notably, both of these studies were conducted with calcium. Following these investigations, studies of α7 activated by low concentrations of ACh revealed brief and infrequent channel openings without calcium, which contrasted with longer and more frequent openings with calcium [19]. As a corollary to these observations, we find herein that in receptors with one agonist binding site, ACh in combination with PNU elicits brief, unpotentiated channel openings without calcium, while eliciting long clusters of potentiated channel openings with calcium. Thus, whether studied in the presence of ACh alone or together with PNU, binding site reserve in α7 depends on calcium.
Given the all-or-none modulatory effect of calcium on PNU potentiation of α7, we sought to identify the corresponding calcium binding site. Early investigations, using a chimeric α7 receptor, identified several anionic residues whose mutation reduced the ability of calcium to enhance the whole cell response to ACh alone [17]. Recent cryo-EM studies of α7 structure revealed that these and additional electron-rich residues flank a density consistent with a calcium ion situated between the ligand binding and transmembrane domains of each subunit [14]. Similarly, within the analogous region of the heteromeric α4β2 nAChR, several electron rich residues were found to coordinate a calcium ion density and were required for stoichiometry-selective potentiation of ACh-elicited channel openings [41]. This structural transition zone between the ligand binding and transmembrane domains is also pivotal in transducing conformational changes due to agonist binding to gating of the ion channel [42]. Specifically, several electron-rich residues equivalent to those surrounding the calcium binding site also interact with a conserved arginine residue in mediating the agonist-binding-transduction process in muscle nAChRs [43, 44]. We find that mutation of E45 within this functionally pivotal region affects PNU potentiation such that the impact depends jointly on the number of E45V mutations per pentamer and the presence of calcium. Receptor pentamers with four copies of E45V show no potentiation without calcium, but show full potentiation with calcium. Our observation that one wild type E45 per pentamer is sufficient to restore calcium-dependent PNU potentiation suggests that calcium does not associate with sites harboring E45V. In contrast, the mutations D42N, D44N and E173Q do not impact PNU potentiation regardless of the presence of calcium. Thus, the four residues that form the calcium binding site in α7 differ in their contributions to both the binding-transduction process and calcium-dependent PNU potentiation.
We find that reducing the number of agonist binding sites unmasks contributions of the four residues flanking the calcium binding site to PNU potentiation. Among these contributions, the impact of mutation of E45 is the greatest. Without calcium, PNU either fails to potentiate or exhibits markedly reduced potentiation for receptors with agonist to calcium site ratios from 1:4 to 5:0 (Fig. 6b), in contrast to receptors with ratios from 2:5 to 5:5 (Fig. 6a). These observations highlight the contributions of both the agonist and calcium sites to the binding-transduction process underpinning PNU potentiation, even without calcium. On the other hand, with calcium, PNU potentiates receptors with ratios from 1:4 to 3:2. However, an abrupt transition occurs when the ratio of agonist to calcium sites is shifted from 3:2 to either 4:1 or 5:0, resulting in brief unpotentiated channel openings despite the presence of calcium (Fig. 6b). These observations underscore the interdependence between agonist and calcium binding sites in mediating both the binding-transduction process and the modulatory effect of calcium on PNU potentiation.
Reducing the number of agonist binding sites also unmasks contributions of D42 and D44 to calcium-independent PNU potentiation. In receptors with five agonist binding sites, without calcium, mutations of either D42 or D44 enable complete PNU potentiation (Figs. 5, S3). In contrast, in receptors with only two agonist binding sites, without calcium, mutations of either D42 or D44 prevent PNU potentiation (Fig. 6c, d). This suppression of calcium-independent potentiation further underscores the interdependence between agonist and calcium binding sites in contributing to the binding-transduction process underpinning PNU potentiation. However, when these receptors are tested with calcium, full PNU potentiation is restored, indicating that these mutations do not interfere with the permissive effect of calcium on PNU potentiation. Thus, altering the ratio of agonist to calcium sites reveals differences among residues flanking the calcium binding site in contributing to calcium-independent PNU potentiation.
Our findings provide physiological and therapeutic insights into α7 signaling potentially applicable to a spectrum of conditions including schizophrenia, Parkinson’s and Alzheimer’s disease, neuropathic pain, inflammation, sepsis, and neuroprotection [45,46,47,48,49,50]. Allosteric potentiators have emerged as promising therapeutic avenues owing to their improved selectivity compared to agonists along with their ability to preserve the temporal and spatial dynamics of endogenous neurotransmitter activation [12, 51]. Despite the considerable number of α7 ligands and potentiators developed, some of which have advanced to clinical trials, mechanisms behind potentiation have remained obscure. Our mechanistic investigations show that under conditions of low agonist occupancy, mimicked by inactivation of agonist binding sites, PNU potentiation depends on calcium. This becomes physiologically relevant as α7, in addition to being present at pre- and post-synaptic sites, is found extra-synaptically and in non-neuronal cells [11, 52, 53]. At these non-synaptic sites, α7 is thought to activate via volume transmission, where the concentration of ACh reaching the receptor is low [54], thus achieving only partial occupancy of its five identical agonist binding sites. In extracellular spaces with limited volumes, calcium concentration could transiently fluctuate over a physiological range [55] in response to changes in calcium channel or transporter activity, thereby providing an additional dimension for modulation. Thus, our findings suggest that under physiological conditions, drug potentiation of α7 depends not only on the neurotransmitter concentration but also on the concentration of calcium.
Availability of data and material
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
Abbreviations
- ACh:
-
Acetylcholine
- nAChR:
-
Nicotinic acetylcholine receptor
- PNU:
-
PNU-120596
- HC:
-
High conductance
- LC:
-
Low conductance
References
Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP (2009) Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 8:733–750. https://doi.org/10.1038/nrd2927
Wonnacott Nicotinic S (2014) ACh Receptors. Tocris bioscience scientific reviews series. 1–31.33.
Zoli M, Pistillo F, Gotti C (2015) Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96:302–311. https://doi.org/10.1016/j.neuropharm.2014.11.003
Becchetti A, Aracri P, Meneghini S, Brusco S, Amadeo A (2015) The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front Physiol 6:22. https://doi.org/10.3389/fphys.2015.00022
Bye LJ, Finol-Urdaneta RK, Tae HS, Adams DJ (2023) Nicotinic acetylcholine receptors: key targets for attenuating neurodegenerative diseases. Int J Biochem Cell Biol. 157:106387. https://doi.org/10.1016/j.biocel.2023.106387
Engel AG, Shen XM, Selcen D, Sine SM (2015) Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 14:420–434. https://doi.org/10.1016/S1474-4422(14)70201-7
Dineley KT, Pandya AA, Yakel JL (2015) Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci. 36:96–108. https://doi.org/10.1016/j.tips.2014.12.002
Ito T, Inden M, Ueda T, Asaka Y, Kurita H, Hozumi I (2020) The neuroprotective effects of activated alpha7 nicotinic acetylcholine receptor against mutant copper-zinc superoxide dismutase 1-mediated toxicity. Sci Rep 10:22157. https://doi.org/10.1038/s41598-020-79189-y
Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97:463–472. https://doi.org/10.1016/j.bcp.2015.07.032
Yang X, Zhao C, Chen X, Jiang L, Su X (2017) Monocytes primed with GTS-21/alpha7 nAChR (nicotinic acetylcholine receptor) agonist develop anti-inflammatory memory. QJM. 110:437–445. https://doi.org/10.1093/qjmed/hcx014
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 421:384–388. https://doi.org/10.1038/nature01339
Bouzat C, Lasala M, Nielsen BE, Corradi J, Esandi MDC (2018) Molecular function of alpha7 nicotinic receptors as drug targets. J Physiol. 596:1847–1861. https://doi.org/10.1113/JP275101
Morales-Perez CL, Noviello CM, Hibbs RE (2016) X-ray structure of the human alpha4beta2 nicotinic receptor. Nature 538:411–415. https://doi.org/10.1038/nature19785
Noviello CM, Gharpure A, Mukhtasimova N, Cabuco R, Baxter L, Borek D, Sine SM, Hibbs RE (2021) Structure and gating mechanism of the alpha7 nicotinic acetylcholine receptor. Cell. 184:2121-2134e2113. https://doi.org/10.1016/j.cell.2021.02.049
Rahman MM, Teng J, Worrell BT, Noviello CM, Lee M, Karlin A, Stowell MHB, Hibbs RE (2020) Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins. Neuron 106:952-962 e955. https://doi.org/10.1016/j.neuron.2020.03.012
Zhao Y, Liu S, Zhou Y, Zhang M, Chen H, Eric Xu H, Sun D, Liu L, Tian C (2021) Structural basis of human alpha7 nicotinic acetylcholine receptor activation. Cell Res. 31:713–716. https://doi.org/10.1038/s41422-021-00509-6
Galzi JL, Bertrand S, Corringer PJ, Changeux JP, Bertrand D (1996) Identification of calcium binding sites that regulate potentiation of a neuronal nicotinic acetylcholine receptor. Embo J. 15:5824–5832. https://doi.org/10.1002/j.1460-2075.1996.tb00969.x
Burke SM, Avstrikova M, Noviello CM, Mukhtasimova N, Changeux JP, Thakur GA, Sine SM, Cecchini M, Hibbs RE (2024) Structural mechanisms of alpha7 nicotinic receptor allosteric modulation and activation. Cell. https://doi.org/10.1016/j.cell.2024.01.032
Natarajan K, Mukhtasimova N, Corradi J, Lasala M, Bouzat C, Sine SM (2020) Mechanism of calcium potentiation of the alpha7 nicotinic acetylcholine receptor. J Gen Physiol. https://doi.org/10.1085/jgp.202012606
Andersen ND, Nielsen BE, Corradi J, Tolosa MF, Feuerbach D, Arias HR, Bouzat C (2016) Exploring the positive allosteric modulation of human alpha7 nicotinic receptors from a single-channel perspective. Neuropharmacology. 107:189–200. https://doi.org/10.1016/j.neuropharm.2016.02.032
Bouzat C, Sine SM (2018) Nicotinic acetylcholine receptors at the single-channel level. Br J Pharmacol. 175:1789–1804. https://doi.org/10.1111/bph.13770
daCosta CJ, Free CR, Corradi J, Bouzat C, Sine SM (2011) Single-channel and structural foundations of neuronal alpha7 acetylcholine receptor potentiation. J Neurosci. 31:13870–13879. https://doi.org/10.1523/JNEUROSCI.2652-11.2011
Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffmann WE, Piotrowski DW, Groppi VE, Allaman G, Ogier R, Bertrand S, Bertrand D, Arneric SP (2005) A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 25:4396–4405. https://doi.org/10.1523/JNEUROSCI.5269-04.2005
Sine SM (1993) Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci U S A. 90:9436–9440. https://doi.org/10.1073/pnas.90.20.9436
Andersen N, Corradi J, Sine SM, Bouzat C (2013) Stoichiometry for activation of neuronal alpha7 nicotinic receptors. Proc Natl Acad Sci U S A. 110:20819–20824. https://doi.org/10.1073/pnas.1315775110
Nielsen BE, Minguez T, Bermudez I, Bouzat C (2018) Molecular function of the novel alpha7beta2 nicotinic receptor. Cell Mol Life Sci. 75:2457–2471. https://doi.org/10.1007/s00018-017-2741-4
Pear WS, Nolan GP, Scott ML, Baltimore D (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 90:8392–8396. https://doi.org/10.1073/pnas.90.18.8392
Zhang G, Gurtu V, Kain SR (1996) An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun. 227:707–711. https://doi.org/10.1006/bbrc.1996.1573
Sine SM, Claudio T, Sigworth FJ (1990) Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 96:395–437. https://doi.org/10.1085/jgp.96.2.395
daCosta CJ, Sine SM (2013) Stoichiometry for drug potentiation of a pentameric ion channel. Proc Natl Acad Sci U S A. 110:6595–6600. https://doi.org/10.1073/pnas.1301909110
Rayes D, De Rosa MJ, Sine SM, Bouzat C (2009) Number and locations of agonist binding sites required to activate homomeric Cys-loop receptors. J Neurosci. 29:6022–6032. https://doi.org/10.1523/JNEUROSCI.0627-09.2009
Lasala M, Corradi J, Bruzzone A, Esandi MDC, Bouzat C (2018) A human-specific, truncated alpha7 nicotinic receptor subunit assembles with full-length alpha7 and forms functional receptors with different stoichiometries. J Biol Chem. 293:10707–10717. https://doi.org/10.1074/jbc.RA117.001698
Hill AV (1913) The combinations of haemoglobin with oxygen and with carbon monoxide. I. Biochem J. 7:471–480. https://doi.org/10.1042/bj0070471
Clark AJ (1926) The reaction between acetyl choline and muscle cells. J Physiol. 61:530–546. https://doi.org/10.1113/jphysiol.1926.sp002314
Stephenson RP (1956) A modification of receptor theory. Br J Pharmacol Chemother. 11:379–393. https://doi.org/10.1111/j.1476-5381.1956.tb00006.x
Del Castillo J, Katz B (1957) Interaction at end-plate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci. 146:369–381. https://doi.org/10.1098/rspb.1957.0018
Costa T, Herz A (1989) Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci U S A. 86:7321–7325. https://doi.org/10.1073/pnas.86.19.7321
Katz B, Thesleff S (1957) A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol 138:63–80. https://doi.org/10.1113/jphysiol.1957.sp005838
Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol. 12:88–118. https://doi.org/10.1016/s0022-2836(65)80285-6
Koshland DE Jr, Nemethy G, Filmer D (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 5:365–385. https://doi.org/10.1021/bi00865a047
Mazzaferro S, Kang G, Natarajan K, Hibbs RE, Sine SM (2024) Structural bases for stoichiometry-selective calcium potentiation of a neuronal nicotinic receptor. Br J Pharmacol. https://doi.org/10.1111/bph.16321
Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM (2004) Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 430:896–900. https://doi.org/10.1038/nature02753
Lee WY, Sine SM (2005) Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 438:243–247. https://doi.org/10.1038/nature04156
Mukhtasimova N, Sine SM (2013) Nicotinic receptor transduction zone: invariant arginine couples to multiple electron-rich residues. Biophys J. 104:355–367. https://doi.org/10.1016/j.bpj.2012.12.013
Bitner R, Anderson D, Drescher K, Kohlhaas K, Gronlien H, Hu M, Li J, Markosyan S, Marsh K, Mohler E, Nikkel A, Radek R, Robb H, Schrimpf M, Waring J, Lee CH, Gopalakrishnan M (2013) P4–310: Preclinical characterization of a selective alpha-7 neuronal nicotinic acetylcholine receptor agonist ABT-126: a novel therapeutic agent for the treatment of cognitive impairment in Alzheimer’s disease and schizophrenia. Alzheimer’s Dementia. 9:817–818. https://doi.org/10.1016/j.jalz.2013.05.1755
Hone AJ, McIntosh JM (2018) Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. Febs Lett. 592:1045–1062. https://doi.org/10.1002/1873-3468.12884
Liu Q, Liu C, Jiang L, Li M, Long T, He W, Qin G, Chen L, Zhou J (2018) alpha7 Nicotinic acetylcholine receptor-mediated anti-inflammatory effect in a chronic migraine rat model via the attenuation of glial cell activation. J Pain Res. 11:1129–1140. https://doi.org/10.2147/JPR.S159146
Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al-Abed Y (2007) Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. 35:1139–1144. https://doi.org/10.1097/01.CCM.0000259381.56526.96
Preskorn SH, Gawryl M, Dgetluck N, Palfreyman M, Bauer LO, Hilt DC (2014) Normalizing effects of EVP-6124, an alpha-7 nicotinic partial agonist, on event-related potentials and cognition: a proof of concept, randomized trial in patients with schizophrenia. J Psychiatr Pract. 20:12–24. https://doi.org/10.1097/01.pra.0000442935.15833.c5
Uteshev VV (2014) The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol 727:181–185. https://doi.org/10.1016/j.ejphar.2014.01.072
Chatzidaki A, Millar NS (2015) Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 97:408–417. https://doi.org/10.1016/j.bcp.2015.07.028
Duffy AM, Zhou P, Milner TA, Pickel VM (2009) Spatial and intracellular relationships between the α7 nicotinic acetylcholine receptor and the vesicular acetylcholine transporter in the prefrontal cortex of rat and mouse. Neuroscience. 161:1091–1103
Jones IW, Wonnacott S (2004) Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 24:11244–11252. https://doi.org/10.1523/JNEUROSCI.3009-04.2004
Sarter M, Parikh V, Howe WM (2009) Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci. 10:383–390. https://doi.org/10.1038/nrn2635
Egelman DM, Montague PR (1999) Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys J. 76:1856–1867. https://doi.org/10.1016/s0006-3495(99)77345-5
Acknowledgements
We thank Dr. Isabel Bermudez for generously providing the cDNA encoding concatemeric α7.
Funding
This work was supported by NIH grant NS031744 to SMS and grants from Universidad Nacional del Sur (PGI 24/B298), Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (PICT 2020–00936) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PIP11220200102356), Argentina to C.B.
Author information
Authors and Affiliations
Contributions
NM, CB and SMS designed the research; NM performed the electrophysiological experiments and SMS the molecular biology; NM and CB analyzed the data; SMS wrote the paper; NM and CB reviewed and edited the paper; SMS supervised the study.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mukhtasimova, N., Bouzat, C. & Sine, S.M. Novel interplay between agonist and calcium binding sites modulates drug potentiation of α7 acetylcholine receptor. Cell. Mol. Life Sci. 81, 332 (2024). https://doi.org/10.1007/s00018-024-05374-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05374-1