
Abstract
Mutations in cysteine and glycine-rich protein 3 (CSRP3)/muscle LIM protein (MLP), a key regulator of striated muscle function, have been linked to hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) in patients. However, the roles of CSRP3 in heart development and regeneration are not completely understood. In this study, we characterized a novel zebrafish gene-trap line, gSAIzGFFM218A, which harbors an insertion in the csrp3 genomic locus, heterozygous fish served as a csrp3 expression reporter line and homozygous fish served as a csrp3 mutant line. We discovered that csrp3 is specifically expressed in larval ventricular cardiomyocytes (CMs) and that csrp3 deficiency leads to excessive trabeculation, a common feature of CSRP3-related HCM and DCM. We further revealed that csrp3 expression increased in response to different cardiac injuries and was regulated by several signaling pathways vital for heart regeneration. Csrp3 deficiency impeded zebrafish heart regeneration by impairing CM dedifferentiation, hindering sarcomere reassembly, and reducing CM proliferation while aggravating apoptosis. Csrp3 overexpression promoted CM proliferation after injury and ameliorated the impairment of ventricle regeneration caused by pharmacological inhibition of multiple signaling pathways. Our study highlights the critical role of Csrp3 in both zebrafish heart development and regeneration, and provides a valuable animal model for further functional exploration that will shed light on the molecular pathogenesis of CSRP3-related human cardiac diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cysteine and glycine-rich protein 3 (CSRP3), also known as muscle LIM protein (MLP), expresses in both the myocardium and skeletal muscles and serves as a scaffold protein and a transcription cofactor important for the maintenance of normal muscle structure and function [1]. Evidence has highlighted the role of CSRP3 as a mechanosensor capable of converting extracellular mechanical stimuli into intracellular biochemical signals, and triggering downstream signals for myocyte growth and survival [2, 3]. Mutations in the CSRP3 gene have been linked to dilated cardiomyopathies (DCM) and hypertrophic cardiomyopathies (HCM) [4,5,6]. While considerable efforts have been undertaken to understand the underlying mechanisms, the role of CSRP3 in heart development and regeneration as well as in the etiology of relevant diseases still warrants further investigation.
Zebrafish are excellent model animals for studies of organ development and human diseases, and novel genetic tools are continuously being developed. Taking advantage of Tol2 transposable elements and Gal4-UAS systems, various gene-trap lines have been generated in which Gal4 is inserted into proper intron regions in the genome mediated by Tol2 transposase [7, 8]. When gene-trap lines were crossed with transgenic fish carrying fluorescent reporter genes downstream of UAS, the expression of the targeted gene can be visualized. Heterozygous fish can be used as reliable reporter lines for endogenous gene expression, whereas homozygous fish represent mutants of the targeted genes. Collections of gene-trap lines have proven to be valuable resources for studying gene function [9, 10].
In the present study, we characterized a novel zebrafish gene-trap line, gSAIzGFFM218A, which harbors an insertion in the csrp3 genomic locus and we further explored the role of Csrp3 in heart development and regeneration. We revealed that Csrp3 deficiency in zebrafish led to excessive trabeculation and impeded zebrafish ventricle regeneration. csrp3 expression was dynamic during heart development and regeneration, and was influenced by hemodynamic alteration, Notch signaling, and other pathways vital for heart regeneration. Csrp3 overexpression in zebrafish promoted CM proliferation after injury and heart regeneration. Our study highlights the critical role of Csrp3 in zebrafish heart development and regeneration, and provides a valuable animal model for further functional exploration which will facilitate the understanding of the molecular pathogenesis of CSRP3-related human cardiac diseases.
Materials and methods
Zebrafish husbandry
Zebrafish were raised and maintained under standard conditions. Zebrafish used in this study included wild-type AB strain, transgenic lines Tg(cmlc2:nDsRed), Tg(cmlc2:mCherry), Tg(flk:mCherry), Tg(vmhc:mCherry-NTR), Tg(tp1:d2GFP), Tg(cmlc2:Csrp3-EGFP), and gene-trap line Tg(gSAIzGFFM218A;UAS:GFP). To prevent pigmentation, embryos were incubated with 0.003% PTU (1-phenyl-2-thiourea) in E3 water from 24 hpf. All experiments were performed in accordance with institutional and national animal welfare guidelines.
Chemical treatment
To perform ventricular CM ablation, Tg(vmhc:mCherry-NTR) larvae were treated with 6 mM MTZ (metronidazole, Sigma-Aldrich) in E3 water for 4 h at 3 dpf as previously described [11]. To modulate signaling pathways, larvae were incubated with the following chemicals for the indicated time period: 100 μM DAPT (Sigma-Aldrich), 12 μM AG1478 (Sigma-Aldrich), 5 μM cardiomogen-1 (Sigma-Aldrich), 7.5 μM dorsomorphin (Sigma-Aldrich), 5 μM LDN193189 (Selleck), or 10 μM rapamycin (Cell Signaling Technology). To stop blood flow, larvae were treated with 1.8 mM tricaine (3-aminobenzoic acid ethyl ester, Sigma-Aldrich) or 10 mM BDM (2,3-butanedione monoxime, Sigma-Aldrich) in E3 water for the indicated time period, and then washed with fresh E3 water.
Cryoinjury of adult zebrafish heart
The ventricular cryoinjury was performed according to a previously established procedure [12]. Briefly, adult zebrafish at 6–12 months of age were anesthetized and placed ventral side up in a moistened sponge. Pericardial sac was exposed by removing surface scales and a small piece of skin with an incision. The apex of the ventricle was gently pulled up and further frozen with a precooled cryoprobe for 5 s. The fish were then placed back into a water tank, and water was puffed over the gills with a plastic pipette until they breathed and swam regularly.
Morpholino injection
Morpholino injections were performed as previously described [13]. The morpholino against tnnt2a (5′-CATGTTTGCTCTGATCTGACACGCA-3′) was purchased from Gene-tools, dissolved in nuclease-free water containing 10% phenol red, then injected into one-cell stage embryos. All injected embryos and larvae were used for relevant experiments at indicated stages.
Generation of Tg(cmlc2:Csrp3-EGFP) zebrafish
To generate Tg(cmlc2:Csrp3-EGFP) zebrafish, HiFi DNA Assembly kit (NEB) was used to simultaneously clone cmlc2 promoter, full length csrp3 CDS (without a stop codon) and EGFP fragments into pDESTol2pA2 destination vector. After sequence verification, DNA construct was co-injected with Tol2 transposase mRNA into embryos at the one-cell stage. Injected embryos were raised to adult, positive founders were outcrossed with wild-type fish to generate the stable transgenic line.
Immunofluorescence
For PCNA staining, adult zebrafish hearts were extracted and fixed in 4% paraformaldehyde for 1 h at room temperature, and subsequently dehydrated in 30% sucrose, then embedded in OCT solution and further sectioned at 10 μm thickness with a Leica cryostat. Immunostaining on cryosections was performed as previously described [14], administrated with antigen retrieval at 98 °C for 20 min in the citric acid buffer. CM proliferation index was calculated as the number of PCNA+Mef2C+ cells over the number of total Mef2C+ cells. Whole-mount larvae immunofluorescence was performed as previously described [15]. The primary and secondary antibodies used in this study were listed in Supplementary Table 2. Images were obtained using a Leica SP8 confocal microscope.
Histochemical staining
Injured and control zebrafish hearts were extracted, fixed, dehydrated, and embedded in paraffin and then sectioned at 5 μm thickness. Histochemical staining was performed on paraffin-embedded heart sections according to the manufacturer protocol of hematoxylin–eosin staining kit (Beyotime Biotech). Acid Fuchsin Orange-G (AFOG) staining for fibrin and fibrotic scar analyses was performed on 10 μm cryosections at 30 dpa as previously described [16]. Images were taken using an Olympus IX83 Microscope.
Western blotting
Larvae were homogenized in cold RIPA lysis buffer supplemented with complete proteinase inhibitors. Protein lysates were separated on 12% SDS-PAGE and transferred to PVDF membranes according to standard protocol [15]. The membranes were then incubated with anti-Csrp3 or anti-α-tubulin primary antibody, and further with HRP-conjugated anti-rabbit IgG or anti-mouse IgG secondary antibody (CWBio). Signals were visualized with Clarity Western ECL Substrate using a ChemiScope series system (Clinx).
in situ hybridization
Whole-mount in situ hybridization (WISH) was performed as previously described [11], using the following probes: csrp3, nkx2.5, hand2, tp53, fosl2, piezo1, ilk. Antisense riboprobes were synthesized in vitro using T7 RNA polymerase (NEB) with DIG RNA Labeling Mix. Transcription templates were amplified from cDNA using specific primers listed in Supplementary Table 1. The signal was detected using anti-digoxigenin-AP (Roche) and visualized by NBT/BCIP substrate (Roche). Images were taken by a Nikon SMZ18 stereo microscope. Fluorescence in situ hybridization (FISH) of csrp3 expression on cryosections was performed as previously described [17], signals were detected using anti-digoxigenin-POD (Roche) and visualized by TSA plus fluorescence kit (PerkinElmer), subsequently conducted immunofluorescence with Tnnt2 antibody as described above. FISH images were obtained using a Leica SP8 confocal microscope.
Quantification and statistical analysis
ImageJ software was used to measure the size of the scar area. Statistical tests and p-values are indicated in the figure legends. All statistical values were presented as mean ± SD and the statistical significance was defined as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 determined by Student’s t-test or the chi-squared test using SPSS and GraphPad Prism 9.0 software.
Results
gSAIzGFFM218A directs GFP expression in zebrafish hearts and harbors an insertion in the csrp3 genomic locus
gSAIzGFFM218A was generated through the Tol2 transposon-mediated gene-trap approach described previously that drives the expression of Gal4FF, an engineered Gal4 transcription activator in specific cell types [7, 18]. To visualize the expression of Gal4FF, gSAIzGFFM218A was mated with Tg(UAS:GFP) to obtain the double transgenic line Tg(gSAIzGFFM218A;UAS:GFP), referred to as 218A hereafter. Heterozygous 218A (+ /218A) fish exhibited GFP expression in the heart and gut and faintly in the somites (Figure S1A). Interestingly, the fluorescence pattern in the heart was restricted to the ventricle during early larval development and was noted as early as 2 days post-fertilization (dpf). By 10 dpf, the fluorescence displayed a dynamic mosaic pattern, gradually increased over time, and became widespread throughout the adult ventricle at 3 months post-fertilization (mpf), with scattered fluorescence seen in the adult atrium occasionally (Fig. 1A). To precisely determine the localization of GFP fluorescence in the ventricle of + /218A, we further bred this strain with transgenic lines Tg(cmlc2:mCherry) and Tg(flk:mCherry), in which the myocardium and endocardium are labeled, respectively. We also labeled the epicardium with an anti-pan-cytokeratin (Pck) antibody [19]. We observed that the GFP signal in + /218A larval hearts specifically overlapped with the myocardial marker, but not the endocardial or epicardial markers (Fig. 1B), which indicated that GFP expression in + /218A ventricle is specific to CMs.

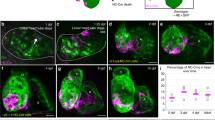
gSAIzGFFM218A directs GFP expression in zebrafish hearts and harbors an insertion in the csrp3 genomic locus. A Mosaic GFP expression in the hearts of heterozygous gSAIzGFFM218A (+ /218A) larvae and juveniles at indicated stages. Scale bars, 50 μm. B GFP in + /218A overlapped with the myocardial marker (cmlc2:mCherry reporter), but neither with the endocardial marker (flk:mCherry reporter) nor the epicardial marker (anti-Pck staining). Scale bars, 20 μm. C Schematic diagram illustrated the components of the gene-trap construct and its insertion in intron 3 of the csrp3 locus, which was predicted to result in a truncated csrp3 transcription. D Sequencing analysis of csrp3 cDNA from wild-type or homozygous 218A larvae. E RT-PCR analysis of csrp3 transcripts from wild-type, + /218A, and 218A larvae using specific primers indicated in C. M, molecular weight markers. F Western blotting revealed a dramatic reduction in Csrp3 protein level in 218A mutants. G Whole-mount in situ hybridization (WISH) showed the endogenous csrp3 expression in wild-type or 218A larval hearts. Scale bars, 50 μm
The transposon integration site in 218A was analyzed via Southern blotting and inverse PCR, and was mapped in intron 3 of the csrp3 genomic locus. This was predicted to result in truncated csrp3 transcription due to the SV40-poly A element (Fig. 1C). RT-PCR and sequencing analysis further confirmed the existence of hybrid transcripts consisting of truncated csrp3 fused with the insertion sequence (Fig. 1D) and the absence of wild-type full-length csrp3 transcripts in 218A homozygous fish, while + /218A heterozygous fish possessed both wild-type and hybrid csrp3 transcripts (Fig. 1E). Zebrafish csrp3 encodes a polypeptide of 193 amino acids that shares highly conserved sequence and functional domains with its human and mouse orthologs (Figure S2). Whole-mount in situ hybridization (WISH) analysis revealed that endogenous csrp3 was also specifically expressed in the zebrafish ventricle during early larval development (Fig. 1G), in line with the expression pattern of GFP in + /218A. Further investigation showed that wild-type csrp3 transcription was abolished in 218A homozygous fish, as validated by the lack of wild-type Csrp3 protein (Fig. 1F, G). These data suggested that gSAIzGFFM218A harbors an insertion in the csrp3 genomic locus and that heterozygous + /218A can be used as a reliable reporter line of endogenous csrp3 expression, while homozygous 218A serves as a csrp3 mutant line.
Csrp3 is involved in trabeculation, and its expression is regulated by hemodynamics and Notch signaling during heart development
The 218A larvae were visually indistinguishable in gross morphology from wild-type and heterozygous larvae and grew normally to adulthood with comparable fertility (Figure S1A). Given the cardiac expression of endogenous csrp3 and the + /218A GFP fluorescence, we further focused on the heart development of 218A fish. They displayed regular heart shape, similar heart rates and ventricular size compared to wild types (Figure S1B–H). Although Csrp3 is suggested to be involved in myofibril organization and the maintenance of the contractile apparatus [20], we did not observe apparent structural abnormalities by anti-Tnnt2 (CT3) immunostaining (Figure S1D). Instead, we observed excessive trabeculation in 218A larval and adult hearts. A slightly greater number of trabeculae appeared in the ventricle wall at 3 and 5 dpf when trabeculation was initiated. Later, a higher myofibril density of the trabeculae region was observed at 7 dpf and in adult hearts of 218A fish than in those of wild-type fish at the corresponding stages (Fig. 2A, B), indicating that Csrp3 deficiency caused excessive growth of trabeculae during heart development. Meanwhile, we also observed a marked reduction of N-cadherin (CDH2) on the surface of outer layer CMs in the 218A larval hearts (Figure S3), implying the impairment of cell–cell adhesion which may facilitate the delamination and growth of trabeculae.

Csrp3 is involved in trabeculation, and its expression is regulated by hemodynamics and Notch signaling during heart development. A Upper: visualization of trabeculae using Tg(vmhc:mCherry-NTR) displayed increased trabeculation in 218A larval hearts. Scale bars, 20 μm. Bottom: Quantification of the number or density of trabeculae in wild-type and 218A larval hearts at indicated stages. N = 9 each. Data are presented as mean ± SD, Student’s t-test, **, p < 0.01. B Upper: HE staining showed an increased myocardial density in 218A adult hearts. Scale bars, 100 μm. Bottom: Quantification of the myocardial density in wild-type and 218A adult hearts. N = 6 each. Data are presented as mean ± SD, Student’s t-test, ***, p < 0.001. C Comparison of the fluorescence pattern in the hearts of + /218A and Tg(tp1:d2GFP) larvae. Scale bars, 20 μm. D, E WISH and confocal images showed that inhibition of Notch signaling with DATP treatment enhanced both endogenous csrp3 expression in wild-type hearts (D) and GFP fluorescence in + /218A hearts (E). Scale bars, 20 μm. F Confocal and WISH images showed that reducing blood flow via tnnt2a MO injection markedly suppresses both GFP fluorescence in + /218A hearts and endogenous csrp3 expression in wild-type hearts. Scale bars, 20 μm
Previous studies have elucidated the critical role of Notch signaling in trabeculation [21, 22]. Interestingly, the dynamic mosaic pattern of GFP in + /218A hearts resembled the fluorescence pattern in the heart of Tg(tp1:d2GFP) (Fig. 2C), a reporter line expressing destabilized GFP in Notch-activated cells [21]. To determine whether Notch signaling acts as a regulator of csrp3 expression, we inhibited Notch signaling with the specific inhibitor DAPT from 60 to 72 h post-fertilization (hpf). The results showed that inhibition of Notch signaling enhanced both GFP fluorescence in + /218A hearts and endogenous csrp3 expression in wild-type hearts during larval stages (Fig. 2D, E).
Blood flow also plays a crucial role during trabeculation [23, 24]. We next examined the impact on csrp3 expression when hemodynamics was altered via tnnt2a morpholino (MO) injection at one-cell stage. Compared with control MO, injection of tnnt2a MO completely abolished the expression of both GFP in + /218A hearts and endogenous csrp3 in wild-type hearts (Fig. 2F). Since tnnt2a knockdown disturbed zebrafish cardiac development, we sought to further validate this observation by treating the larvae with two anesthetics, tricaine and 2,3-butanedione monoxime (BDM), for a short period to temporarily reduce blood flow. Consistently, we observed that both the GFP signal in + /218A hearts and endogenous csrp3 expression in wild-type hearts were markedly suppressed (Figure S4). Overall, these results suggested that Csrp3 is involved in zebrafish cardiac trabeculation, and that blood flow and Notch signaling play opposing roles in the regulation of csrp3 expression during heart development.
csrp3 expression increases in response to zebrafish heart injury
To investigate whether Csrp3 also plays a role in heart regeneration, we first examined its spatiotemporal expression profiles in various zebrafish heart injury models. We mated + /218A with Tg(vmhc:mCherry-NTR), a well-established ventricle ablation line [11], and treated with metronidazole (MTZ) to ablate ventricular CMs at 3 dpf. Instead of restricted ventricle expression in uninjured control hearts, the GFP signal was strongly upregulated in the atrium of ablated hearts from 1 day post-ablation (dpa) (Fig. 3A, upper panel). The GFP signal gradually expanded over the atrium and later increased in the ventricle from 2 to 4 dpa, and was restricted in the CMs in both chambers, as it overlapped with immunostaining for the myocardial marker MHC (MF20 antibody) (Fig. 3B). WISH analysis revealed a more dynamic endogenous csrp3 expression in wild-type ablated hearts during larval ventricle regeneration. The staining was first dramatically increased in the atrium at 1 dpa, and gradually extended to the ventricle at later stages (Fig. 3A, lower panel).

csrp3 expression increases in response to zebrafish heart injury. A The temporal expression profiles of GFP in + /218A hearts (upper) and endogenous csrp3 in wild-type hearts (bottom) after ventricle ablation. Scale bars, 20 μm. B Immunostaining showed the colocalization of GFP fluorescence with MF20-marked myocardium in + /218A larval hearts after ventricle ablation. Scale bar, 20 μm. C Confocal images illuminated a significant increase of GFP fluorescence in TnnT2(CT3)-marked myocardial cells at the border zone of the cryoinjured + /218A adult ventricle. Areas of dashed boxes are magnified. Scale bars, 100 μm. D Immunostaining revealed that GFP fluorescence of + /218A was specifically expressed in myocardial (Actn1+) cells, while absent in the endocardial (Flk+) and epicardial (Pck+) cells. Areas of dashed boxes are magnified. Scale bars, 100 μm. E Fluorescence in situ hybridization (FISH) analysis corroborated a robust upregulation of csrp3 expression at the border zone of cryoinjured ventricle, compared to the faint and dispersive signal in the uninjured adult ventricle. Scale bars, 100 μm
Additionally, we performed cryoinjury on the ventricle of adult + /218A fish at 6 months of age. We observed a significant increase in GFP fluorescence at the border zone of the injured area at 3 days post-cryoinjury (dpci), which became more extensive at 7 dpci (Fig. 3C). Fluorescence in situ hybridization (FISH) analysis corroborated a robust upregulation of csrp3 expression in the injury border zone of the ventricle, compared to the faint and dispersed csrp3 expression in the uninjured adult hearts (Fig. 3E). Colocalization of GFP with the myocardial marker Actn1 in the injured + /218A hearts further affirmed the myocardium-specific expression of csrp3, while GFP was absent in the endocardial and epicardial cells, marked by Flk and Pck, respectively (Fig. 3D). These findings indicated that csrp3 expression increased in response to heart injury in zebrafish, and may play a role in heart regeneration.
Csrp3 deficiency impedes zebrafish heart regeneration by reducing CM proliferation and enhancing apoptosis
After ventricle ablation the injured hearts regenerated through CM proliferation, dedifferentiation/transdifferentiation and migration, and full contractile function was restored at 4 dpa [13, 25]. However, we found that csrp3 deficiency notably impeded ventricle regeneration. Quantification of the heart regeneration ratio (number of recovered larvae over total injured larvae) revealed that the percentage of recovered larvae was significantly reduced to 49.7% (N = 201) in the ablated 218A group compared to 79.7% (N = 312) in the ablated wild-type group and 74.3% (N = 300) in the ablated + /218A group (Fig. 4A, B). We then assessed CM proliferation in 218A larvae using immunostaining of the mitotic marker phospho-histone H3 (pH3) and observed a significant decrease in the number of pH3-positive CMs in 218A ventricles compared to wild-type ventricles. The number of pH3-positive CMs was reduced from 6.2 ± 0.3 and 8.8 ± 0.3 per ventricle in wild type to 3.3 ± 0.3 and 3.5 ± 0.2 per ventricle in the 218A mutant at 1 and 2 dpa, respectively (Fig. 4C, D).

Csrp3 deficiency reduces CM proliferation and impedes zebrafish heart regeneration. A Representative images of the recovered/ unrecovered heart of 218A mutant larvae at 4 dpa/7 dpf. Scale bars, 20 μm. B Quantification of the regeneration ratio of ablated wild-type, + /218A, and 218A hearts at 4 dpa/7 dpf. The numbers of larvae analyzed are indicated. Binomial test, ****P < 0.0001, NS, non-significant. C Immunostaining of the mitotic marker phospho-histone H3 (pH3) revealed a significant decrease of proliferating CMs in 218A larval hearts at indicated stages. Scale bars, 20 μm. D Quantification of the number of pH3+ CMs in ablated wild-type and 218A hearts. N = 18 each. Data are presented as mean ± SD, Student’s t-test, ****, p < 0.0001. E Representative AFOG staining of cryoinjured ventricles from wild-type, + /218A, and 218A fish at 30 dpci. Scale bars, 100 μm. F Quantification of ventricular scar area ratio in wild-type, + /218A, and 218A fish at 30 dpci. N = 7, 6, 7, respectively. Student’s t-test, ***, p < 0.001, NS, non-significant. G Representative confocal images of cryoinjured ventricle sections from wild-type and 218A adult fish at 7 dpci stained with anti-PCNA (green) and anti-Mef2c (red) antibodies. Box areas are amplified. Arrowheads indicate proliferating CMs. Scale bars, 100 μm. H Quantification of CM proliferation index at border zone and injury site of ventricle sections at 7 dpci. N = 6 each. Student’s t-test, ***, p < 0.001
Furthermore, hearts of 218A adult fish at 6 mpf were subjected to ventricular cryoinjury and then assayed for fibrin and fibrotic scar tissue using acid fuchsin-orange G (AFOG) staining at 30 dpci. We observed that 218A hearts retained large unrecovered wounds with prominent fibrin or collagen deposits in comparison to minimal scar patches in injured wild-type and + /218A hearts accompanied by substantial new cardiac myofiber formation (Fig. 4E, F). We next evaluated injury-induced CM proliferation by conducting immunostaining of the DNA replication marker PCNA and CM marker Mef2c at 7 dpci. Notably, the number of PCNA-positive CMs was markedly decreased in 218A mutant hearts. Quantification revealed an approximately 56% (0.18 ± 0.03 to 0.09 ± 0.01) decrease in the CM proliferation index (PCNA+Mef2C+ cells/Mef2C+ cells) in 218A hearts compared to wild-type hearts (Fig. 4G, H).
In addition, the apoptosis of CMs after ventricle ablation was also assessed using TUNEL assays, which revealed that apoptotic signals were profoundly increased in 218A larval hearts, from 7.6 ± 2.3, 11.9 ± 2.7 and 11.3 ± 2.5 per wild-type ventricle to 21.2 ± 5.5, 40.4 ± 12.9 and 41.4 ± 9.9 per 218A larval ventricle at 5, 10 and 24 h post-ablation (hpa), respectively (Fig. 5A, B). WISH also revealed the upregulation of the apoptosis-related gene tp53 as well as the downregulation of antiapoptotic gene fosl2 (Fig. 5C, D). In general, these results elucidated the crucial role of Csrp3 in zebrafish heart regeneration, indicating that Csrp3 deficiency impeded ventricle regeneration by reducing CM proliferation and enhancing apoptosis.

Csrp3 deficiency results in elevated CM apoptosis. A TUNEL assay showed significantly increased apoptosis signals in the 218A larval hearts at the early stages after ablation. Scale bars, 20 μm. B Quantification of TUNEL-positive CMs in the ablated ventricle of wild-type and 218A larval at indicated stages. N = 10 each. Student’s t-test, ****, p < 0.0001. C, D WISH showed the upregulation of apoptosis-related gene tp53 and the downregulation of antiapoptotic gene fosl2 in 218A larval hearts. Scale bars, 50 μm
Csrp3 deficiency impairs injury-induced CM dedifferentiation and sarcomere reassembly
Sarcomere disassembly and re-expression of key early cardiac transcriptional regulators are essential processes for the dedifferentiation and subsequent proliferation of preexisting CMs during zebrafish heart regeneration [19, 26]. Immunostaining of cardiac troponin T (cTnT) showed that sarcomere disassembly of 218A CMs was not blocked at 1 dpa but was instead exacerbated during regeneration, characterized by disarrayed, and even collapsed arrangements. This in turn hindered sarcomere reassembly at later stages of regeneration (Fig. 6A). Meanwhile, we further examined the expression of several cardiogenic factors. In line with the effect of csrp3 deficiency on CM proliferation during regeneration, csrp3 deficiency sharply reduced the expression of nkx2.5 and hand2 in ablated 218A larval hearts (Fig. 6B). The level of embryonic CM marker Myh7 (stained with the N2.261 antibody) was also decreased in the border zone of injured 218A adult ventricle compared to that in the wild-type hearts (Fig. 6C). These results suggested that Csrp3 may have an important role in CM dedifferentiation and is indispensable for sarcomere reassembly during zebrafish heart regeneration.

Csrp3 deficiency impairs injury-induced CM dedifferentiation and sarcomere reassembly. A Immunostaining of cTnT indicated that sarcomere disassembly of 218A CMs was not blocked but instead exacerbated during regeneration while sarcomere reassembly was hindered. Scale bars, 20 μm. B WISH showed reduced expression of cardiogenic factors nkx2.5 and hand2 in ablated 218A larval hearts during regeneration. Scale bars, 50 μm. C Immunostaining showed reduced embryonic Myh7 signal in the border zone of injured 218A adult ventricle at 7 dpci. Scale bars, 100 μm
Previous studies demonstrated that Csrp3 played an essential role in the CM stretch sensor machinery [2], and was required for mechanical stability in skeletal muscles [27]. We recently showed mechanotransduction is critical for zebrafish heart regeneration [28]. Thus, we also assessed the expression of several genes involved in mechanotransduction process in ablated 218A larval hearts. We observed a significant downregulation of peizo1 and a slight upregulation of integrin-linked kinase (ilk) in the 218A ablated hearts at 2 dpa compared to wild-type ablated hearts (Figure S5), indicating a disturbance of mechanotransduction. These data implied that Csrp3 might also play a crucial role in mechanotransduction to facilitate zebrafish heart regeneration, while the exact mechanisms warrant further exploration.
Multiple signaling pathways regulate csrp3 expression in response to zebrafish larval heart ablation
Studies have shown that multiple signaling pathways are activated to orchestrate the regeneration of injured zebrafish hearts [29,30,31], including hemodynamics, Notch signaling, ErbB signaling, BMP signaling, etc. Pharmacological experiments were conducted to explore whether csrp3 expression responded to the suppression of these crucial signaling pathways which resulted in a failure of zebrafish heart regeneration (Figure S6A). Blood flow reduction by tricaine or inhibition of endocardial Notch activation by DAPT markedly blocked the appearance of GFP signal in the atrium of ablated + /218A hearts (Fig. 7A). WISH also revealed dismissed endogenous csrp3 expression in the atrium and reduced expression in the ventricle of ablated wild-type hearts treated with tricaine or DAPT (Fig. 7B). Pharmacological inhibition of ErbB signaling with AG1478 treatment induced a similar effect on + /218A GFP fluorescence and csrp3 expression following heart ablation (Fig. 7A, B), while inhibitors for Wnt signaling (cardiomogen-1), BMP signaling (dorsomorphin, LDN193189), and mTOR signaling (rapamycin) showed negligible impacts on GFP intensity in ablated + /218A hearts and endogenous csrp3 expression in ablated wild-type hearts (Figure S6B, C).

csrp3 overexpression relieves the inhibitory effects of multiple signaling blockage on zebrafish heart regeneration. A, B Comparative analysis of the expression changes of GFP in ablated + /218A hearts and endogenous csrp3 in ablated wild-type hearts after inhibiting blood flow (tricaine), Notch (DAPT), and ErbB (AG1478) signaling. Scale bars, 20 μm. C Fluorescence pattern of Tg(cmcl2:Csrp3-EGFP) at 4 dpf. Scale bars, 20 μm. D Confocal images showed that Csrp3 overexpression (OE) ameliorated the impairment of heart regeneration resulting from inhibiting blood flow (tricaine), Notch (DAPT), and ErbB (AG1478) signaling. Scale bars, 20 μm. E Quantification of the regeneration ratio of ablated wild-type and Csrp3 overexpressed larvae after treatment with indicated inhibitors at 4 dpa/7 dpf. N = 481, 548, 487, 411, 689, 524, 312, 328, respectively. Data are presented as mean ± SD, Student’s t-test, **P < 0. 01, ***P < 0.001, ****P < 0.0001
csrp3 overexpression relieves the inhibitory effects of multiple signaling blockage on zebrafish heart regeneration
To investigate the potential contribution of Csrp3 gain of function to zebrafish heart regeneration, we generated a stable transgenic line Tg(cmlc2:Csrp3-EGFP), that specifically overexpresses Csrp3 in CMs. Tg(cmlc2:Csrp3-EGFP) larvae exhibited normal gross morphology and intact heart structure with striated myofibers (Fig. 7C). After ventricle ablation, Csrp3 overexpression promoted CM proliferation (Figure S7) and further increased the heart regeneration ratio at 4 dpa from 78.5% (N = 481) to 90.7% (N = 548) (Fig. 7D, E). Intriguingly, Csrp3 overexpression partially ameliorated the impairment of heart regeneration resulting from the suppression of blood flow, Notch signaling, and ErbB signaling (Fig. 7D, E). The heart regeneration ratio at 4 dpa increased from 27.5% (N = 487), 41.6% (N = 689) and 18.7% (N = 312) in tricaine-treated, DAPT-treated and AG1478-treated ablated wild-type larvae to 57.7% (N = 411), 80.1% (N = 524) and 39.8% (N = 328) in ablated Tg(cmlc2: Csrp3-EGFP) larvae with corresponding inhibitor treatments. Csrp3 overexpression also reversed the altered expressions of mechanotransduction-related genes in 218A ablated hearts (Figure S5).
Discussion
In the current study, through analysis of the novel gene-trap line gSAIzGFFM218A, we revealed the dynamic patterns and essential roles of csrp3 in zebrafish heart development and regeneration. The 218A fish survive to adulthood with normal gross morphology, while the CSRP3/MLP-null mice exhibit severe heart failure leading to mortality [32]. This discrepancy may be partially attributed to the residual functions of truncated Csrp3 peptides in 218A fish. Besides, as a critical scaffold protein for maintaining cardiac muscle structure and function, the dysfunction of Csrp3 would likely have a more severe impact on mice hearts than on zebrafish hearts since mice have much higher heart rates and stronger cardiac contractions compared to zebrafish, which indicates that the mouse heart is subjected to greater mechanical forces and requires higher rigidity. Previously we showed by WISH that csrp3 expression was restricted in larval ventricle with a mosaic pattern [27]. Colocalization analysis with markers of different cardiac layers in the current study revealed that the GFP fluorescence of heterozygous + /218A larvae was restricted to the ventricular CMs, resembling the fluorescence pattern in the Notch signaling reporter line Tg(tp1:d2GFP) [21]. Interestingly, both GFP fluorescence in + /218A fish and endogenous csrp3 expression in wild-type fish changed in response to alterations in hemodynamic force and Notch signaling activity, two well-known factors regulating cardiac trabeculation [22, 33, 34]. Indeed, homozygous 218A fish with Csrp3 deficiency caused excessive growth of trabeculae during heart development, as evidenced by the increased myofibril density in the trabecular area, which may be partially attributed to the reduction and mislocalization of certain junctional proteins. We did not observe such phenotype in our previously reported csrp3 knockout fish [27], which may be due to the genetic compensation response triggered in many CRISPR knockout [35,36,37]. However, abnormal trabeculation was found in human patients with left ventricular non-compaction (LVNC) in which CSRP3 defect is believed to contribute to the etiology [38]. Taken together, our results indicated that csrp3 is involved in trabeculation during heart development, yet the exact molecular mechanism and its relationship with Notch signaling warrants further investigation.
The role of csrp3 in zebrafish heart regeneration has not been examined before. We observed a significant reduction of regeneration ratio after larval ventricle ablation or impeded regeneration with elevated scar tissue after adult cryoinjury in 218A fish compared to wild type. Csrp3 deficiency also blocked CM proliferation in both injury models. We further generated a myocardial-specific Csrp3 overexpression line and found that OE-Csrp3 increased CM proliferation and regeneration ratio after larval ventricle ablation. Previous studies have proven that autophagy is involved in zebrafish heart regeneration [39,40,41]. Through CSRP3 silence, autophagy impairment also resulted in increased apoptosis in both myoblasts and myotubes [42, 43]. We indeed observed elevated apoptosis after heart injury in 218A fish by TUNEL staining. WISH results showed increased expression of apoptosis-related gene tp53 and reduced expression of antiapoptotic gene fosl2 [44]. CMs spared from apoptosis will undergo dedifferentiation-proliferation-redifferentiation to replace the lost myocardium [45,46,47]. Disassembly and reassembly of sarcomere is a critical step of this process [26, 48]. We found in 218A fish, sarcomere disassembly was significantly aggravated at 1 dpa, which in turn hindered the sarcomere reassembly at later stages of regeneration. We also found altered expressions of mechanotransduction-related genes in ablated 218A hearts. Overall, our results revealed the essential roles of csrp3 in zebrafish heart regeneration.
Meanwhile, csrp3 displayed a dynamic expression pattern during heart regeneration. GFP signal in + /218A larvae was strongly upregulated in the atrium of ablated hearts, gradually expanded over the atrium, and later increased in the ventricle. Multiple signaling pathways orchestrated heart regeneration [30], and some of them were shown to regulate csrp3 expression in the current study. We found that csrp3 activation in the atrium was eliminated with the treatment of inhibitors to block hemodynamics, Notch, and ErbB signaling pathways, but remained no change with the treatment of inhibitors for Wnt, BMP, and mTOR signaling. Whether other players in mechanotransduction also regulate csrp3 activation remains to be explored. Furthermore, OE-Csrp3 ameliorated the impairment of heart regeneration resulting from the blockage of blood flow, Notch, and ErbB signaling pathways by pharmacological inhibitors. We previously showed lepb-linked enhancer sequence (LEN) was activated in the endocardium of ablated hearts and its activity was also regulated by hemodynamic forces and Notch signaling [31]. With the restricted myocardial expression during heart regeneration and dynamic response to certain signaling pathways, + /218A would be a valuable addition to our toolsets for dissecting the epistasis and cross-talking of signaling networks during heart regeneration.
Data availability
All data supporting the findings of this study are available within the article and the Supplementary Materials.
References
Vafiadaki E, Arvanitis DA, Sanoudou D (2015) Muscle LIM protein: master regulator of cardiac and skeletal muscle functions. Gene 566(1):1–7
Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111(7):943–955
Rashid MM, Runci A, Polletta L, Carnevale I, Morgante E, Foglio E, Arcangeli T, Sansone L, Russo MA, Tafani M (2015) Muscle LIM protein/CSRP3: a mechanosensor with a role in autophagy. Cell Death Discov 1:15014
Geier C, Perrot A, Ozcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PF, Furst DO, Vornwald A, von Hodenberg E, Nurnberg P, Scheffold T, Dietz R, Osterziel KJ (2003) Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107(10):1390–1395
Riaz M, Park J, Sewanan LR, Ren Y, Schwan J, Das SK, Pomianowski PT, Huang Y, Ellis MW, Luo J, Liu J, Song L, Chen IP, Qiu C, Yazawa M, Tellides G, Hwa J, Young LH, Yang L, Marboe CC, Jacoby DL, Campbell SG, Qyang Y (2022) Muscle LIM protein force-sensing mediates Sarcomeric biomechanical signaling in human familial hypertrophic cardiomyopathy. Circulation 145(16):1238–1253
Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, Chrisco MA, Murphy RT, Lurie PR, Schwartz RJ, Elliott PM, Vatta M, McKenna W, Towbin JA, Bowles NE (2003) Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 80(1–2):207–215
Asakawa K, Kawakami K (2009) The Tol2-mediated Gal4-UAS method for gene and enhancer trapping in zebrafish. Methods 49(3):275–281
Abe G, Suster ML, Kawakami K (2011) Tol2-mediated transgenesis, gene trapping, enhancer trapping, and the Gal4-UAS system. Zebrafish Genetics Genom Inform 104:23–49
Brown EA, Kawakami K, Kucenas S (2022) A novel gene trap line for visualization and manipulation of erbb3b+ neural crest and glial cells in zebrafish. Dev Biol 482:114–123
Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, Urasaki A, Kishimoto Y, Hibi M, Kawakami K (2008) Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A 105(4):1255–1260
Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, Yelon D, Chi NC (2013) In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 498(7455):497–501
Gonzalez-Rosa JM, Mercader N (2012) Cryoinjury as a myocardial infarction model for the study of cardiac regeneration in the zebrafish. Nat Protoc 7(4):782–788
Li X, Lu Q, Peng Y, Geng F, Shao X, Zhou H, Cao Y, Zhang R (2020) Primary cilia mediate Klf2-dependant Notch activation in regenerating heart. Protein Cell 11(6):433–445
She P, Zhang H, Peng X, Sun J, Gao B, Zhou Y, Zhu X, Hu X, Lai KS, Wong J, Zhou B, Wang L, Zhong TP (2020) The Gridlock transcriptional repressor impedes vertebrate heart regeneration by restricting expression of lysine methyltransferase. Development 147(18):190678
Ma J, Shao X, Geng F, Liang S, Yu C, Zhang R (2022) Ercc2/Xpd deficiency results in failure of digestive organ growth in zebrafish with elevated nucleolar stress. iScience 25(9):104957
Liang S, Shi X, Yu C, Shao X, Zhou H, Li X, Chang C, Lai KS, Ma J, Zhang R (2020) Identification of novel candidate genes in heterotaxy syndrome patients with congenital heart diseases by whole exome sequencing. Biochim Biophys Acta Mol Basis Dis 1866(12):165906
Munch J, Grivas D, Gonzalez-Rajal A, Torregrosa-Carrion R, de la Pompa JL (2017) Notch signalling restricts inflammation and serpine1 expression in the dynamic endocardium of the regenerating zebrafish heart. Development 144(8):1425–1440
Kawakami K, Asakawa K, Hibi M, Itoh M, Muto A, Wada H (2016) Gal4 driver transgenic zebrafish: powerful tools to study developmental biology, organogenesis, and neuroscience. Adv Genet 95:65–87
Peng X, Lai KS, She P, Kang J, Wang T, Li G, Zhou Y, Sun J, Jin D, Xu X, Liao L, Liu J, Lee E, Poss KD, Zhong TP (2021) Induction of Wnt signaling antagonists and p21-activated kinase enhances cardiomyocyte proliferation during zebrafish heart regeneration. J Mol Cell Biol 13(1):41–58
Hoshijima M (2006) Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am J Physiol Heart Circ Physiol 290(4):H1313–H1325
Han P, Bloomekatz J, Ren J, Zhang R, Grinstein JD, Zhao L, Burns CG, Burns CE, Anderson RM, Chi NC (2016) Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature 534(7609):700–704
Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Perez-Pomares JM, de la Pompa JL (2007) Notch signaling is essential for ventricular chamber development. Dev Cell 12(3):415–429
Samsa LA, Givens C, Tzima E, Stainier DY, Qian L, Liu J (2015) Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 142(23):4080–4091
Rasouli SJ, Stainier DYR (2017) Regulation of cardiomyocyte behavior in zebrafish trabeculation by Neuregulin 2a signaling. Nat Commun 8:15281
Galvez-Santisteban M, Chen D, Zhang R, Serrano R, Nguyen C, Zhao L, Nerb L, Masutani EM, Vermot J, Burns CG, Burns CE, Del Alamo JC, Chi NC (2019) Hemodynamic-mediated endocardial signaling controls in vivo myocardial reprogramming. Elife 8:44816
Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC (2010) Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464(7288):606–609
Chang Y, Geng F, Hu Y, Ding Y, Zhang R (2019) Zebrafish cysteine and glycine-rich protein 3 is essential for mechanical stability in skeletal muscles. Biochem Biophys Res Commun 511(3):604–611
Yu C, Li X, Ma J, Liang S, Zhao Y, Li Q, Zhang R (2024) Spatiotemporal modulation of nitric oxide and Notch signaling by hemodynamic-responsive Trpv4 is essential for ventricle regeneration. Cell Mol Life Sci 81(1):60
D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E (2015) ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol 17(5):627–638
Gong R, Jiang Z, Zagidullin N, Liu T, Cai B (2021) Regulation of cardiomyocyte fate plasticity: a key strategy for cardiac regeneration. Signal Transduct Target Ther 6(1):31
Geng F, Ma J, Li X, Hu Z, Zhang R (2021) Hemodynamic forces regulate cardiac regeneration-responsive enhancer activity during ventricle regeneration. Int J Mol Sci 22(8):44816
Arber S, Hunter JJ, Ross J Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P (1997) MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88(3):393–403
Lee J, Vedula V, Baek KI, Chen J, Hsu JJ, Ding Y, Chang CC, Kang H, Small A, Fei P, Chuong CM, Li R, Demer L, Packard RRS, Marsden AL, Hsiai TK (2018) Spatial and temporal variations in hemodynamic forces initiate cardiac trabeculation. JCI Insight 3(13):96672
Del Monte-Nieto G, Ramialison M, Adam AAS, Wu B, Aharonov A, D’Uva G, Bourke LM, Pitulescu ME, Chen H, de la Pompa JL, Shou W, Adams RH, Harten SK, Tzahor E, Zhou B, Harvey RP (2018) Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 557(7705):439–445
Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, Stainier DY (2015) Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524(7564):230–233
Ma Z, Zhu P, Shi H, Guo L, Zhang Q, Chen Y, Chen S, Zhang Z, Peng J, Chen J (2019) PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568(7751):259–263
El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Gunther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, Fukuda R, Gerri C, Giraldez AJ, Stainier DYR (2019) Genetic compensation triggered by mutant mRNA degradation. Nature 568(7751):193–197
Sasse-Klaassen S, Probst S, Gerull B, Oechslin E, Nurnberg P, Heuser A, Jenni R, Hennies HC, Thierfelder L (2004) Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15. Circulation 109(22):2720–2723
Chávez MN, Morales RA, López-Crisosto C, Roa JC, Allende ML, Lavandero S (2020) Autophagy activation in zebrafish heart regeneration. Sci Rep 10(1):2191
Sciarretta S, Maejima Y, Zablocki D, Sadoshima J (2018) The role of autophagy in the heart. Annu Rev Physiol 80:1–26
Ye S, Zhao T, Zhang W, Tang Z, Gao C, Ma Z, Xiong JW, Peng J, Tan WQ, Chen J (2020) p53 isoform Δ113p53 promotes zebrafish heart regeneration by maintaining redox homeostasis. Cell Death Dis 11(7):568
Cui C, Han S, Tang S, He H, Shen X, Zhao J, Chen Y, Wei Y, Wang Y, Zhu Q, Li D, Yin AH (2020) The Autophagy regulatory molecule CSRP3 interacts with LC3 and protects against muscular dystrophy. Int J Mol Sci 21(3):749
Rashid MM, Runci A, Russo MA, Tafani M (2015) Muscle Lim Protein (MLP)/CSRP3 at the crossroad between mechanotransduction and autophagy. Cell Death Dis 6(10):e1940
Jahangiri L, Sharpe M, Novikov N, Gonzalez-Rosa JM, Borikova A, Nevis K, Paffett-Lugassy N, Zhao L, Adams M, Guner-Ataman B, Burns CE, Burns CG (2016) The AP-1 transcription factor component Fosl2 potentiates the rate of myocardial differentiation from the zebrafish second heart field. Development 143(1):113–122
Jopling C, Boue S, Belmonte JCI (2011) Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol 12(2):79–89
Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR, Zeng C (2017) Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 136(9):834–848
Zhang Y, Gago-Lopez N, Li N, Zhang Z, Alver N, Liu Y, Martinson AM, Mehri A, MacLellan WR (2019) Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discovery 5(1):30
Martin TG, Kirk JA (2020) Under construction: the dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J Mol Cell Cardiol 148:89–102
Acknowledgements
We thank Haitao Zhou and Hongbo Lv for zebrafish care, Drs. Kaa Seng Lai, Yabo Fang, and other lab members for technical support and in-depth discussion.
Funding
This study was supported by the National Key R&D Program of China grant (2020YFA0803900), NSFC grant (32170852), and the Fundamental Research Funds for the Central Universities (2042023kf0205) to R.Z. The collaboration was supported by a 2015 Collaborative Research (A1)8 grant from the National Institute of Genetics, Japan to R.Z and K.K, and the generation and maintenance of the transgenic resource was supported by the National BioResource Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and JSPS KAKENHI JP21H02463 to K.K.
Author information
Authors and Affiliations
Contributions
S.L. and R.Z. conceived and designed the project. S.L., Y.Z., Y.C., J.L., M.Z., P.G., Q.L., H.Y., and J.M. conducted the experiments and analyzed the data. K.K. provided key resources. S.L., H.Y., J.M., and R.Z. wrote and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Ethics statement
All experiments were performed according to institutional and national animal welfare guidelines.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, S., Zhou, Y., Chang, Y. et al. A novel gene-trap line reveals the dynamic patterns and essential roles of cysteine and glycine-rich protein 3 in zebrafish heart development and regeneration. Cell. Mol. Life Sci. 81, 158 (2024). https://doi.org/10.1007/s00018-024-05189-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05189-0