
Abstract
Background
Acetaminophen (APAP) overdose is a significant contributor to drug-induced liver injury worldwide. G-protein–coupled receptor 116 (GPR116) is an important homeostatic maintenance molecule in the body, but little is known about its role in APAP-induced liver injury (AILI).
Methods
GPR116 expression was determined in both human and mouse AILI models. Hepatic function and damage response were analyzed in hepatocyte-specific GPR116 deletion (GPR116△HC) mice undergoing APAP challenge. RNA-sequencing, immunofluorescence confocal, and co-immunoprecipitation (CO-IP) were employed to elucidate the impact and underlying mechanisms of GPR116 in AILI.
Results
Intrahepatic GPR116 was upregulated in human and mice with AILI. GPR116△HC mice were vulnerable to AILI compared to wild-type mice. Overexpression of GPR116 effectively mitigated AILI in wild-type mice and counteracted the heightened susceptibility of GPR116△HC mice to APAP. Mechanistically, GPR116 inhibits the binding immunoglobulin protein (BiP), a critical regulator of ER function, through its interaction with β-arrestin1, thereby mitigating ER stress during the early stage of AILI. Additionally, the activation of GPR116 by ligand FNDC4 has been shown to confer a protective effect against early hepatotoxicity caused by APAP in murine model.
Conclusions
Upregulation of GPR116 on hepatocytes inhibits ER stress by binding to β-arrestin1, protecting mice from APAP-induced hepatotoxicity. GPR116 may serve as a promising therapeutic target for AILI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug-induced liver injury is a common cause of acute liver failure and frequently leads to the withdrawal of drugs from the market. Acetaminophen (APAP) is a widely used antipyretic and analgesic. While generally safe at recommended doses, APAP can be toxic to the liver at high doses, leading to liver injury and failure [1]. In fact, APAP overdose is currently the leading cause of ALF in the United States, resulting in approximately 30,000 hospitalizations each year [2]. The development of strategies for the treatment of APAP-induced liver injury (AILI) has been extensive in recent years, but pharmacological options remain limited, providing an avenue for discovering new therapeutic targets.
In hepatocytes, APAP is converted by cytochrome P450 enzyme 2E1 (CYP2E1) to a toxic intermediate, N-acetyl-p-benzoquinone imine (NAPQI), which rapidly consumes glutathione (GSH) and binds to intracellular proteins to induce a series of signaling events, including endoplasmic reticulum (ER) stress [3,4,5,6,7]. ER stress has been shown to be closely associated with different stages of liver injury, and the death of hepatocytes triggered by ER stress may be crucial in drug-induced liver injury [8]. In the early stage of APAP hepatotoxicity, ER stress can be initiated when the influx of unfolded or misfolded proteins exceeds the folding capacity of the ER, triggering the terminal unfolded protein response (UPR) pathway, potentially leading to hepatocyte cell necrosis if excessively severe or prolonged [9]. However, the endogenous regulating mechanism responsible for alleviating ER stress in AILI remains largely unknown.
Adhesion G protein-coupled receptors (aGPCRs), the second largest GPCR class with exceptionally long ectodomains, are highly amenable to modulation by pharmaceuticals [10]. A wide variety of diseases have been linked to aGPCRs, which are involved in many physiological processes throughout the body [11]. Multiple aGPCRs are expressed in hepatocytes, mainly regulating fatty liver disease and diabetes [12]. G protein-coupled receptor 116 (GPR116), an aGPCR involved in tumorigenesis and cancer progression, is an important predictor of prognosis in cancers and is emerging as a promising therapeutic target [13,14,15,16]. Expressed in a variety of tissues, GPR116 plays a crucial role in regulating pulmonary surfactant homeostasis [17, 18], maintaining vascular endothelial junction integrity [19, 20], regulating kidney urine acid-base balance [21], and mediating insulin-sensitizing in systemic glucose homeostasis [22]. Despite our growing understanding of the biological roles of GPR116 in the lungs and other tissues, its function in the AILI remains unclear.
This study utilized hepatocyte-specific GPR116 knockout mice to establish an AILI model in order to investigate the specific role of GPR116 in AILI, elucidate its underlying mechanism, and identify a new intervention target for the treatment of AILI.
Materials and methods
Human samples
The human liver samples were obtained from patients with acute liver failure induced by APAP overdose who underwent liver transplantation (n = 5) in Renji Hospital of Shanghai Jiao Tong University (China), and healthy controls (HCs, n = 5) were obtained from liver donors. Liver tissues were collected during the transplantation procedure, and after fixation in formalin, embedded in paraffin for histological assessment. The informed consent was obtained from each subject. The study was carried out under the principles of the Declaration of Helsinki and approved by the research ethics boards of Renji Hospital. Demographic features of enrolled subjects are shown in Table 1.
Animal experiments
Male C57BL/6J mice (aged 8–12 weeks) were provided by the Experimental Animal Center of the Naval Military Medical University. Hepatocyte-specific GPR116 knockout (GPR116△HC) mice were generated by crossing GPR116flox/flox mice with Alb-Cre mice (B6.Cg- Speer6-ps1Tg(Alb-cre) 21Mgn/J, 003574) purchased from Jackson Laboratory as previously reported [23]. The above-mentioned mice were backcrossed with the C57BL/6J strain for nine generations, of which the healthy male knockout mice aged 8–12 weeks were used in the experiment. All knockout mice were constructed and identified by the Shanghai Model Organisms Center Inc. (Shanghai, China). All mice were housed in a specific pathogen-free environment at 23–25 °C and a 12/12 h light/dark cycle and were provided with water and chow ad libitum.
APAP (103-90-2, MCE, Monmouth Junction, NJ, USA) was dissolved in warm saline (0.9% NaCl; 55℃) and cooled to 37℃ before injection. To induce AILI, a single dose of 250 or 500 mg/kg body weight APAP was intraperitoneally (IP) injected in mice after overnight fasting (Fig. S1A) [24]. In ER stress inhibitor experiments, tauroursodeoxycholic acid (TUDCA; 14605-22-2, Selleck, Houston, TX, USA), dissolved in saline (250 mg/kg), was IP administered 3 times every 12 h along with APAP injection (Fig. S1B) [25]. In GPR116 ligand experiments, recombinant fibronectin type III domain containing 4 (FNDC4) (AG-40B-0124-C010, AdipoGen, Switzerland), dissolved in PBS (0.2 mg/kg), was IP administered 1 h after APAP injection (Fig. S1C) [22].
Adenovirus and peptides
The mouse GPR116 gene (https://www.uniprot.org/uniprotkb?query =NM001081178.1& view=%20cards) was cloned into H340-pIRES2-EGFP-3xFLAG to generate GPR116-FLAG, and vectors were generated and packaged by Obio Technology (Shanghai, China). For ADV experiments, mice were intravenously injected with 109 pfu of control (ADV-RAM) or GPR116-expressing (ADV-GPR116) ADVs 5 days before APAP treatment. GAP16-activating peptide was synthesized by GenScript Probio (Nanjing, China). Amino acid sequences for peptides: GAP16 (TSFSILMSPDSPDPGS), SCR (IFSDSTPSPDGLSMSP).
Analysis of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
Serum ALT and AST levels were measured using ALT (MAK052, Sigma-Aldrich, St. Louis, MO, USA) and AST colorimetric assay kits (MAK055, Sigma-Aldrich), respectively, according to the manufacturer’s instructions.
Western blotting and co-immunoprecipitation (CO-IP) analysis
For western blotting analysis, total protein was isolated from tissue or cell samples using RIPA lysis buffer (20–188, Sigma-Aldrich) with protease inhibitors. Samples were incubated for 10 min at 4 °C, vortexed, and centrifuged at 12 000 rpm for 10 min. Supernatants were collected and quantified for protein concentration using a BCA Protein Assay Kit (23,225, Thermo Fisher Scientific, Waltham, MA, USA). The protein samples were separated by 5–10% SDS-PAGE. Western blotting was performed as previously described, images were digitally captured using a ChemiScope 6 000 Exp (Clinx Science, Shanghai, China), and optical density was estimated using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The antibodies used in this study are listed in Table 2.
For CO-IP analysis, lysates that contain 500–1000 µg proteins were incubated with the primary antibody against GPR116, β-arrestin1 or IgG control (2 µg) overnight at 4 °C, followed by incubation with 25 µl of protein A/G magnetic beads. The beads were washed 5 times and eluted. The eluents were analyzed by western blotting.
Quantitative real-time PCR (Q-PCR)
Total mRNA was extracted using a rapid extraction kit (220,010; Fastegen, Shanghai, China) according to the manufacturer’s instructions. cDNA was synthesized from 6.5 µg of mRNA using a PrimeScript RT-PCR Kit (RR014A, Takara Bio, Maebashi, Japan). mRNA levels were quantified by Q-PCR using TB Green Premix Ex Tap (RR420A, Takara Bio). Gene expression was normalized to that of the housekeeping gene B2M according to the 2−ΔΔCt method. The primers used in this study are listed in Table 3.
Histology and immunostaining analysis
Liver tissues were fixed in 10% formalin for 24 h before processing. Section (5-µm thick) from paraffin-embedded liver tissue were subjected to hematoxylin and eosin (H&E) staining, histological analysis, and various other types of staining. A standard H&E staining protocol was used. Sectioned slides were immunohistochemically stained for GPR116 and CHOP using previously described techniques [7, 14]. TdT-mediated dUTP nick-end labelling (TUNEL) was conducted using a fluorescein TUNEL Cell Apoptosis Detection Kit (G1501-50T, Servicebio, Wuhan, China) according to the manufacturer’s instructions. TUNEL and H&E staining signals were quantified using ImageJ software. The number of TUNEL+ cells was quantified in 10 random fields in each slide collected from per animal. Immunofluorescence (IF) staining was performed as described previously [26]. The cells were observed using a laser scanning confocal microscope (Leica, Mannheim, Germany).
Cell isolation and treatment
Primary mouse hepatocytes were isolated from 8 weeks old mice as described previously [27], and were cultured in Williams’ medium E with 10% FBS without other supplements for 2 h to allow for attachment. AML-12 cells were cultured in DMEM/F-12 medium with 1% penicillin and streptomycin, 1% ITS (insulin-selenium-transferrin) and 10% FBS. AML-12 cells were transfected with control or GPR116-overexpressing (ov-GPR116) plasmid and incubated for 24 h. Cultured cells were incubated at 37 ℃ with 5% CO2. For APAP treatment, cells were treated in 12-well plates with APAP (10 mM) diluted in DMSO. For tunicamycin treatment, primary mouse hepatocytes were treated in 12-well plates with tunicamycin (10 µg/ml) diluted in DMSO.
GSH levels
GSH levels were determined by using a GSH ELISA Kit (E-EL-0026c, Elabscience, Wuhan, China). One hundred milligrams of liver tissue from each mouse were taken and homogenized together with the lysis buffer from the kit. After centrifugation at 12 000×g for 10 min, the supernatant was collected, and the total concentration of GSH was measured according to the manufacturer’s instructions.
CCK-8 assay
CCK-8 assay was carried out for detection of cell viability in AML-12 cells according to the manufacturer’s instructions. AML-12 cells were inoculated in 96-well plates with 2 × 103 cells per well. Absorbance at 450 nm was recorded at specified time points using the CCK-8 kit, based on which the viability curve was plotted.
APAP-cysteine protein adducts
APAP-Cysteine protein adducts were determined as previously described [28]. Briefly, liver tissues were homogenized and dialyzed to remove the free APAP-cysteine, then digested with protease, and the amount of APAP-cysteine was measured using high performance liquid chromatography-tandem mass spectrometry. Hepatic H2O2 levels were accessed using the Peroxidde Assay kit (Sigma-Aldrich, MAK311) according to the manufacturer’s protocol.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 9.0; GraphPad Software, San Diego, CA, USA). Data were analyzed using two-tailed Student’s t test or one-way ANOVA followed by post hoc t tests. The results of survival experiments were analyzed using a Log-Rank test. Data are presented as the mean ± standard deviation (SD). Statistical significance was set at P < 0.05.
Results
Hepatic GPR116 expression is upregulated in human and mouse AILI
To explore the potential involvement of GPR116 in AILI, we assessed the hepatic expression of GPR116 in both patients and mice with AILI. Immunohistochemistry (IHC) analysis revealed a significant increase in GPR116 expression in liver tissues from AILI patients compared to healthy controls (Fig. 1A). Additionally, protein expression of GPR116 (Fig. 1B) was found to be elevated in the livers of AILI mice. IHC and immunofluorescence (IF) analyses further confirmed the upregulation of GPR116 in parenchymal cells (hepatocytes) of AILI mice (Fig. 1C, D). Moreover, IF analysis revealed that GPR116 did not exhibit an increased expression in non-parenchymal cells, including macrophages (F4/80), neutrophils (myeloperoxidase, MPO), and hepatic stellate cells (α-smooth muscle actin, α-SMA) following APAP challenge (Fig. S1D-F). These findings collectively suggest a potential involvement of hepatocyte GPR116 in the pathogenesis of AILI.

Hepatic expression of GPR116 is increased in human and mouse AILI. (A) Representative immunohistochemical (IHC) staining and quantification of GPR116 in livers of healthy controls and patients with AILI (n = 5 per group, scale bar: 200 μm). (B-D) C57BL/6J mice were injected with either APAP (250 mg/kg, IP) or an equal volume of saline (IP) as a vehicle control, and the livers of mice were harvested at different timepoints (n = 6 per group). (B) The protein level of GPR116 in the liver tissue homogenates was determined by western blotting and relative GPR116 level compared with β-Actin were shown as line graphs. (C) Representative IHC staining and quantification of GPR116 in liver tissues of mice treated with APAP for 4 h (scale bar: 200 μm). (D) Representative immunofluorescence (IF) staining of GPR116 (red) in liver tissues of mice treated with APAP for 4 h. Shown on the right are the quantifications of relative GPR116 signals. (scale bar: 100 μm). Statistical analysis was performed by Student’s t-test. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. The experiments were repeated three times independently with similar results, and the data of one representative experiment was shown
Hepatocyte-specific knockout of GPR116 exacerbates AILI in mice
In order to examine the functional significance of hepatocyte GPR116 in AILI, we created GPR116 hepatocyte-specific knockout (GPR116△HC) mice, as outlined in a previous publication [23]. The efficacy of GPR116 knockout at the hepatic protein level was validated through western blot analysis, with no discernible knockout observed in the lung, kidney, or heart tissues (Fig. S2A). The GPR116△HC mice exhibited normal health and fertility under basal conditions, displaying no overt phenotypic abnormalities in terms of gross organ appearance or weight (Fig. S2B, C). In comparison to GPR116Hep+/+ (wildtype, WT) mice, the absence of GPR116 exacerbated the elevation of serum ALT and AST levels following APAP administration in a time-dependent manner (Fig. 2A). Additionally, GPR116△HC mice displayed increased levels of liver necrosis as determined by histological analysis (Fig. 2B) and TUNEL staining (Fig. 2C). Furthermore, GPR116△HC mice exhibited a significantly higher mortality rate compared to WT mice when administered a lethal dose of APAP (Fig. 2D). Together, these findings suggest that GPR116 may play a protective role in the context of AILI.

Hepatocyte-specific knockout of GPR116 exacerbates AILI. (A-C) WT and GPR116△HC mice were injected with either APAP (250 mg/kg, IP) or an equal volume of saline (IP) as a vehicle control (n = 6–12 per group). (A) Serum levels of ALT and AST in mice measured at different timepoints. (B) Left, representative H&E staining (scale bar: 200 μm) of liver tissues from mice harvested at 24 h post-APAP injection; right, quantification of necrotic areas by ImageJ software (n = 3 per group). (C) Left, representative TUNEL staining (scale bar: 50 μm) of liver tissues from mice harvested at 24 h post-APAP injection; right, quantification of the number of TUNEL-positive cells per field by ImageJ software (n = 3 per group). (D) The survival rate of WT (n = 11) and GPR116△HC (n = 12) mice for 72 h after injection with a lethal dose of APAP (500 mg/kg, IP). Statistical analysis was performed by Student’s t-test and one-way ANOVA. The survival rates were analyzed by the Log-Rank test. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. Data are pooled from at least two independent experiments
Hepatocyte-specific knockout of GPR116 aggravates ER stress in APAP-challenged mice
We further conducted a detailed investigation into the intrahepatic signaling pathways of GPR116 in the context of AILI. Our findings suggested that GPR116 did not play a significant role in APAP metabolism, as evidenced by no significant difference in APAP-Cysteine protein adducts, glutathione (GSH) consumption, and CYP2E1 expression observed in GPR116△HC and wild-type (WT) mice following APAP overdose (Fig. 3A, Fig. S3A, B).

Hepatocyte-specific knockout of GPR116 aggravates APAP-induced ER stress. (A-F) WT and GPR116△HC mice were treated with either APAP (250 mg/kg, IP) or an equal volume of saline (IP) as a vehicle control (n = 4–8 per group) for 4 h. (A) The APAP-CYS level, CYP2E1 protein level and GSH level in the liver tissue homogenates of mice. (B) KEGG analysis showed the top 20 enriched pathways in the GPR116△HC group versus WT controls (n = 3 per group). (C) GO functional enrichment analysis of RNA-seq data. (D) Western blotting of ER stress markers in liver tissue homogenates of mice. (E) mRNA levels of ER stress markers in liver tissue homogenates of mice. (F) Representative IHC staining and quantification of CHOP in liver tissues of mice (scale bar: 200 μm). Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. Data of A, D-F are pooled from at least two independent experiments
Since GPR116 began to be upregulated significantly after 4 h APAP treatment, and there were already differences in ALT and AST levels of GPR116△HC and WT mice at this timepoint, liver tissues harvested from two groups of mice treated with APAP for 4 h were subjected to RNA-seq analysis. The results revealed 24 differentially expressed genes, of which 20 genes were upregulated following GPR116 deletion (Fig. S4A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that the interacting proteins related to the dominant genes were enriched in ER protein processing-related signaling pathways (Fig. 3B). Additionally, Gene Ontology (GO) enrichment analysis showed that GPR116 deficiency upregulated protein folding and unfolded protein binding (Fig. 3C). An overload of ER stress is associated with APAP hepatotoxicity and multiple molecules, including CHOP, play vital roles in AILI. GPR116 deletion markedly exacerbated the increase in ER stress markers at the protein (e.g., PERK-eIF2α-ATF4-CHOP, s-XBP1, and t-XBP1) and mRNA (e.g., PDI, PERK, ATF4, CHOP, ATF6, s-XBP1, and t-XBP1) levels in the livers of mice 4 h post APAP injection (Fig. 3D, E), indicating that different branches of the ER stress pathway are involved. IHC analysis showed that CHOP expression was increased in the livers of APAP-treated GPR116△HC mice compared with WT mice (Fig. 3F). In summary, the intolerance of APAP challenge exhibited by GPR116△HC mice may be originated from uncontrolled ER stress.
Targeting ER stress rescues GPR116 deletion-induced deterioration of APAP-induced hepatotoxicity
To determine the role of ER stress in GPR116 signaling following APAP challenge, in vivo rescue assays were performed (Fig. 4A). Treatment with TUDCA (an ER stress antagonist, Fig. S4B-D) resulted in a decrease in serum ALT and AST, as well as a reduction in liver tissue necrosis, in WT mice 24 h post APAP injection. Notably, TUDCA reversed the disease phenotype of GPR116△HC APAP mice effectively (Fig. 4B, C). Consistent with these findings, western blot and IHC staining demonstrated that TUDCA attenuated the expression of ER stress marker-CHOP in liver tissues of both GPR116△HC and WT mice (Fig. 4D, E). Consequently, it can be concluded that GPR116 acts as a protective factor against APAP-induced hepatotoxicity by impeding ER stress.

Targeting ER stress rescues GPR116 deletion-induced deterioration of APAP-induced hepatotoxicity. (A) Serum levels of ALT and AST in mice. (B) Representative H&E staining of liver tissues from mice (scale bar: 200 μm) and quantification of necrotic areas (n = 3 per group). (C) Western blotting of CHOP in liver tissue homogenates of mice and relative CHOP level compared with β-Actin were shown as line graphs. (D) Representative IHC staining and quantification of CHOP in liver tissues of mice (scale bar: 200 μm). Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; no significance (ns) P > 0.05 as indicated. Data are pooled from at least two independent experiments
Re-expression of GPR116 in mice alleviates APAP-induced hepatotoxicity
In order to ascertain the hepatoprotective role of GPR116 in vivo, we conducted an assessment on the impact of GPR116 overexpression on APAP-induced hepatotoxicity. C57BL/6J mice were administered a GPR116-expressing adenovirus (ADV-GPR116) or control virus (ADV-RAM) prior to APAP administration (Fig. 5A, Fig. S5A-C). It was worth noting that GPR116 overexpression provided protection against AILI in the mice, as evidenced by the reduction in serum ALT and AST levels (Fig. 5B), and the mitigation of pathological liver injury (Fig. 5C). Moreover, GPR116 overexpression also lowered the elevation of APAP-induced ER stress marker protein levels (Fig. 5D). Correspondingly, IHC staining revealed that GPR116 overexpression weakened CHOP expression in liver tissues (Fig. 5E). To provide additional evidence for the protective role of GPR116, we administered ADV-GPR116 to GPR116△HC mice. Consistent with our expectations, the overexpression of GPR116 mitigated the hepatotoxicity induced by APAP in GPR116△HC mice (Fig. 6A, B) and counteracted the elevated expression of CHOP resulting from GPR116 knockout (Fig. 6C, D). In summary, our findings establish GPR116 as a crucial hepatoprotective gene in the context of APAP-induced hepatotoxicity.

Re-expression of GPR116 alleviates APAP-induced hepatotoxicity. (A-E) C57BL/6J mice were injected with control (ADV-RAM) or GPR116-expressing (ADV-GPR116) adenovirus 5 days before APAP treatment (250 mg/kg, IP) (n = 9 per group). (A) The protein level of GPR116 in the liver tissue homogenates was determined by western blotting. (B) Serum levels of ALT and AST in mice 24 h after APAP injection. (C) Representative H&E staining of liver tissues from mice (scale bar: 200 μm) and quantification of necrotic areas 24 h after APAP injection (n = 3 per group). (D) Western blotting of ER stress markers in liver tissue homogenates of mice 4 h after APAP injection. (E) Representative IHC staining and quantification of CHOP in liver tissues of mice 4 h after APAP injection (scale bar: 200 μm). Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. Data are pooled from at least two independent experiments

Re-expression of GPR116 in GPR116△HC mice reverse exacerbation of APAP-induced hepatotoxicity. (A-E) WT and GPR116△HC mice were injected with control (ADV-RAM) or GPR116-expressing (ADV-GPR116) adenovirus 5 days before APAP treatment (250 mg/kg, IP) (n = 3–9 per group). (A) Serum levels of ALT and AST in mice 24 h after APAP injection. (B) Representative H&E staining of liver tissues from mice (scale bar: 200 μm) and quantification of necrotic areas 24 h after APAP injection (n = 3 per group). (C) Western blotting of CHOP in liver tissue homogenates of mice 4 h after APAP injection and relative CHOP level compared with β-Actin were shown as line graphs. (D) Representative IHC staining and quantification of CHOP in liver tissues of mice 4 h after APAP injection (scale bar: 200 μm). Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. Data are pooled from at least two independent experiments
GPR116 modulates ER stress in APAP-challenged mouse hepatocytes via β-arrestin1
We analyzed above RNA-seq data (Fig. 3) and found that knocking out GPR116 did not affect the expression of classical G protein pathway-related genes during AILI (Fig. 7A), indicating that GPR116 may influence AILI through non-classical pathways. Recent research has revealed the role of the β-arrestin1 non-classical pathway in downstream signaling of GPR116 [29]. Therefore, we first analyzed the correlation between GPR116 and β-arrestin1 and found a positive correlation (R = 0.46) between the expression of GPR116 and β-arrestin1 mRNA expression in human normal liver tissue sequencing data from the GTEx database (Fig. 7B). Subsequent double IF staining and CO-IP experiments confirmed an interaction between GPR116 and β-arrestin1 in primary mouse hepatocytes following APAP challenge (Fig. 7C, D). The roles of β-arrestin1 were determined in vitro using small-interfering RNA (siRNA), si-β-arrestin1, to knock down the expression of β-arrestin1 in GPR116 signaling in mouse hepatocytes, and hepatocyte toxicity of APAP was detected using cell counting kit-8 (CCK-8) assay. si-β-arrestin1 administration substantially diminished the protective effect induced by FNDC4, a ligand of GPR116, during APAP challenge (Fig. 7E), suggesting that GPR116 activation protects hepatocytes from APAP toxicity through β-arrestin1.
Studies have highlighted the inhibitory role of β-arrestin1 in ER stress, which may be achieved through negative regulation of BiP by β-arrestin1 [30]. BiP, a member of the heat shock protein 70 (Hsp70) family located in the ER, plays a crucial role in regulating ER functions [31]. Upon ER stress, BiP associates with unfolded or misfolded proteins in the ER, leading to the activation of PERK and IRE1α and ATF6 [32]. GO enrichment analysis revealed that GPR116 significantly affected chaperone/heat shock protein-related protein folding after APAP challenge (Fig. 3C, red arrow). Besides, we found that the level of BiP mRNA in APAP-stimulated GPR116△HC primary hepatocytes was significantly higher than that in WT mice (Fig. 7F). In contrast, in primary hepatocytes obtained from WT mice, si-β-arrestin1 reversed the FNDC4-induced decline in BiP and CHOP expression during the APAP challenge (Fig. 7G). In summary, GPR116 activation inhibits ER stress through the β-arrestin1-BiP signaling pathway in the APAP challenge.

GPR116 inhibit ER stress in APAP-challenged mouse hepatocytes via β-arrestin1. (A) Heatmap of the differentially expressed genes in the RNA-seq data. (B) Co-expression analysis of GPR116 and β-arrestin1 in normal livers based on Q-PCR data. The value of each gene represents the relative expression normalized to HEK293T cells. Correlation coefficients (R) and P values were calculated by Spearman’s correlation analysis. (C) Representative double IF staining images and quantification showing the colocalization of GPR116 (green) and β-arrestin1 (red) in mouse primary hepatocytes treated with 10 mM APAP for 2 h (scale bar: 50 μm). (D) CO-IP assays showed that GPR116 interacted with β-arrestin1. Immunoprecipitations were performed by using anti-GPR116 (left panel) and anti-β-arrestin1 (right panel). (E) Primary mouse hepatocytes were transfected with si-NC or si-β-arrestin1 for 24 h before and subjected to 30 min of pretreatment with FNDC4 (200 nM) or PBS before APAP treatment (10 mM, 2 h). Cell viability was assessed using CCK-8. (F) BiP mRNA expression in primary mouse hepatocytes treated with 10 mM APAP for 2 h. (G) Primary mouse hepatocytes were transfected with si-NC or si-β-arrestin1 for 24 h before and subjected to 30 min of pretreatment with FNDC4 (200 nM) or PBS before APAP treatment (10 mM, 2 h). Protein levels of β-arrestin1, BiP and CHOP. Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ns P > 0.05 as indicated. Data are pooled from three independent experiments
The therapeutic role of GPR116 activation in APAP-induced hepatotoxicity in mice
Considering the clinical significance of therapeutic administration in APAP overdose, previous reported synthetic peptides GAP16 and FNDC4 that activate GPR116 were administered in AILI. Notably, GAP16 did not alleviate hepatocellular toxicity of APAP in vitro or in vivo (Fig. S6A-C). Remarkably, our findings indicated that recombinant FNDC4 effectively alleviated APAP-induced hepatocellular toxicity in vitro (Fig. 8A). To investigate whether FNDC4 can alleviate AILI by activating GPR116 in vivo, we used ADV to overexpress GPR116 in GPR116△HC mice. Interestingly, FNDC4 reduced AILI in WT mice, but did not improve liver damage in GPR116△HC mice at 4 h post-APAP. More importantly, FNDC4 significantly alleviated liver injury in GPR116 re-expressing GPR116△HC mice (Fig. 8B, C). Consistent with these findings, FNDC4 attenuated the upregulation of CHOP protein levels in WT and GPR116 re-expressing GPR116△HC mice (Fig. 8D). However, 12 to 24 h post-APAP, FNDC4 administration failed to show protection in WT-APAP mice (Fig. S7A). The collective administration of GPR116 stimulation demonstrated protective effects against APAP in mice. Therefore, GPR116 holds promise as a valuable therapeutic approach for the management of AILI.

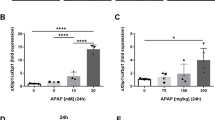
The therapeutic role of GPR116 activation in APAP-induced hepatotoxicity in mice. (A) Primary mouse hepatocytes were treated with FNDC4 (100/200/500 nM) or PBS 30 min after stimulated with APAP (10 mM). Cell viability was assessed using CCK-8 after 2 h (n = 3 independent experiments). (B-D) WT and GPR116△HC mice were injected with ADV-RAM or ADV-GPR116 5 days before and subjected to FNDC4 (0.2 mg/kg, IP) or vehicle (PBS) 1 h after APAP treatment (250 mg/kg, IP) (n = 3–4 per group). (B) Serum levels of ALT and AST in mice 4 h after APAP injection. (C) Representative H&E staining of liver tissues from mice (scale bar: 200 μm) and quantification of necrotic areas 4 h after APAP injection (n = 3 per group). (D) Western blotting of CHOP in liver tissue homogenates of mice 4 h after APAP injection. Statistical analysis was performed by one-way ANOVA. Data are expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 as indicated. Data are pooled from at least two independent experiments
Discussion
The aim of this research was to examine the intrinsic protective mechanisms of AILI and lay a rational groundwork for the advancement of novel therapeutic strategies. This investigation represents the initial documentation of the protective role of GPR116 in AILI progression. The upregulation of GPR116 in the liver was observed in both human subjects and murine models with AILI. The targeted deletion of GPR116 in hepatocytes notably exacerbated APAP-induced liver injury and mortality in mice. Mechanistically, GPR116 protected hepatocytes from APAP-induced toxicity by suppressing ER stress via the β-arrestin1-BiP signaling pathway.
The expression of GPR116 was increased by APAP in the early stages. Given the acute nature of APAP overdose and the observed effects of GPR116 activation in WT-APAP mice, it is possible that APAP-induced GPR116 expression serves as a protective feedback mechanism. GPR116 owns a variety of important physiological homeostasis maintenance effects [17, 21, 22, 29], which is consistent with our results. Prior research has demonstrated that GPR116 in alveolar macrophages and neutrophils has the ability to suppress exaggerated inflammatory reactions [18, 33]. However, our IF staining revealed a notable enrichment of GPR116 in hepatocytes compared to non-parenchymal cells. These findings serve as a compelling rationale for further investigation into the role of GPR116 in hepatocytes.
During AILI, the main target of NAPQI is mitochondrial proteins prompting mechanism studies on AILI to focus on mitochondrial oxidative stress and dysfunction [34]. N-acetylcysteine (NAC) has been recommended by the FDA as the only treatment option for patients with APAP overdose; however, the application of this drug is very limited due to its side effects and narrow treatment window [35]. Thus, there is a need to explore possible treatments for other cellular events. Various hepatic pathologies, including drug toxicity, have been established to be linked to ER stress [36]. Usually, activation of UPR is sufficient to handle transient and mild forms of ER stress [37]. Nonetheless, if ER stress is severe and unresolved, it can lead to persistent activation of the UPR signaling pathway, resulting in cell death and accelerated organ damage. ER stress plays a key role in the early stage of hepatotoxicity of APAP, making it crucial to study the mechanism of UPR activation and homeostasis maintenance during AILI. The activation of GPCRs expressed in various cell types has been found to regulate ER stress [38], although the relationship between GPR116 and ER stress has not been previously explored. Following stimulation with tunicamycin, the expression of GPR116 in primary mouse hepatocytes remained unaltered (Fig. S5C), indicating that GPR116 is upstream of ER stress. Our research discovered, for the first time, the negative regulatory effect of GPR116 on ER stress. This finding demonstrates the critical role of GPR116 in maintaining ER homeostasis and provides a theoretical basis for further exploring the role of GPR116 in other liver diseases.
Following stimulation with APAP, we noted an increase in β-arrestin1 expression in hepatocytes, which co-localized with the upregulated GPR116. The inhibition of ER stress by GPR116 was found to be dependent on β-arrestin1. β-arrestin1, a widely expressed intracellular regulator of GPCR trafficking and desensitization, has been recognized as an independent mediator of GPCR signaling [39]. Through interactions with various signaling proteins, β-arrestin1 facilitates the formation of signaling protein complexes and the activation of downstream kinase cascades [40]. It has been reported that β-arrestin1 can inhibit ER stress in intestinal stem cell proliferation induced by radiation [41]. The role of BiP as a crucial regulator of ER stress makes it a potential therapeutic target in various physiological and pathological processes [42, 43]. Particularly, precision treatment approaches that focus on specific disease states or cell types can benefit from the therapeutic potential of BiP [44]. Our findings supported that targeting the GPR116-β-arrestin1-BiP may present a novel therapeutic strategy for the management of AILI. CHOP is a key regulator of AILI. During AILI, CHOP upregulation may compromise hepatocyte survival via various mechanisms [7]. Accordingly, inhibiting CHOP could mitigate further injury to the liver following APAP toxicity. Our study demonstrated that GPR116 inhibited CHOP expression enhancement, resulting in decreased hepatocyte necrosis.
GPR116, an aGPCR, features a conserved GPS. Previous studies have demonstrated receptor activation by its corresponding tethered agonist GAP16 in vitro [45]. However, despite these findings, GAP16 failed to alleviate hepatocyte toxicity of APAP both in vivo and in vitro, which was unexpected. A functional analysis of the stachel sequences and derived peptides revealed agonist promiscuity both within and between aGPCR subfamilies [46]. Hence, potential functional overlap must be considered for in vitro and in vivo studies. Nevertheless, the functional antibody (anti-GPR116) targeting the extracellular part of GPR116 blocks a wider range of sites and effectively prevented the inhibitory effect of GPR116 on PERK-CHOP signal channel (Fig. S6E).
This study focused on investigating the role and mechanism of GPR116 during the early stage of AILI. The expression of GPR116 increased shortly after APAP stimulation, which is consistent with the occurrence of ER stress during the early phase of APAP-induced hepatotoxicity. In contrast, the mechanism of AILI during later stages is complex [47]. It involves processes such as inflammation, mitochondrial damage, and liver repair, which are not the focal point of this study. Our study found that GPR116 overexpression can alleviate the degree of liver injury in mice. However, the up-regulation of endogenous GPR116 expression in mice stimulated by APAP under physiological conditions did not significantly protect against liver injury. This is potentially due to that endogenous GPR116 expression was up-regulated by 2–3 times after APAP stimulation, while the ADV-induced GPR116 overexpression was up-regulated by up to 800 times. The significant difference in the effect caused by such a huge disparity in expression level was understandable.
FNDC4, a type of hepatokine, has been reported to exert anti-inflammatory effects on macrophages and osteoclasts, promoting survival in response to severe chronic inflammation [48, 49]. Furthermore, FNDC4 suppresses ER stress in adipocytes, which reduces insulin resistance caused by hyperlipidemia [50]. In white adipose tissue, GPR116 has been discovered to be a receptor for FNDC4. Upon direct binding of FNDC4 to GPR116, insulin signaling and insulin-mediated glucose uptake are promoted in white adipocytes [22]. Our study found that FNDC4 could not improve APAP-induced hepatocyte toxicity in mice lacking GPR116 specifically in hepatocytes, it did alleviate liver injury and ER stress to some extent in GPR116 re-expressing GPR116△HC mice, supporting the notion that FNDC4 primarily targets hepatocyte GPR116. Notably, FNDC4 administration exhibited initial liver protection, however, its efficacy diminished between 12 and 24 h post-APAP injection. This observation aligns with the role of GPR116 as an early protective response in AILI. The intricate nature of AILI during later stages, characterized by inflammation, mitochondrial damage, and liver repair processes, may contribute to the waning protective effects of FNDC4.
Extensive necrosis in cases of APAP overdose leads to sterile inflammation and the subsequent recruitment of inflammatory cells [51]. While our findings indicate a lack of significant alteration in the expression of GPR116 in macrophages, neutrophils, and hepatic stellate cells, further investigation is needed to elucidate the role of GPR116 in APAP-induced inflammation. Recent advancements in technology have enabled the direct dynamic observation of mouse liver microenvironments [52, 53]. By introducing fluorescently labeled cells, real-time monitoring within the live mouse liver can track the migration and distribution of inflammatory cells [54]. This technique will be utilized in our future studies to gain a more comprehensive understanding of the effects of GPR116 on the mouse liver, including the distribution of inflammatory cells, the progression of hepatic pathologies, and the evaluation of therapeutic effectiveness.
Conclusions
Our study has provided evidence in both mice and humans indicating that GPR116 serves as a crucial endogenous protective factor in AILI, protecting against hepatocyte necrosis and liver damage induced by excessive APAP through the inhibition of ER stress via the β-arrestin1-BiP signaling pathway. These findings have the potential to enhance our understanding of the mechanisms underlying AILI.
Data availability
The raw data that support the findings of this study are available from the corresponding author, Jinjun Bian upon reasonable request.
Abbreviations
- APAP:
-
Acetaminophen
- AILI:
-
APAP-induced liver injury
- aGPCRs:
-
Adhesion GPCRs
- GPR116:
-
G protein-coupled receptor 116
- ADV:
-
Adenoviruses
- ER:
-
Endoplasmic reticulum
- BiP:
-
Binding immunoglobulin protein
- FNDC4:
-
Fibronectin type III domain containing 4
- CYP2E1:
-
Cytochrome P 450 enzymes 2E1
- NAPQI:
-
N-acetyl-p-benzoquinone imine
- GSH:
-
Glutathione
- UPR:
-
Unfolded protein response
- IHC:
-
Immunohistochemistry
- IP:
-
Intraperitoneally
- TUDCA:
-
Tauroursodeoxycholic acid
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- Q-PCR:
-
Quantitative real time polymerase chain reaction
- H&E:
-
Hematoxylin and Eosin
- TUNEL:
-
TdT-mediated dUTP Nick-end labeling
- IF:
-
Immunofluorescence
- SD:
-
Standard deviation
- MPO:
-
Myeloperoxidase
- α-SMA:
-
Alpha-smooth muscle actin
- WT:
-
Wild type
- APAP-CYS:
-
APAP-cysteine protein adducts
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene ontology
- Hsp70:
-
Heat shock protein 70
- CO-IP:
-
Co-Immunoprecipitation
- ATF4:
-
Activating transcription factor 4
- ATF6:
-
Activating transcription factor 6
- PDI:
-
Protein disulfide isomerase
- XBP-1:
-
X-box binding protein-1
- CHOP:
-
C/EBP-homologous protein
- eIF2α:
-
Eukaryotic initiation factor 2α
- IRE1α:
-
Inositol-requiring enzyme 1α
- PERK:
-
Protein kinase R-like ER kinase
- CCK-8:
-
Cell counting kit-8
- NAC:
-
N-acetyl cysteine
- GPS:
-
GPCR proteolysis site
References
Lee WM (2013) Drug-induced acute liver failure. Clin Liver Dis 17(4):575–586 viii
Bunchorntavakul C, Reddy KR (2018) Acetaminophen (APAP or N-Acetyl-p-Aminophenol) and Acute Liver failure. Clin Liver Dis 22(2):325–346
Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ (1996) Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem 271(20):12063–12067
Chen C, Krausz KW, Idle JR, Gonzalez FJ (2008) Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem 283(8):4543–4559
Roberts DW, Pumford NR, Potter DW, Benson RW, Hinson JA (1987) A sensitive immunochemical assay for acetaminophen-protein adducts. J Pharmacol Exp Ther 241(2):527–533
Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA (2006) Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131(1):165–178
Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A et al (2013) CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol 59(3):495–503
Zhang J, Guo J, Yang N, Huang Y, Hu T, Rao C (2022) Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. ;13(12)
Wang X, Thomas B, Sachdeva R, Arterburn L, Frye L, Hatcher PG et al (2006) Mechanism of arylating quinone toxicity involving Michael adduct formation and induction of endoplasmic reticulum stress. Proc Natl Acad Sci U S A 103(10):3604–3609
Araç D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC et al (2012) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. Embo j 31(6):1364–1378
Purcell RH, Hall RA (2018) Adhesion G protein-coupled receptors as drug targets. Annu Rev Pharmacol Toxicol 58:429–449
Olaniru OE, Persaud SJ (2019) Adhesion G-protein coupled receptors: implications for metabolic function. Pharmacol Ther 198:123–134
Zheng T, Sun M, Liu L, Lan Y, Wang L, Lin F (2021) GPR116 overexpression correlates with poor prognosis in gastric cancer. Medicine 100(48):e28059
Tang X, Jin R, Qu G, Wang X, Li Z, Yuan Z et al (2013) GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Galphaq-p63RhoGEF-Rho GTPase pathway. Cancer Res 73(20):6206–6218
Wang C, Fan H, Zhang Y (2019) MiR-511-5p functions as a tumor suppressor and a predictive of prognosis in colorectal cancer by directly targeting GPR116. Eur Rev Med Pharmacol Sci 23(14):6119–6130
Muthiah I, Rajendran K, Dhanaraj P (2021) In silico molecular docking and physicochemical property studies on effective phytochemicals targeting GPR116 for breast cancer treatment. Mol Cell Biochem 476(2):883–896
Fukuzawa T, Ishida J, Kato A, Ichinose T, Ariestanti DM, Takahashi T et al (2013) Lung surfactant levels are regulated by Ig-Hepta/GPR116 by monitoring surfactant protein D. PLoS ONE 8(7):e69451
Ariestanti DM, Ando H, Hirose S, Nakamura N (2015) Targeted disruption of Ig-Hepta/Gpr116 causes Emphysema-like symptoms that are Associated with alveolar macrophage activation. J Biol Chem 290(17):11032–11040
Niaudet C, Petkova M, Jung B, Lu S, Laviña B, Offermanns S et al (2019) Adgrf5 contributes to patterning of the endothelial deep layer in retina. Angiogenesis 22(4):491–505
Niaudet C, Hofmann J, Mäe M, Jung B, Gaengel K, Vanlandewijck M et al (2015) Gpr116 receptor regulates distinctive functions in Pneumocytes and Vascular Endothelium. PLoS ONE 10(9):e0137949
Zaidman NA, Tomilin VN, Hassanzadeh Khayyat N, Damarla M, Tidmore J, Capen DE et al (2020) Adhesion-GPCR Gpr116 (ADGRF5) expression inhibits renal acid secretion. Proc Natl Acad Sci U S A 117(42):26470–26481
Georgiadi A, Lopez-Salazar V, Merahbi RE, Karikari RA, Ma X, Mourão A et al (2021) Orphan GPR116 mediates the insulin sensitizing effects of the hepatokine FNDC4 in adipose tissue. Nat Commun 12(1):2999
Wang Y, Wang T, Xiang Q, Li N, Wang J, Liu J et al (2023) GPR116 promotes ferroptosis in sepsis-induced liver injury by suppressing system Xc–/GSH/GPX4. Cell Biol Toxicol 39(6):3015–3030
Chen D, Ni HM, Wang L, Ma X, Yu J, Ding WX et al (2019) p53 Up-regulated modulator of apoptosis induction mediates Acetaminophen-Induced Necrosis and Liver Injury in mice. Hepatology 69(5):2164–2179
Torres S, Baulies A, Insausti-Urkia N, Alarcon-Vila C, Fucho R, Solsona-Vilarrasa E et al (2019) Endoplasmic reticulum stress-Induced Upregulation of STARD1 promotes Acetaminophen-Induced Acute Liver failure. Gastroenterology 157(2):552–568
Wang LX, Zhu XM, Luo YN, Wu Y, Dong N, Tong YL et al (2020) Sestrin2 protects dendritic cells against endoplasmic reticulum stress-related apoptosis induced by high mobility group box-1 protein. Cell Death Dis 11(2):125
Severgnini M, Sherman J, Sehgal A, Jayaprakash NK, Aubin J, Wang G et al (2012) A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology 64(2):187–195
Cheng J, Ma X, Krausz KW, Idle JR, Gonzalez FJ (2009) Rifampicin-activated human pregnane X receptor and CYP3A4 induction enhance Acetaminophen-Induced toxicity. Drug Metab Dispos 37(8):1611–1621
Sénéchal C, Fujita R, Jamet S, Maiga A, Dort J, Orfi Z et al (2022) The adhesion G-protein-coupled receptor Gpr116 is essential to maintain the skeletal muscle stem cell pool. Cell Rep 41(7):111645
Tan S, Li L, Chen T, Chen X, Tao L, Lin X et al (2015) β-Arrestin-1 protects against endoplasmic reticulum stress/p53-upregulated modulator of apoptosis-mediated apoptosis via repressing p-p65/inducible nitric oxide synthase in portal hypertensive gastropathy. Free Radic Biol Med 87:69–83
Pobre KFR, Poet GJ, Hendershot LM (2019) The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J Biol Chem 294(6):2098–2108
Hetz C, Zhang K, Kaufman RJ (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21(8):421–438
Wang T, Wang Y, Xiang Q, Lin S, Jin P, Wang J et al (2022) Deletion of G protein-coupled receptor 116 enhances neutrophil function and aggravates lung injury in mice. Genes & Diseases 10(4):1214–1216
Jaeschke H, McGill MR, Ramachandran A (2012) Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44(1):88–106
Du K, Ramachandran A, Jaeschke H (2016) Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol 10:148–156
Ajoolabady A, Kaplowitz N, Lebeaupin C, Kroemer G, Kaufman RJ, Malhi H et al (2023) Endoplasmic reticulum stress in liver diseases. Hepatology 77(2):619–639
Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B (2018) Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69(4):927–947
Maeyashiki C, Melhem H, Hering L, Baebler K, Cosin-Roger J, Schefer F et al (2020) Activation of pH-Sensing receptor OGR1 (GPR68) induces ER stress Via the IRE1α/JNK pathway in an intestinal epithelial cell model. Sci Rep 10(1):1438
Smith JS, Rajagopal S (2016) The β-Arrestins: multifunctional regulators of G protein-coupled receptors. J Biol Chem 291(17):8969–8977
Liu X, Zhao X, Zeng X, Bossers K, Swaab DF, Zhao J et al (2013) β-arrestin1 regulates γ-secretase complex assembly and modulates amyloid-β pathology. Cell Res 23(3):351–365
Liu Z, Jiang J, He Q, Liu Z, Yang Z, Xu J et al (2019) β-Arrestin1-mediated decrease in endoplasmic reticulum stress impairs intestinal stem cell proliferation following radiation. Faseb j 33(9):10165–10176
Kawaguchi Y, Hagiwara D, Miyata T, Hodai Y, Kurimoto J, Takagi H et al (2020) Endoplasmic reticulum chaperone BiP/GRP78 knockdown leads to autophagy and cell death of arginine vasopressin neurons in mice. Sci Rep 10(1):19730
Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU (2019) UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol 26(11):1053–1062
Li T, Fu J, Cheng J, Elfiky AA, Wei C, Fu J (2023) New progresses on cell surface protein HSPA5/BiP/GRP78 in cancers and COVID-19. Front Immunol 14:1166680
Bridges J, Safina C, Pirard B, Brown K, Filuta A, Panchanathan R et al (2022) Regulation of pulmonary surfactant by the adhesion GPCR GPR116/ADGRF5 requires a tethered agonist-mediated activation mechanism. eLife. ;11
Demberg LM, Winkler J, Wilde C, Simon KU, Schön J, Rothemund S et al (2017) Activation of adhesion G protein-coupled receptors: AGONIST SPECIFICITY OF STACHEL SEQUENCE-DERIVED PEPTIDES. J Biol Chem 292(11):4383–4394
Yan M, Huo Y, Yin S, Hu H (2018) Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol 17:274–283
Bosma M, Gerling M, Pasto J, Georgiadi A, Graham E, Shilkova O et al (2016) FNDC4 acts as an anti-inflammatory factor on macrophages and improves colitis in mice. Nat Commun 7:11314
Lv ZT, Liang S, Chen K, Zhang JM, Cheng P, Guo JC et al (2018) FNDC4 inhibits RANKL-Induced Osteoclast formation by suppressing NF-κB activation and CXCL10 expression. Biomed Res Int 2018:3936257
Lee W, Yun S, Choi GH, Jung TW (2018) Fibronectin type III domain containing 4 attenuates hyperlipidemia-induced insulin resistance via suppression of inflammation and ER stress through HO-1 expression in adipocytes. Biochem Biophys Res Commun 502(1):129–136
Xu L, Yang Y, Wen Y, Jeong JM, Emontzpohl C, Atkins CL et al (2022) Hepatic recruitment of eosinophils and their protective function during acute liver injury. J Hepatol 77(2):344–352
Lu Z, Liu Y, Jin M, Luo X, Yue H, Wang Z et al (2023) Virtual-scanning light-field microscopy for robust snapshot high-resolution volumetric imaging. Nat Methods 20(5):735–746
Lu Z, Cai Y, Nie Y, Yang Y, Wu J, Dai Q (2022) A practical guide to scanning light-field microscopy with digital adaptive optics. Nat Protoc 17(9):1953–1979
Chen S, Lu Z, Zhao Y, Xia L, Liu C, Zuo S et al (2024) Myeloid-Mas Signaling modulates pathogenic crosstalk among MYC(+)CD63(+) endothelial cells, MMP12(+) macrophages, and monocytes in Acetaminophen-Induced Liver Injury. Adv Sci (Weinh) 11(16):e2306066
Acknowledgements
Thanks to technician Kangqing Fang of our laboratory for her contributions.
Funding
This work was supported by grants from the National Natural Science Foundation of China (NO. 82272205, NO. 81771697) and the Clinical Medicine Plus X - Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (PKU2024LCXQ003).
Author information
Authors and Affiliations
Contributions
Qian Xiang, Na Li, Yan Zhang: Conceptualization, Methodology, Writing- Original draft preparation. Ting Wang, Ying Wang: Data curation, Software. Jinjun Bian: Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were reviewed, approved, and supervised by the Medical Ethics Committee of the Experimental Animal Center of the Naval Military Medical University (Shanghai, China, CHEC2021-021). These experiments were performed in strict compliance with the ARRIVE guidelines and were carried out in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, Q., Li, N., Zhang, Y. et al. GPR116 alleviates acetaminophen-induced liver injury in mice by inhibiting endoplasmic reticulum stress. Cell. Mol. Life Sci. 81, 299 (2024). https://doi.org/10.1007/s00018-024-05313-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05313-0