
Abstract
Copper is a trace element essential for numerous biological activities, whereas the mitochondria serve as both major sites of intracellular copper utilization and copper reservoir. Here, we investigated the impact of mitochondrial copper overload on the tricarboxylic acid cycle, renal senescence and fibrosis. We found that copper ion levels are significantly elevated in the mitochondria in fibrotic kidney tissues, which are accompanied by reduced pyruvate dehydrogenase (PDH) activity, mitochondrial dysfunction, cellular senescence and renal fibrosis. Conversely, lowering mitochondrial copper levels effectively restore PDH enzyme activity, improve mitochondrial function, mitigate cellular senescence and renal fibrosis. Mechanically, we found that mitochondrial copper could bind directly to lipoylated dihydrolipoamide acetyltransferase (DLAT), the E2 component of the PDH complex, thereby changing the interaction between the subunits of lipoylated DLAT, inducing lipoylated DLAT protein dimerization, and ultimately inhibiting PDH enzyme activity. Collectively, our study indicates that mitochondrial copper overload could inhibit PDH activity, subsequently leading to mitochondrial dysfunction, cellular senescence and renal fibrosis. Reducing mitochondrial copper overload might therefore serve as a strategy to rescue renal fibrosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Kidney fibrosis is the common pathway and main histological manifestation underlying the progression of chronic kidney disease (CKD) to end-stage kidney disease [1]. The pathogenic mechanisms of renal fibrosis are diverse and involve a wide range of signalling pathways. Recent studies have shown that copper over-accumulation is associated with fibrosis in tissues such as the lung, the liver and the oral mucosa [2,3,4]. Copper is an essential micronutrient in living organisms and participates in many essential cellular processes [5, 6]. In Wilson’s disease, the functional loss of ATPase copper transporting β (ATP7B) induces excessive copper accumulation in the liver, leading to liver cirrhosis, which can be ameliorated by copper chelator like penicillamine [7]. In our previous studies, we demonstrated that intracellular copper overload, driven by high expression of copper transporter 1 (CTR1), activated lysyl oxidase, enhanced matrix crosslinking and promoted renal fibrosis [8].
Mitochondria are crucial in eukaryotic cells due to their metabolic and biosynthetic functions. Mitochondria are not only vital intracellular copper reservoirs but are also the main copper utilizers [9]. Pyruvate dehydrogenase complex (PDHC) is a rate-limiting enzyme of glucose oxidation in mitochondrial matrix, converting pyruvate into acetyl-CoA and linking glycolysis to tricarboxylic acid (TCA) cycle [10]. Dihydrolipoamide acetyltransferase (DLAT), the E2 component of the PDHC, requires lipoylation for enzymatic function and exhibits a high affinity for copper within its lipoyl moiety [11]. A novel cell death pathway triggered by copper ionophore elesclomol, which is distinct from known death mechanisms and involves a 15–60 fold increase in intracellular copper levels, is discovered and coined “cuproptosis” by Tsvetkov et al. [12]. Our previous studies have shown that a mild accumulation of intracellular and mitochondrial copper, which is much lower than the fold increase mentioned in “cuproptosis”, induces cell damage and renal fibrosis [8, 13]. However, the underlying mechanism requires further investigation.
In this study, we demonstrated that mitochondrial copper was elevated in fibrotic kidney tissues and TGF-β1-stimulated tubular epithelial cells. We further showed that mitochondrial copper might bind to the lipoyl moiety of lipoylated DLAT, leading to altered protein subunit interaction, lipoylated DLAT dimerization, and inhibited pyruvate dehydrogenase activity, thereby resulting in mitochondrial dysfunction and cellular senescence. Reducing mitochondrial copper levels by downregulating CTR1 expression or using a copper chelator were shown to restore mitochondrial function and attenuate renal fibrosis.
Materials and methods
Human kidney tissues
Kidney biopsy tissues were acquired from the Department of Nephrology, Tongji Hospital, Shanghai, China. Control samples were obtained from patients with minimal change disease (MCD) without fibrosis, while fibrotic samples were collected from CKD patients with IgA nephropathy (IgAN) or diabetic nephropathy (DN). Masson trichrome staining, as previously reported [14], was used to evaluate the severity of renal fibrosis. The study was approved by the Human Subjects Committee of Tongji Hospital (No. K-W-2021-012), and written informed consent was obtained by all patients.
Animals
CTR1+/− mice and the control mice (WT) were purchased from the Model Animal Research Center of Nanjing University and were routinely maintained on the C57BL/6 mice background. Polymerase chain reaction analysis of tail DNA was used to determine the targeted genes. All animals were housed in a room with controlled temperature (23 ± 2 ℃) and constant humidity (40–60%). All animals were provided with water and chow ad libitum. All animal experiments were approved by the Animal Ethics Committee of Tongji Hospital (No. 2020-DW-003).
For the unilateral renal ischaemia/reperfusion injury with contralateral nephrectomy (uIRIx) model, C57BL/6 male mice aged 6 to 8 weeks were used for the in vivo analysis. uIRIx models were established by clamping the left renal pedicle for 30 min using nontraumatic microvascular clamps at 37 °C. 7 days later, the mice were anaesthetized and the right kidney was removed. Twenty-eight days after the ischaemia, the mice (n = 5 per group) were killed, and the kidneys were harvested for analysis. Four groups of mice were prepared, namely, sham-operated WT mice (WT, sham), sham-operated CTR1+/− mice (CTR1+/−, sham), IRI 28d WT mice (WT, IRI 28d), and IRI 28d CTR1+/− mice (CTR1+/−, IRI 28d). For copper chelating experiments, copper chelating was achieved by daily oral treatment with TM (0.7 mg/day, Sigma-Aldrich) [15, 16] for 28 days following the IRI operation. Mice were prepared into groups of sham-operated (Sham), sham-operated + TM-treated(Sham + TM), IRI 28d, and IRI 28d + TM-treated (IRI 28d + TM).
For the unilateral ureteral obstruction (UUO) model, C57BL/6 male mice aged 6 to 8 weeks were subjected to UUO operation as described previously [17]. Left ureter was exposed and ligated by surgical 5 − 0 silk. Mice (n = 4 per group) were sacrificed on day 14 after UUO surgery for biochemical and histological analyses.
For the folic acid (FA) model, C57BL/6 male mice aged 6 to 8 weeks were injected with 250 mg/kg of FA diluted in 300 mM NaHCO3 intraperitoneally. Mice (n = 4 per group) were killed on day 28 after FA injection for biochemical and histological analyses.
Cell culture and treatment
Rat renal tubular epithelial cells (NRK-52E) were obtained from the Chinese Academy of Sciences (Shanghai, China). Cells were grown in DMEM (Gibco, #10,569,044) supplemented with 4% FBS and maintained at 37 °C in a 5% CO2 air incubator. NRK-52E cells were treated with 5 ng/ml TGF-β1 (R&D Systems, USA), 10 µmol/L CuSO4 (Sangon Biotech, Shanghai, China), with or without TM. A CTR1 shRNA plasmid was used to generate a lentivirus, which was then transfected to cells to downregulate CTR1.
Western blotting
NRK-52E cells and kidney tissues were lysed in RIPA buffer. Protein samples were separated by SDS–PAGE gels and transferred to polyvinylidene fluoride (PVDF) membrane. Subsequently, the membranes were blocked with 5% skim milk for 1 h and incubated with primary antibodies at 4 °C overnight, followed by incubation with a horseradish peroxidase-conjugated secondary antibody. The primary antibodies used were as follows: anti-GAPDH (Proteintech, #60004-1-AP), anti-Collagen I (Abcam, #ab-21,286), anti-α-SMA (Abcam, #ab-124,964), anti-CTR1 (Abcam, #ab-129,067), anti-p16INK4A (Santa Cruz, #sc-1661), anti-γH2AX (Abcam, ab26350;), anti-DLAT (Proteintech, #13426-1-AP), anti-p21 (Proteintech, #10355-1-AP), anti-p53 (Cell Signaling Technology, #2524).
Reverse transcription and quantitative real-time PCR
RNA was extracted from cells and kidney tissues with TRIzol reagent (Invitrogen, Carlsbad, CA) as previously reported [18]. RNA was then transcribed into cDNA using a PrimeScript RT Reagent kit (Takara). Subsequently, cDNA served as a template for real-time PCR, which was conducted with SYBR Green PCR Master Mix (Roche, Mannheim, Germany). The primers used in this study targeted mouse GAPDH, Collagen I, CTR1 and α-SMA (Table 1), along with rat GAPDH and CTR1 (Table 2).
Renal histology and immunohistochemistry
Renal tissues were fixed in 4% neutral-buffered paraformaldehyde and paraffin-embedded according to a standard procedure. Kidney Sect. (4-µm thickness) were subjected to Masson trichrome staining, HE staining, or immunohistochemistry following the protocol [19]. For immunochemical staining, after the tissue sections were dewaxed and subjected to antigen retrieval, the sections were blocked with 3% hydrogen peroxidase for 10 min, followed by a 60-min incubation with 5% goat serum at room temperature. Next, the sections were incubated with a primary antibody against CTR1 (Abcam, #ab-129,067). Following incubation with an HRP-conjugated secondary antibody for 1 h at room temperature, a 3,3′-diaminobenzidine (DAB) kit (Vector, #SK4100) was used in accordance with the manufacturer’s instructions.
Transmission electron microscopy
To examine mitochondrial structure, transmission electron microscopy (TEM) was used. Kidney cortexes and NRK-52E cells were fixed in 2.5% glutaraldehyde/0.1 M phosphate buffer at 4 °C overnight. Subsequently, ultrathin renal sections were stained with uranyl acetate and lead citrate, then embedded in copper grid according to routine procedures. Mitochondria were photographed under an electron microscope (JEOL JEM-1010, Tokyo, Japan).
Isolation of mitochondria
Mitochondria from the kidney cortex and NRK-52E cells were isolated using commercial Mitochondria Isolation kit for Tissue (Abcam, #ab-110,168) and Mitochondria Isolation Kit for Cultured Cells (Abcam, #ab-110,170) following the manufacturer’s instructions respectively. The isolated mitochondria were then resuspended in mitochondrial storage buffer and BCA method was used to assess the protein concentration of the isolated mitochondria.
Copper content determination
Subcellular fractions of renal tissues and cells were treated with 65% nitric acid at 100 °C for 1 h, followed by diluted with ultrapure water. Copper levels were measured using inductively coupled plasma-mass spectrometry (ICP-MS) (NexION 300D, PerkinElmer, USA) as previously described [20]. Standard samples were also employed for quantification [21].
PDH enzyme activity
PDH activity in vitro experiment was measured using the PDH Enzyme Activity Assay Kit (Abcam, #ab-109,902) according to the manufacturer’s protocol. PDH activity in vivo experiment was assayed by PDH Enzyme Activity Assay Kit (Solarbio, # BC0385) as previously described [22].
Measurement of oxygen consumption rate
Seahorse XFe96 Extracellular Flux Analyzer (Agilent, CA) was used to detect oxygen consumption rate (OCR) according to the instructions provided by Seahorse Biosciences. Briefly, NRK-52E cells were seeded at a density of 6,000 cells per well in XF96 cell culture miniplates and incubated at 37 ℃ for 24 h. The cells were washed with XF assay medium supplemented with 2 mM glutamine, 1 mM pyruvate and 10 mM glucose, and equilibrated in 180 µL assay medium per well in a 37℃ incubator without CO2 for 1 h. OCR was analyzed by injections of 1.5 µM oligomycin, 1µM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 µM rotenone plus antimycin A to assess a mitochondrial stress test using the XF Extracellular Flux Analyzer. Following each assay, cell numbers were recounted for normalization of the OCR.
SA-β-gal activity assay
SA-β-gal staining was examined according to the manufacturer’s instructions (Beyotime, #C0602). Frozen kidney tissue sections were fixed with fixative solution at room temperature for 15 min and then washed three times with PBS. The fixed sections were incubated with a staining solution mix overnight at 37 ℃ without CO2. SA-β-Gal quantification was performed by Image J software [23].
ATP
ATP levels were detected using the ATP Assay Kit (Beyotime, #S0027) according to the manufacturer’s protocol. Briefly, cells and kidney tissues were homogenized in ATP lysis buffer and then centrifuged at 12,000 ×g and 4 ℃ for 5 min. 100 µL of ATP detection working solution was mixed with 20 µL of supernatant in 96-well plates. Luminance was then assayed and normalized to protein concentration.
Protein structure
The protein structure of DLAT was downloaded from the AlphaFold2 modelling results of the Uniprot website [24], where the UniProt number of DLAT is Q8BMF4. And lipoylation was performed on amino acids 131 and 258 of the DLAT. The pymol.edit module was used for initial structure establishment.
Molecular dynamics simulations (MDS)
To examine the interaction between lipoylation site and copper, wild-type and lipoylated DLAT protein structures underwent dynamics simulations. Gromacs 2019.6 was used as the dynamics simulation software [25] and GROMOS96 54a7 was used as the dynamic force field [26]. TIP3P water model was used along with NaCl and then one copper was added to construct control (DLAT-Cu1+(1)) and experimental (Lipoylated DLAT-Cu1+(1)) simulation system. Verlet, cg algorithms were used in the elastic simulation, with PME handling electrostatic interactions. Energy minimization used the steepest descent method, and the system was balanced using canonical (NVT) and isothermal-isobaric (NPT) ensembles. A 100 ns MD simulation at 300 K and normal pressure.
We took another simulation to examine the role of copper in lipoylation DLAT protein. TIP3P water model was used along with NaCl and then one copper was added to lipoylated DLAT system to construct one experimental simulation system (Lipoylated DLAT-Cu1+(1)). Lipoylated DLAT-Cu1+(0) were used as control group. The simulation, based on previously set parameters, involved a 10 ns MD simulation. Mirror the relaxed protein. SMD simulations on heavy atoms used a constraint force of 0.5 kcal/ (mol Å2) to get initial coordinates for umbrella sampling along the unbinding pathway. A virtual spring constant of 5000 kcal/ (mol Å2) was applied to the ligand’s centre of mass with a velocity of 0.01 Å/ps.
Determination of blood urea nitrogen and serum creatinine
The serum of the mice was isolated by centrifugation. Serum creatinine and blood urea nitrogen (BUN) levels were assessed using an automatic chemistry analyser according to the manufacturer’s manual. The levels of serum creatinine and BUN were expressed as µmol/L and mmol/L respectively.
Statistical methods
All results were presented as the mean ± SEM. Differences between two groups were analysed using Student’s t-test and one-way analysis of variance (ANOVA) was used for comparisons among multiple groups. Difference at p < 0.05 was considered statistically significant. SPSS 21.0 statistical software (Chicago, IL) was used for statistical analyses.
Results
Mitochondrial copper overload in fibrotic kidneys
Our previous data suggest that increased intracellular copper content contributes to renal fibrosis through lysyl oxidase mediated matrix crosslinking [9]. Here, we further investigated mitochondrial copper content in the fibrotic kidneys. We found that the mitochondrial copper levels but not the cytosolic levels were significantly elevated in the IRI 28d fibrotic kidneys, accompanied by accelerated cellular senescence, which can directly promote fibrogenesis [27] (Fig. 1a-c). Similarly, mitochondrial copper levels were increased in the fibrotic kidneys of UUO-14d mice (Fig. 1d-f) and FA-28d mice (Fig. 1g-i), which were accompanied by elevated cellular senescence. In tubular epithelial cell stimulated by TGF-β1, we also found that the increased mitochondrial copper levels were associated with elevated expression of p16INK4A and γH2AX (Fig. 1j-k). These results indicated the tight association of mitochondrial copper overload with cellular senescence in the fibrotic kidneys.

Mitochondrial copper overload in fibrotic kidneys. (a) The cytosolic and mitochondrial fractions were isolated from kidneys of sham and IRI 28d kidneys. Copper content was detected in the cytosol and mitochondria in the kidneys of the IRI 28d model via ICP-MS. (b) Masson staining, HE staining and SA-β-gal staining in the kidneys of IRI 28d model. Original magnification, ×200. Bar = 50 μm. (c) Western blot analysis of Collagen I, α-SMA, p16INK4A, γH2AX, p21 and p53 expression in sham and IRI 28d kidneys. (d) Copper content was detected in the cytosol and mitochondria in the kidneys of the UUO 14d model via ICP-MS. (e) Masson staining, HE staining and SA-β-gal staining in the kidneys of UUO 14d model. Original magnification, ×200. Bar = 50 μm. (f) Western blot showing the expression of Collagen I, α-SMA, p16INK4A, γH2AX, p21 and p53. (g) The copper content was detected in the cytosol and mitochondria in the kidneys of the FA 28d model. (h) Masson staining, HE staining and SA-β-gal staining in the kidneys of FA 28d model. Original magnification, ×200. Bar = 50 μm. (i) Western blot analysis of Collagen I, α-SMA, p16INK4A, γH2AX, p21 and p53 expression. (j) The copper content was detected in the mitochondria of NRK-52E cells after 48 h of TGF-β1 treatment. (k) Western blot analysis of Collagen I, α-SMA, p16INK4A and γH2AX expression. Data are the mean ± SEM of at least 3 independent experiments. n = 4 for in vivo experiments and n = 3 for vitro experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
CTR1 is significantly increased in renal fibrosis and contributes to the elevation of mitochondrial copper
CTR1 is the main regulator of copper influx in eukaryotes [28]. We then examined the CTR1 expression in various renal fibrosis models. Notably, renal CTR1 was significantly increased in the fibrotic kidneys of IRI 28d mice by immunohistochemistry analyses (Fig. 2a). Consistent with this, western blot indicated that CTR1 was significantly upregulated in fibrotic renal tissues (Fig. 2b). Furthermore, CTR1 was also highly expressed in the kidneys of UUO 14d and FA 28d fibrotic mice (Fig. 2c-f), in NRK-52E cells treated with TGF-β1 (Fig. 2g), and in fibrotic kidney tissues from diabetic nephropathy and IgA nephropathy patients (Fig. 2h). To explore the effect of CTR1 on mitochondrial copper levels, we generated CTR1 heterozygous mice (CTR1+/−) as CTR1 homozygous deletion mice (CTR1−/−) were embryonic lethal and constructed a CTR1 knockdown cell lines in vitro. We found CTR1 knockdown markedly reduced the levels of the mitochondrial copper but not the cytosolic copper in kidneys of the IRI 28d mice and in NRK-52E cells treated with TGF-β1 (Fig. 2i, j, Supplementary Figure S1a-d). CTR1 thus was highly expressed in renal fibrosis and CTR1 deficiency alleviated mitochondrial copper accumulation.

CTR1 is highly expressed in renal fibrosis and induces mitochondrial copper overload. (a) Immunohistochemical staining of CTR1 in low magnification (left panel: magnification,×40; bar = 200 μm) and high magnification (right panel: magnification,×200; bar = 50 μm) in the kidneys of IRI 28d model. (b) Western blot analysis of CTR1 expression in sham and IRI 28d kidneys. (c) Immunohistochemical staining of CTR1 in low magnification (left panel: magnification,×40; bar = 200 μm) and high magnification (right panel: magnification, ×200; bar = 50 μm) in the kidneys of UUO 14d model. (d) Western blot analysis of CTR1 expression in sham and UUO 14d kidneys. (e) Immunohistochemical staining of CTR1 in low magnification (left panel: magnification,×40; bar = 200 μm) and high magnification (right panel: magnification,×200; bar = 50 μm) in the kidneys of FA 28d model. (f) Western blot analysis of CTR1 expression in sham and FA 28d kidneys. (g) Western blot analysis of CTR1 expression in NRK-52E cells treated with TGF-β1. (h) Immunohistochemical staining of CTR1, Masson staining and HE staining in the kidneys of patients with or without fibrosis. Original magnification, ×200. Bar = 50 μm. (i) The cytosolic and mitochondrial fractions were isolated from kidneys of wild-type control mice, wild-type mice that received IRI surgery, CTR1+/− control mice and CTR1+/− mice that received IRI surgery. Copper concentrations in the subcellular fractions were assessed via ICP-MS. (j) Copper content was detected in the mitochondria of control, CTR1-knockdown cell lines treated with TGF-β1(n = 3). Data are the mean ± SEM of at least 3 independent experiments. n = 5 for in vivo experiments and n = 3 for vitro experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. MCD, minimal change disease; DN, diabetic nephropathy; IgAN, IgA nephropathy
Reducing mitochondrial copper overload by knocking-down CTR1 mitigates cellular senescence and renal fibrosis
We then investigated the effects of reducing mitochondrial copper through inhibiting CTR1 on cellular senescence and renal fibrosis. Our results showed that the degree of renal fibrosis in the CTR1+/− IRI 28d mice was markedly ameliorated, based on Masson trichrome staining, HE staining, mRNA and protein expression levels of Collagen I and α-SMA (Fig. 3a-d). Moreover, with the knockdown of CTR1, the level of SA-β-gal activity and the expression of p16INK4A, γH2AX, p21 and p53 in IRI 28d mice kidneys were significantly reduced (Fig. 3e-f). We also found the knockdown of CTR1 mitigated p16INK4A and γH2AX expression in NRK-52E cells treated with TGF-β1 (Fig. 3g). Collectively, these results suggest that mitochondrial copper overload promotes cellular senescence and renal fibrosis.

Reducing mitochondrial copper overload by knockdown of CTR1 alleviates cellular senescence and renal fibrosis. (a) Masson and HE staining of renal sections among indicated groups. Original magnification, ×200. Bar = 50 μm. (b, c) RT–PCR showing the changes in Collagen I and α-SMA mRNA levels. (d) Western blot showing the expression of α-SMA and Collagen I among the groups as indicated. (e)Representative staining micrographs showing SA-β-gal activity in different groups. Original magnification, ×200. Bar = 50 μm. (f) Western blot showing the expression of p16INK4A, γH2AX, p21 and p53. (g) Western blot showing the expression of p16INK4A and γH2AX. Data are the mean ± SEM of at least 3 independent experiments. n = 5 for in vivo experiments and n = 3 for vitro experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.05 versus TGF-β1-treated cells or IRI 28d groups, #P < 0.01, ###P < 0.001
Reducing mitochondrial copper overload ameliorates mitochondrial dysfunction
To further examine the effect of mitochondrial copper overload on mitochondrial function, we assessed mitochondrial morphological changes following CTR1 knockdown. In the IRI 28d kidneys, we observed that following the knockdown of CTR1, mitochondrial swelling(with a loss of cristae)was improved (Fig. 4a). Moreover, downregulation of CTR1 increased ATP generation (Fig. 4b). In vitro in the NRK-52E cells, the deformation of mitochondria induced by TGF-β1 was also ameliorated after knockdown of CTR1 (Fig. 4c). In the Seahorse tests, TGF-β1-induced alterations in the basal respiration, ATP-production dependent respiration, maximal respiration were all partially reversed in CTR1 knockdown cells (Fig. 4d-e). ATP levels were also restored after knockdown of CTR1 (Fig. 4f). Together, these data indicated that reducing mitochondrial copper overload attenuated TGF-β1-induced mitochondrial dysfunction.

Reducing mitochondrial copper overload alleviates mitochondrial dysfunction. (a) Representative images of mitochondria morphology detected by transmission electron microscopy in low magnification (upper panel: magnification, ×6800; bar = 1 μm) and high magnification (bottom panel, × 13,000; bar = 500 nm). (b) ATP content detection of kidney tissues. (c) Representative electron micrographs of mitochondria in NRK-52E cells of the indicated groups in low magnification (upper panel: magnification, ×7000; bar = 2 μm) and high magnification (bottom panel: magnification, ×15,000; bar = 1 μm). (d, e) Measurements of the OCR in NRK-52E cells among different groups. (f) ATP content detection of cultured cells. Data are the mean ± SEM of at least 3 independent experiments. n = 5 for in vivo experiments and n = 3 for vitro experiments. *P < 0.05, **P < 0.01
Mitochondrial copper overload inhibits PDH enzyme activity via inducing the dimerization of lipoylated DLAT
We then performed a series of further experiments to explore the mechanism by which mitochondrial copper overload induces mitochondrial dysfunction and cellular senescence. Through molecular dynamics simulations that had been verified to be stable and viable (Supplementary Figure S2), we found that copper ions were in close proximity to the lipoylated DLAT protein but distant from the non-lipoylated DLAT protein (Fig. 5a). According to the trajectory, copper ions bound to the lipoylated site of lipoylated DLAT with a tendency of forming stable postures (Fig. 5b), enhancing the interactions between protein subunits (Fig. 5c) and reducing resistance energy between protein subunits (from − 268.9 kal/mol to -65.42 kal/mol) (Fig. 5d), thereby facilitating protein dimerization. Additionally, we examined lipoylated DLAT dimer in fibrotic kidneys and found lipoylated DLAT dimer was significantly increased in the renal tissues of IRI 28d mice. In CTR1+/− IRI 28d mice, however, the levels of DLAT dimer were significantly decreased (Fig. 5e). Since DLAT is a crucial component of the PDHC, we further explored whether the dimerization of lipoylated DLAT could inhibit PDH enzyme activity. On one hand, downregulation of CTR1 markedly ameliorated IRI-induced decrease in PDH enzyme activity (Fig. 5f). On the other hand, in tubule cells treated with TGF-β1, downregulation of CTR1 led to a reduced level of lipoylated DLAT dimer and increased PDH enzyme activity (Fig. 5g, h). These findings suggest that mitochondrial copper overload probably inhibits PDH enzyme activity in part through the dimerization of lipoylated DLAT.

Mitochondrial copper induces the dimerization of lipoylated DLAT and inhibits PDH enzyme activity.(a) Molecular dynamics simulations of the distance between copper ions and lipoylated DLAT/non-lipoylated DLAT. (b) Molecular dynamics simulations showing the effect between copper and modification site of lipoylated DLAT. (c) Molecular dynamics simulations showing the forces between lipoylated DLAT protein subunits with/without copper. (d) Energy between lipoylated DLAT protein subunits with/without copper. (e) Western blot analysis of the dimer of lipoylated DLAT proteins among different groups in vivo. (f) PDH enzyme activity detection. (g) Western blot analysis of the dimer of lipoylated DLAT proteins among different groups in vitro. (h) PDH enzyme activity detection. Data are the mean ± SEM of at least 3 independent experiments. n = 5 for in vivo experiments and n = 3 for vitro experiments. *P < 0.05, **P < 0.01, **P < 0.001
Treatment with copper chelator tetrathiomolybdate (TM) improves PDH enzyme activity and mitochondrial dysfunction
To further affirm the role of mitochondrial copper over-accumulation in mitochondrial function, we used a copper chelation agent, TM. In comparison with the untreated IRI 28d mice, TM treatment significantly restored the PDH enzyme activity in renal tissues of the IRI 28d mice (Fig. 6a). In addition, mitochondrial swelling and fragmentation of cristae in the IRI 28 model were improved after TM treatment (Fig. 6b). The reduction of ATP levels in the IRI 28d mice was mitigated after TM stimulation (Fig. 6c). Furthermore, we found that the lipoylated DLAT dimer significantly increased in tubule cells stimulated with TGF-β1 and markedly decreased after TM treatment (Fig. 6d). TM treatment restored TGF-β1-stimulated reduction in PDH enzyme activity (Fig. 6e). TGF-β1-induced abnormal mitochondrial morphology, declined maximal respiration, and decreased ATP levels were improved as a consequence of TM treatment (Fig. 6f-i). In line with above observations, copper levels were markedly increased in the mitochondria after CuSO4 treatment (Supplementary Figure S3a). Mitochondrial shrinkage and disorganized cristae in CuSO4-treated NRK-52E cells were mitigated following TM treatment (Supplementary Figure S3b).

Copper chelator TM treatment improves PDH enzyme activity and mitochondrial dysfunction. (a) PDH enzyme activity detection. (b) Representative electron microscopy images of mitochondria in renal tubular cells among the indicated groups in low magnification (upper panel: magnification, ×2500; bar = 5 μm) and high magnification (bottom panel: magnification, ×15,000; bar = 1 μm). (c) ATP content detection. (d) Western blot analysis of the dimer of lipoylated DLAT proteins among different groups in vitro. (e) PDH enzyme activity detection. (f) Representative electron microscopy images of mitochondria in renal tubular cells among the indicated groups in low magnification (upper panel: magnification, ×7000; bar = 2 μm) and high magnification (bottom panel: magnification, ×15,000; bar = 1 μm). (g, h) Measurements of the OCR in NRK-52E cells among different groups. (i) ATP content detection. Data are the mean ± SEM of at least 3 independent experiments. n = 4 for in vivo experiments and n = 3 for vitro experiments. * P < 0.05, **P < 0.01
TM treatment mitigates renal cellular senescence and fibrosis
We finally verified the effects of copper chelation on cellular senescence and renal fibrosis. As evidenced by histological examination, and Collagen I and α-SMA expression, TM treatment significantly reduced renal fibrosis (Fig. 7a-d). In addition, copper chelation markedly attenuated the abnormal high level of SA-β-gal activity and the increased expression of p16INK4A, γH2AX, p21 and p53 (Fig. 7e, f). The IRI-induced upregulation of serum creatinine and BUN in the IRI 28d mice model were profoundly attenuated after TM treatment (Fig. 7g, h). In cultured tubule epithelial cells, TM treatment also markedly reduced TGF-β1 or CuSO4-induced overexpression of p16INK4A and γH2AX (Fig. 7i, Supplementary Figure S3c). Together, these results indicated that copper chelating could ameliorate cellular senescence and renal fibrosis.

TM treatment improves cellular senescence and renal fibrosis. IRI 28d or sham-operated mice were divided into four groups: (1) sham mice treated with saline; (2) sham mice treated with TM (0.7 mg/day); (3) IRI 28d mice treated with saline; and (4) IRI 28d mice treated with TM. (a) Masson and HE staining of renal sections from the indicated groups. (b-d) Western blot and QPCR showing the expression of α-SMA and Collagen I among the groups as indicated. (e) Representative staining micrographs showing SA-β-gal activity. (f) Western blot analyses of the expression of p16INK4A and γH2AX in different groups. (g) Serum creatinine and (h) blood urea nitrogen levels in different groups. (i) Western blot analyses of the expression of p16INK4A and γH2AX in different groups. Data are the mean ± SEM of at least 3 independent experiments. n = 5 for in vivo experiments and n = 3 for vitro experiments. **P < 0.01, *** P < 0.001, **** P < 0.0001; #P < 0.05 versus TGF-β1-treated cells, ##P < 0.01 versus IRI 28d groups, ###P < 0.001
Discussion
The present study demonstrates mitochondrial copper overload in experimental CKD models. The mitochondrial copper is shown to bind directly to lipoylated DLAT protein, induces lipoylated DLAT protein dimerization, and inhibits PDH enzyme activity, which leads to mitochondrial dysfunction, cellular senescence and renal fibrosis.
Tsvetkov et al. challenged traditional cell death dogmas and proposed “Cuproptosis” in Science, suggesting that pulse treatment with the copper ionophore elesclomol results in a 15–60 fold increase in intracellular copper level, triggering proteotoxic stress and ultimately leading to cell death [12]. Similar to the proposed cuproptosis, the effects of copper overload on renal cells as we have demonstrated are also mediated at least in part through the mitochondria. Distinct differences, however, exist between cuproptosis and copper overload-induced renal cellular senescence and fibrosis. Firstly, in our models, the increase in intracellular or mitochondrial copper is relatively small and probably not sufficient to cause cuproptosis. In our studies, a mild increase (approximately 1.5-2 fold) in copper ions level within renal tubular epithelial cells and mitochondria, which is significantly lower than the fold-increase of copper ions in cuproptosis, is sufficient to cause cell damage, renal senescence and fibrosis [8, 13].
Cellular senescence permanently arrests cell proliferation, accompanied by the development of senescence-associated secretory phenotype (SASP) [29]. It is showed that decreased respiratory capacity, elevated ADP/ATP and AMP/ATP ratios, reduced mitochondrial membrane potential, increased production of oxygen free radicals, serves as both a cause and a consequence of cellular senescence [30]. In current study we pinpoint that mitochondrial copper overload could promote renal senescence and fibrosis in part by inhibiting mitochondrial PDH activity. Malthankar et al. reported previously that elevated levels of Cu2+, Mn2+ and Zn2+ can inhibit the activities of TCA cycle enzymes, such as citrate synthase and ketoglutarate dehydrogenase complex in neuronal cells and fungal cells [31,32,33]. In line with this, in our study involving CKD mouse models and TGF-β1-stimulated NRK-52 cells, we observed that mitochondrial copper overload leads to a reduction in PDH activity within the TCA cycle, thereby impeding ATP generation, inducing cellular senescence and promoting renal fibrosis. Reducing mitochondrial copper content (using copper chelation with TM or CTR1 knockdown), on the other hand, rescued PDH enzyme activity, replenished ATP production, and ameliorated renal fibrosis. Wilson’s Disease (WD), a rare hereditary condition, is characterized by hepatic mitochondrial copper accumulation and lethal liver failure. Unlike the mechanisms in our research, in WD, hepatic copper accumulation induces mitochondrial membrane crosslinking and destruction. Using copper-cheating agents can reverse copper levels, preserve mitochondrial structural integrity and function, and thereby alleviate liver damage [34].
Our results also suggest a novel mechanism through which mitochondrial copper overload inhibits PDH enzyme activity. Transition metal ions, such as copper ions, are potent mediators for protein aggregation, as they can facilitate stable and rigid interactions between proteins on a small surface area [35, 36]. Martina G et al. reported that copper ions mediate protein aggregation and are implicated in the progression of neurodegenerative diseases [37]. In Alzheimer’s disease, it has been discovered that copper ions bind to amyloid-β peptide (Aβ), inducing the formation of Aβ oligomers and ultimately exacerbating the neurotoxicity of Aβ-aggregation [38]. Pyruvate dehydrogenase complex (PDHC) is a mitochondrial matrix enzyme that catalyses the irreversible conversion of pyruvate generated by glycolysis into acetyl-CoA (coenzyme A), which can then be fuelled into TCA cycle [39]. Zhang et al. reported that PDH enzyme activity was reduced in the UUO model and in tubular epithelial cells incubated with TGF-β1 [40]. The PDHC consists of three different enzymes, including the E2 subunit, DLAT, which requires lipoylation (lipoic acid acylation) for its enzymatic function. The possibility of copper to bind to lipoylated DLAT directly is supported by the observed binding of copper to free lipoic acid, with a measured dissociation constant of 10− 17 [11]. Our results, for the first time, demonstrated mitochondrial copper bound to lipoylated site of lipoylated DLAT, which further altered interactions between DLAT protein subunits and induced protein dimerization, thereby inhibiting the activity of pyruvate dehydrogenase and ultimately resulting in cellular senescence and renal fibrosis.
Regarding the uptake of copper ions by the mitochondria, preliminary studies suggested that copper chaperone COX17 could ferry cytosolic copper into mitochondria [41, 42]. However, later experiments showed that COX17 functions only in the mitochondrial membrane space and cannot transport copper ions from the cytoplasm to mitochondria, contradicting this notion [43, 44]. Mari et al. proposed that the tripeptide glutathione (GSH) may participate in copper entry into mitochondria, as it can be easily shuttled into mitochondria via porin channels in the mitochondrial outer membrane. Nevertheless, this idea was challenged by studies in GSH deficiency yeast, which showed an unchanged mitochondrial copper level [45]. SLC25A3, a member of the mitochondrial carrier family, transports the copper-ligand (CuL) complex across the inner membrane for storage in the matrix [46]. Dennis R et al. reported that the depletion of SLC25A3 results in a reduction of mitochondrial copper levels [47]. More studies suggested that a non-protein, anionic copper ligand (CuL) might mediate copper transport into mitochondria. Nonetheless, the specific molecular identity of the CuL remains unclear, and further investigations are warranted to validate this concept [6, 45]. CTR1 is a major copper importer for intracellular copper uptake [48]. CTR1 was highly expressed in kidney and regulated by Smad3 signalling [8]. Since the specific mitochondrial copper importer is currently unknown, we used CTR1 knockdown transgenic mice, CTR1 knockdown renal tubular cell lines, and combined with copper chelators to investigate the underlying mechanisms by which mitochondrial copper overload induces cellular damage.
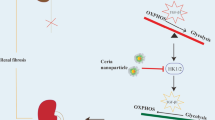
Collectively, our study demonstrates that mitochondrial copper might bind directly to lipoylated DLAT and induces its dimerization, thereby inhibiting PDH enzyme activity and resulting in mitochondrial dysfunction, cellular senescence and renal fibrosis. Reducing mitochondrial copper levels by inhibiting CTR1 or using copper chelation might be an innovate strategy to mitigate the progression of renal fibrosis (Fig. 8).

Schematic illustration of mitochondrial copper overload in renal fibrosis. This study highlights that mitochondrial copper overloaded in fibrotic kidneys will lead to the dimerization of lipoylated DLAT, inhibition of pyruvate dehydrogenase activity, mitochondrial dysfunction, and cellular senescence. On the other hand, reducing mitochondrial copper overload through knocking-down of CTR1 or copper chelator treatment might restore PDH enzyme activity and mitochondrial function
Data availability
Not applicable.
References
Panizo S et al (2021) Fibrosis in chronic kidney disease: Pathogenesis and consequences. Int J Mol Sci, 22(1)
Janssen R et al (2018) Copper as the most likely pathogenic divergence factor between lung fibrosis and emphysema. Med Hypotheses 120:49–54
Yadav A et al (2015) Estimation of serum zinc, copper, and iron in the patients of oral submucous fibrosis. Natl J Maxillofac Surg 6(2):190–193
Dirksen K, Fieten H (2017) Canine Copper-Associated Hepatitis. Vet Clin North Am Small Anim Pract 47(3):631–644
Lutsenko S (2010) Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol 14(2):211–217
Kim BE, Nevitt T, Thiele DJ (2008) Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol 4(3):176–185
Gerosa C et al (2019) Liver pathology in Wilson’s disease: from copper overload to cirrhosis. J Inorg Biochem 193:106–111
Niu YY et al (2020) Elevated intracellular copper contributes a unique role to kidney fibrosis by lysyl oxidase mediated matrix crosslinking. Cell Death Dis 11(3):211
Zischka H, Einer C (2018) Mitochondrial copper homeostasis and its derailment in Wilson disease. Int J Biochem Cell Biol 102:71–75
Saed CT, Dakhili SAT, Ussher JR (2021) Pyruvate dehydrogenase as a therapeutic target for nonalcoholic fatty liver disease. Acs Pharmacol Translational Sci 4(2):582–588
Smirnova J et al (2018) Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. Alpha-lipoic acid as a potential anti-copper agent. Sci Rep 8(1):1463
Tsvetkov P et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375(6586):1254–1261
Zhu SY et al (2023) COX17 restricts renal fibrosis development by maintaining mitochondrial copper homeostasis and restoring complex IV activity. Acta Pharmacol Sin
Wang P et al (2018) Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci Transl Med, 10(462)
Hsu PY et al (2018) Tetrathiomolybdate, a copper chelator inhibited imiquimod-induced skin inflammation in mice. J Dermatol Sci 92(1):30–37
Brewer GJ et al (2003) Tetrathiomolybdate therapy protects against bleomycin-induced pulmonary fibrosis in mice. J Lab Clin Med 141(3):210–216
Livingston MJ et al (2016) Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12(6):976–998
Yu Y et al (2015) Leukemia inhibitory factor attenuates renal fibrosis through Stat3-miR-29c. Am J Physiol Ren Physiol 309(7):F595–603
Han SH et al (2017) PGC-1alpha protects from Notch-Induced kidney Fibrosis Development. J Am Soc Nephrol 28(11):3312–3322
Wu X et al (2016) Potassium and the K+/H + exchanger Kha1p promote binding of copper to ApoFet3p multi-copper ferroxidase. J Biol Chem 291(18):9796–9806
Feng L et al (2017) A novel absolute quantitative imaging strategy of iron, copper and zinc in brain tissues by isotope dilution laser ablation ICP-MS. Anal Chim Acta 984:66–75
Yu LB et al (2022) Thorium inhibits human respiratory chain complex IV (cytochrome c oxidase). J Hazard Mater, 424
Esposito P et al (2024) SA-beta-gal in kidney tubules as a predictor of renal outcome in patients with chronic kidney disease. J Clin Med, 13(2)
Jumper J et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–
Van Der Spoel D et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Schmid N et al (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J 40(7):843–856
Wang WJ, Chen XM, Cai GY (2021) Cellular senescence and the senescence-associated secretory phenotype: potential therapeutic targets for renal fibrosis. Exp Gerontol, 151
Wee NK et al (2013) The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int J Biochem Cell Biol 45(5):960–963
Wiley CD et al (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23(2):303–314
Miwa S et al (2022) Mitochondrial dysfunction in cell senescence and aging. J Clin Invest, 132(13)
Malthankar GV et al (2004) Differential lowering by manganese treatment of activities of glycolytic and tricarboxylic acid (TCA) cycle enzymes investigated in neuroblastoma and astrocytoma cells is associated with manganese-induced cell death. Neurochem Res 29(4):709–717
Brown AM et al (2000) Zn2 + inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J Biol Chem 275(18):13441–13447
Krumova ET et al (2012) Copper stress and filamentous fungus 103 - ultrastructural changes and activities of key metabolic enzymes. Can J Microbiol 58(12):1335–1343
Zischka H et al (2011) Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest 121(4):1508–1518
Ni TW, Tezcan FA (2010) Structural characterization of a microperoxidase inside a metal-directed protein cage. Angew Chem Int Ed Engl 49(39):7014–7018
Salgado EN et al (2009) Control of protein oligomerization symmetry by metal coordination: C2 and C3 symmetrical assemblies through Cu(II) and ni(II) coordination. Inorg Chem 48(7):2726–2728
Quintanar L, Lim MH (2019) Metal ions and degenerative diseases. J Biol Inorg Chem 24(8):1137–1139
Zhao J et al (2019) Nitration of amyloid-beta peptide (1–42) as a protective mechanism for the amyloid-beta peptide (1–42) against copper ion toxicity. J Inorg Biochem 190:15–23
Mattevi A (2022) How evolution dismantles and reassembles multienzyme complexes. Proc Natl Acad Sci USA, 119(1)
Zhang Y et al (2021) Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis 12(9):847
Banci L et al (2010) Affinity gradients drive copper to cellular destinations. Nature 465(7298):645–648
Barros MH, Johnson A, Tzagoloff A (2004) COX23, a homologue of COX17, is required for cytochrome oxidase assembly. J Biol Chem 279(30):31943–31947
Zhu SY et al (2023) COX17 restricts renal fibrosis development by maintaining mitochondrial copper homeostasis and restoring complex IV activity. Acta Pharmacol Sin 44(10):2091–2102
Maxfield AB, Heaton DN, Winge DR (2004) Cox17 is functional when tethered to the mitochondrial inner membrane. J Biol Chem 279(7):5072–5080
Mari M et al (2009) Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 11(11):2685–2700
Baker ZN, Cobine PA, Leary SC (2017) The mitochondrion: a central architect of copper homeostasis. Metallomics 9(11):1501–1512
Winge DR (2018) Filling the mitochondrial copper pool. J Biol Chem 293(6):1897–1898
Ohrvik H, Thiele DJ (2015) The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J Trace Elem Med Biol 31:178–182
Acknowledgements
Not applicable.
Funding
This study is supported by the National Natural Science Foundation of China (No 82170696), SHDC12022104, ITJ(ZD)2201 and 23Y11908900.
Author information
Authors and Affiliations
Contributions
Saiya Zhu conducted experiments, wrote and edited the manuscript. Yangyang Niu conducted experiments while revising the article and reviewed the manuscript. Wenqian Zhou and Yuqing Liu helped conduct animal experiments. Jing Liu and Xi Liu designed the figures. Chen Yu and Limin Lu designed and supervised the study.
Corresponding authors
Ethics declarations
Ethical approval
The authors declared that they have no conflicts of interest related to this work.
Consent for publication
The authors consent to publishing this work.
Competing interests
The authors declare no potential competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, S., Niu, Y., Zhou, W. et al. Mitochondrial copper overload promotes renal fibrosis via inhibiting pyruvate dehydrogenase activity. Cell. Mol. Life Sci. 81, 340 (2024). https://doi.org/10.1007/s00018-024-05358-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05358-1